

Abstract
Purpose
The WEE1 tyrosine kinase regulates G2/M transition and maintains genomic stability, particularly in p53-deficient tumors which require DNA repair after genotoxic therapy. Thus, a need arises to exploit the role of WEE1 inhibition in head and neck squamous cell carcinoma (HNSCC) mostly driven by tumor-suppressor loss. This completed phase I clinical trial represents the first published clinical experience using the WEE1 inhibitor, AZD1775, with cisplatin and docetaxel.
Experimental-Design
We implemented an open-label phase I clinical trial using a 3+3 dose-escalation design for patients with Stage III/IVB HNSCC with borderline-resectable or unresectable disease, but who were candidates for definitive chemoradiation. AZD1775 was administered orally twice a day over 2.5 days on the first week, then in combination with cisplatin (25mg/m2) and docetaxel (35mg/m2) for three additional weeks. The primary outcome measure was adverse events to establish maximum-tolerated-dose (MTD). Secondary measures included response rates, pharmacokinetics (PK), pharmacodynamics, and genomic data.
Results
The MTD for AZD1775 was established at 150mg orally twice per day for 2.5 days. RECISTv1.1 responses were seen in 5 of 10 patients; histological adjustment revealed 3 additional responders. The only drug-limiting toxicity was Grade-3 diarrhea. The PK C8hr target of 240nM was achieved on Day 4 at all three doses tested. Pharmacodynamic analysis revealed a reduction in pY15-Cdk and increases in γH2AX, CC3 and RPA32/RPA2 were noted in responders vs. non-responders.
Conclusion
The triplet combination of AZD1775, cisplatin and docetaxel is safe and tolerable. Preliminary results show promising anti-tumor efficacy in advanced HNSCC, meriting further investigation at the recommended phase 2 dose.
Trial Registration
The trial registry name is ClinicalTrials.gov (www.clinicaltrials.gov); registration # NCT02508246
Keywords: AZD1775, WEE1, head and neck cancer, clinical trial, squamous cell carcinoma
INTRODUCTION
Platinum-based cytotoxic chemotherapy is a standard of care systemic treatment for advanced head and neck squamous cell cancers (HNSCC). Platinum agents induce DNA damage and cause transient cell cycle arrest in proliferating cells that leads to apoptosis in the setting of extensive DNA damage. Therefore, selectively inhibiting checkpoints in cancer cells can enhance the efficacy of DNA damaging agents. HNSCC are characterized by loss of cell cycle checkpoint regulation, with TP53 being one of the most common genetic alterations present in 63-72% of HNSCC patients1–3, and associated with metastatic spread and decreased overall survival4.
Data from our studies and others identified several G2/M checkpoint regulator genes, including WEE1, that are required for the survival of p53 mutant HNSCC cells, but not that of normal cells and furthermore treatment with AZD1775, a WEE1 inhibitor leads to specific killing of HNSCC cells5,6. We have observed that HPV+ tumors were similarly sensitive to AZD1775 suggesting that it is p53 deficiency (whether mutational or due to HPV E6 inactivation) which renders preferential sensitivity to WEE1 inhibition. In addition, we showed synergy with standard of care cisplatin genotoxic therapy in vivo. Thus, preclinical observations provide a strong rationale for exploiting the synthetic lethal interaction between loss of p53 function and WEE1 inhibition, which results in an override of the DNA damage-induced G2/M checkpoint and subsequent unrestrained CDK1-driven premature mitosis and mitotic catastrophe7,8. Furthermore, recent studies including our own also show that WEE1 inhibition can also disrupt progression of DNA replication and induce breakage of nascent DNA, i.e. cause replication stress via over activation of CDK2/cyclin A and cyclin E kinases in a p53-independent manner9–12. This mechanism is thought to underlie the cytotoxic effect of AZD1775 as a single agent13,14. Thus, there is strong rationale to test AZD1775 in tumors with p53 dysfunction as a chemosensitizing agent and in tumors with high genomic instability susceptible to AZD1775-induced replication stress.
Phase I dose-finding studies of AZD1775 as a single agent and in combination with chemotherapy have been completed with encouraging preliminary efficacy and tolerability15,16. In this phase I clinical trial, our objectives were to explore the safety and efficacy of AZD1775 in combination with weekly cisplatin and docetaxel given the neoadjuvant setting prior to curative intent treatment in patients with locally advanced HNSCC (NCT02508246). Correlative biomarker and genomic analysis of premature mitosis and replication stress were obtained.
PATIENTS AND METHODS
Patient Population
Patients with previously untreated, histologically confirmed HNSCC deemed borderline resectable Stage III up to Stage IVb (T1-3, N0-2, M0), or unresectable Stage IV with high nodal status defined as >N2b (by the AJCC 7th Edition Staging) but were amenable to curative treatment were eligible. Surgical unresectability was defined as the combination of the treating surgeon’s judgment of unresectability plus one of the following objective criteria: complete or 75% encasement of the carotid artery by the primary tumor or involved lymph nodes; involvement of prevertebral musculature; bony invasion of the skull base or spine; need for extensive resection where functional outcome was considered unacceptable to surgeon or patient. Borderline resectability status was adjudicated in our multi-disciplinary Tumor Board conference. Patients with locally advanced oropharyngeal squamous cell carcinoma (OPSCC) where a response to upfront AZD1775 and chemotherapy would open a window for organ-sparing surgery such as Trans Oral Robotic Surgery (TORS) were eligible. Patients were required to have an Eastern Cooperative Oncology Group performance status of 0-2, be able to swallow capsules, have no contraindications to docetaxel or cisplatin chemotherapy, and have adequate renal, hepatic and bone marrow function.
The study was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice guidelines, and was approved by relevant regulatory and independent ethics committees. All patients provided written informed consent before study entry.
Study Design
This was an open-label phase I trial of AZD1775 in combination with cisplatin and docetaxel given prior to curative intent therapy using a standard phase I 3+3 dose escalation design as shown in eFigure 1. The primary endpoint was to establish the safety profile and determine the maximal tolerated dose (MTD) of AZD1775. Secondary endpoint included efficacy in terms of response rates based on RECIST v1.1 criteria, successful conversion to surgical resection for patients with borderline resectable disease, pharmacokinetics and correlative pharmacodynamic biomarkers of response. All patients were required to undergo pre-therapy and three additional post-therapy biopsies – after AZD1775 alone; after the first cycle of cisplatin/docetaxel/AZD1775 and at the time of surgical excision.
Treatment and Safety Assessments
Treatment was administered in 28 day cycles. AZD1775 was given orally twice a day on the first week for 5 doses as a single agent lead-in assessment period. This was followed by AZD1775 at the same dose and frequency in combination with weekly cisplatin (25mg/m2) and docetaxel (35 mg/m2) for three consecutive weeks out of a four week cycle. Based on prior phase I published data, AZD1775 was started at a dose of 125 mg and escalated by 50 mg for each subsequent dose level. If at the starting dose of 125 mg there were no drug limiting toxicities (DLTs) and the dose level above at 175 mg exceeded the MTD with 2 or more toxicities, an intermediary dose level of 150 mg would be explored. Patients who demonstrated response and tolerance of this regimen were eligible for proceeding with another 4 week cycle of treatment.
Patients were evaluated by history, physical examinations, laboratory evaluations (CBC and serum chemistries) and toxicity assessments at baseline, after administration of the fifth dose of AZD1775 alone (during the lead-in week) and weekly prior to the administration of the combination regimen with cisplatin/docetaxel/AZD1775. Drug limiting toxicity (DLT) was defined as any adverse event judged by the Principal Investigator to be drug-related and Grade ≥ 3 according to the NCI Common Terminology Criteria for Adverse Events version 4.03 with the following exceptions: nausea, vomiting, diarrhea or hypersensitivity reactions were considered DLT only if persistent despite optimal medical management, neutropenia and thrombocytopenia were considered DLT only if associated with fever or bleeding respectively, and asymptomatic laboratory abnormalities were deemed DLT only if clinically significant.
Pharmacokinetic (PK) Analyses
We utilized a method applicable to the analysis of AZD1775 in human plasma treated with K2EDTA anticoagulant using liquid chromatography with tandem mass spectrometric detection as previously described17. Following the first administration of cisplatin and docetaxel, blood samples were collected at pre-dose (0), 1, 2, 4, 6 and 8 -10 hours after initial AZD1775 administration. An additional sample was drawn prior to the 3rd dose of AZD1775 administered on Day 2 and prior to the final 5th dose on Day 3, and at 1, 2, 4, 6 and 8-10 hours after the 5th dose.
Pharmacodynamic (PD) Analyses
PD biomarkers related to WEE1 inhibition and its associated pathways were analyzed in matched, pre- and post-treatment biopsies. Immunohistochemistry (IHC) and immunofluorescence (IF) assays were performed with the following antibodies: Inhibition of WEE1 was assessed by loss of inhibitory phosphate on pY15-cdc2 using pY15-cdc2 (clone 10A11 Cell Signaling Technology), which recognizes pY15-cdc2 and also pY15-Cdk2 based on sequence similarity18. Cdc2 (Santa Cruz Biotechnology) was used to measure total cdc2 levels. DNA damage and DNA damage response were measured using γH2AX (clone JBW301, EMD Millipore) and pS345-Chk1 (Thermofisher). Replication stress was measured with anti pS4/S8-RPA32 (Abcam). Premature mitosis was measured using pS10-HH3 (EMD Millipore). Lastly, apoptosis was measured with cleaved caspase 3 (Cell Signaling Technology). All sections were then incubated with Catalyzed Signal Amplification (CSA) system (Dako) followed by tertiary Alexa Fluor 647 substrate (Invitrogen) for IF and Leica Refine DAB reagents (Leica Biosystems). Isotype control slides were included for each run. Digital images of stained slides were obtained using whole-slide scanner Aperio ScanScope AT Turbo for IHC, and Aperio FL for IF (Leica Biosystems). Percent positivity was determined using HALO software (Indica Labs, Inc.). Differences in biopsies before and after treatment were tested for significance using Wilcoxon matched-pairs signed rank test (GraphPad PRISM).
Genomic Analyses
Clinically-validated UW-OncoPlex next-generation sequencing (NGS) testing of 262 cancer genes (http://tests.labmed.washington.edu/UW-OncoPlex) on DNA extracted from tumor tissue was performed as previously described19. Post-sequencing, data were de-multiplexed using standard bioinformatics tools. Alignment, variant calling, structural variant determination, copy-number analysis, microsatellite instability (MSI), and ethnicity determination were carried out using a clinically-validated, custom bioinformatics pipeline20,21. HPV-16- and HPV-18-specific sequences were included in the UW-Oncoplex capture design. Post-NGS, custom UNIX-scripts were used to identify unique sequences that contained HPV-16- and HPV-18-sequences. A sample was considered positive if greater than 100 unique HPV sequences were identified.
RESULTS
Patient population
Between December 2015 and January 2017, a total of 12 patients were enrolled in this study with selected demographic and clinical characteristics summarized in eTable 1. The mean age was 56 years (range 46-76). Four patients with p16+ oropharyngeal tumors were enrolled. All patients were evaluable for safety.
Safety
Overall, the combination of AZD1775 and weekly cisplatin and docetaxel was well tolerated. A list of the most common adverse events is shown in Table 1. The most common Grade ≥ 2 toxicities were diarrhea, fatigue and neutropenia. At dose level 2 (DL2) at 175 mg of AZD1775, DLT toxicity of Grade 3 diarrhea was observed in two patients. One of these patients who experienced Grade 3 diarrhea was downgraded promptly to Grade 2 with immodium and supportive treatment and did not meet DLT criteria initially; however, upon resuming therapy developed recurrent Grade 3 diarrhea which was deemed DLT. In these two patients, medical management with antidiarrheals, hydration and drug discontinuation resulted in eventual complete resolution of gastrointestinal toxicity. In general, the diarrhea worsened when AZD1775 was given in combination with chemotherapy. Other instances of Grade 3 diarrhea observed in this study resolved with supportive care and did not meet criteria for DLT. As there were no DLTs noted at 125 mg orally twice daily (DL1), the intermediary dose level at 150 mg was evaluated. No other DLTs were observed in this study. There were no grade 4 adverse events or deaths on study. Therefore, the MTD was established at 150mg orally twice per day for 2.5 days in this setting.
Table 1.
Summary of Adverse events
| Grade
|
|||||||
|---|---|---|---|---|---|---|---|
|
DL1 (125mg) (n=4) |
DL2 (175mg)(n=4) |
DL3 (150mg) (n=4) |
|||||
| Adverse Events | 2 | 3 | 2 | 3 | 2 | 3 | Total (%) |
| Gastrointestinal | |||||||
| Diarrhea¥ | 1 | – | 3 | 3 | 1 | 1 | 9 (75) |
| Nausea | 1 | – | 1 | – | 1 | – | 3 (25) |
| Vomiting | 1 | – | 1 | – | – | – | 2 (17) |
| Flatulence | 1 | – | – | – | – | – | 1 (0.1) |
| Constipation | – | – | – | – | 1 | – | 1 (0.1) |
|
| |||||||
| Hematologic | |||||||
| Neutropenia | – | 1 | 1 | 2 | – | – | 4 (33) |
| Anemia | – | – | 1 | – | – | – | 1 (0.1) |
|
| |||||||
| Metabolism and nutritional | |||||||
| Hypokalemia | – | 1 | – | 1 | – | – | 2 (17) |
| Anorexia | 1 | – | – | – | – | – | 1 (0.1) |
| hypocalcemia | – | – | – | – | 2 | 1 | 3 (25) |
| Hyperglycemia | – | – | – | – | – | 1 | 1 (0.1) |
|
| |||||||
| Skin and subcutaneous | |||||||
| Palmar-plantar erthrodysesthesia syndrome | – | – | – | – | 1 | – | 1 (0.1) |
| Maculopapular rash | 1 | – | – | – | – | – | 1 (0.1) |
|
| |||||||
| Other | |||||||
| Fatigue | – | – | 2 | – | 2 | – | 4 (33) |
| Weight loss | 1 | – | 1 | – | – | – | 2 (17) |
| Dehydration | – | – | 1 | – | – | – | 1 (0.1) |
| Urinary tract infection | 1 | – | 1 | – | 1 | – | 3 (25) |
| Depression | 1 | – | – | – | – | – | 1 (0.1) |
| Sinus pain | – | – | – | – | – | 1 | 1 (0.1) |
| Infusion related reaction | 1 | – | – | – | – | – | 1 (0.1) |
|
| |||||||
| 10 | 2 | 12 | 6 | 9 | 4 | 43 | |
Two patients experienced DLTs of Grade 3 diarrhea during treatment at DL2. One of these patients developed initial Grade 3 at this dose level which promptly resolved to Grade 2 with immodium, but upon resuming therapy subsequently developed recurrent Grade 3 diarrhea meeting DLT criteria.
Efficacy
Ten patients of the 12 patients were evaluable for response. One patient was unevaluable due to an allergic reaction to the first dose of docetaxel; another patient lost the ability to swallow the AZD1775 capsules during the lead-in week. Starting AZD1775 dose and treatment durations for each patient are detailed in eTable 2. Overall, the mean duration of treatment was 34 days. Four patients who completed the first 4 week cycle of therapy (patients #4, 8 and 11 with partial responses and patient #12 with stable disease), went on to receive an additional cycle of AZD1775 in combination with chemotherapy. Patient #8 only received 2 weeks of combination therapy in the subsequent cycle of therapy due to treatment delays from neutropenia, and general tolerability issues. The second cycle of combination therapy elicited additional reductions of 4.5 to 14 percentage points in tumor RECIST measurements with respect to response observed after the first cycle. Although patient #3 responded after cycle 1 without toxicities, a subsequent cycle was not given due to patient financial and transportation issues, patient preference of not delaying surgery and Investigator consideration of reserving full-dose cisplatin for adjuvant use concurrent with radiation after surgery.
Of the 10 evaluable patients, RECIST v1.1 partial responses (PR)s were observed in 5 patients and stable disease (SD) in 4 patients as shown in Figure 1A. One patient (#9) experienced progressive disease (PD) after week 2 of treatment and was started on definitive chemoradiation. The patient progressed and died shortly thereafter.
Figure 1. Clinical response after neoadjuvant therapy with AZD1775.
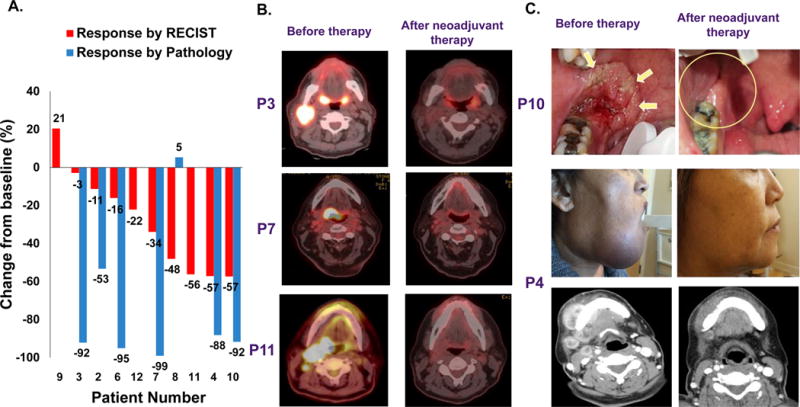
(A) Waterfall plot illustrating change of tumor volume during neoadjuvant therapy by RECIST 1.1 criteria and histological response for those who converted to surgery. (B) Selected PET and (C) clinical responses after neoadjuvant therapy.
Seven patients successfully converted to surgery. Patient #3 presented with a p16+ T2N3M0 of the right tonsil. After one cycle of therapy, this patient demonstrated a remarkable response by PET scan and histologically (no residual carcinoma in N3 neck lymph node and a 3 mm focus of tumor in the right tonsil with clear margins) (Fig. 1B). Of note, despite mild uptake by PET scan on the left tonsil biopsies were negative for malignancy. A complete pathologic response was observed at surgery. Interestingly, due to the post-operative findings, this patient refused adjuvant chemotherapy and only agreed to adjuvant radiation alone. However, after only one week of treatment, this patient discontinued treatment altogether due to local toxicity. The patient was taken to the operating room to evaluate for recurrent disease about 6 months from initiation of treatment, and had no evidence of disease. He continues to be followed closely with imaging and scope examinations and remains free of disease 1.5 years out (539 days). Patient #4 had a T4a/b extensive buccal/alveolar tumor with mandibular invasion. Due to an impressive response after the 4 week cycle at 125mg (DL1) (Fig. 1C), another 3 week cycle of cisplatin/docetaxel/AZD1775 was administered. At the time of surgery, no clinically detectable tumor was observed. Thus, surgery of the primary tumor was limited to a segmental mandibulectomy around the epicenter of the tumor with histology confirming only a 3 mm focus of disease. Two patients (# 6 and 8) with T4 tongue tumors had a hemiglossectomy rather than the total or subtotal glossectomy which would have been required prior to neoadjuvant treatment. One of these, patient #6, had a complete histologic response in the primary tumor and < 5% residual carcinoma in the neck lymph nodes. Despite a RECIST response of -34%, patient #7 with a p16+ T3, N2b of base of tongue tumor had a complete histologic response in the base of tongue after TORS and minimal microscopic disease in one lymph node. Thus, RECIST criteria tended to underestimate pathologic and PET responses: 3 patients with SD by RECIST had histological responses (patient #2, 3 and 6, Fig. 1A) and 3 patients with PR by RECIST had complete PET responses (Fig. 1B).
Pharmacokinetic Analyses
An interim PK analysis was conducted based on preliminary AZD1775 concentration using nominal blood sampling times. Typical mean plasma concentration-time profiles following oral administration of AZD1775 as single-dose (C1D2) and multiple doses (C1D4) in combination with cisplatin and docetaxel are presented in Figure 2. The PK parameters for AZD1775 in are presented in eTable 3.
Figure 2. Pharmacokinetics of AZD1775.
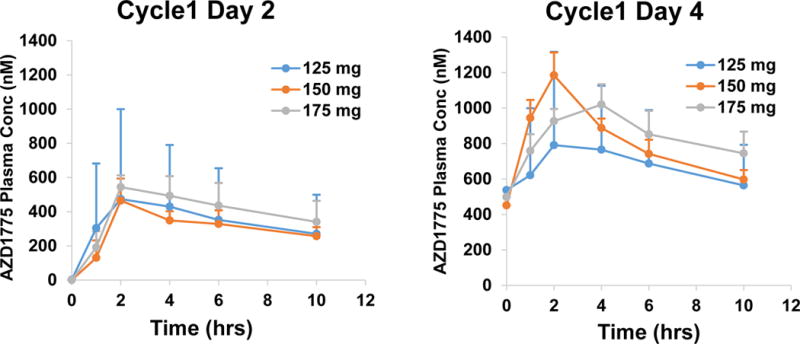
Mean plasma concentration curves after AZD1775 in combination with cisplatin and docetaxel following single (Cycle 1, Day 2) and multiple dose (Cycle 1, Day 4).
After single dose and multiple doses, peak plasma concentrations of AZD1775 were achieved within 1 to 6 hours post dose. The accumulation of drug over 2.5 days on the twice-per-day regimen was consistent with a half-life of approximately 9-12 hours and is in agreement with previously reported data15. As assessed by geometric mean %CV, there was significant inter patient variability in Cmax and AUC at 125 mg dose (%CV, 90 to 192%) and moderate variability (%CV, 11-55 %) at higher doses leading to overlap in exposures. The PK C8hr target of 240 nM, which was associated with maximal efficacy in rat tumor xenograft studies, was achieved on Day 4 at all the three doses. Overall, the PK data was within the range of reported AZD1775 PK estimates.
Pharmacodynamic Analyses
Pre-treatment biopsies were obtained in all 12 patients; 7 patients underwent a biopsy after the lead-in week of AZD1775 alone. Nine patients had biopsy material obtained after completion of the first cycle of AZD1775/chemotherapy. The one patient, who was not biopsied after completion of this first cycle 1, underwent a biopsy at time of definitive therapy. Biomarker data are depicted in Figure 3. We present biomarker data comparing percentage (%) cells staining vs. clinical response in all patients before therapy and after AZD1775 alone or right after the last AZD1775 dose following cisplatin/docetaxel (Fig. 3). In addition, despite limited number of patients, we subdivided the cohort into responders vs. non-responders. For this purpose and genomic analyses below, we sought to account for both RECIST criteria and histologic responses: responders had > 30% reduction in tumor measurements by RECIST, or 0-30% decrease by RECIST with a robust histological response >90%; non-responders had <30% decrease by RECIST and no biopsy material to assess residual tumor mass; (i.e., patients #9 and #12). This is buttressed by scenarios explained above where RECIST response and histology did not correlate well, but a strong PET response was also observed (Fig. 1).
Figure 3. Biomarker response and clinical correlation after neoadjuvant therapy with AZD1775 alone or in combination with chemotherapy.
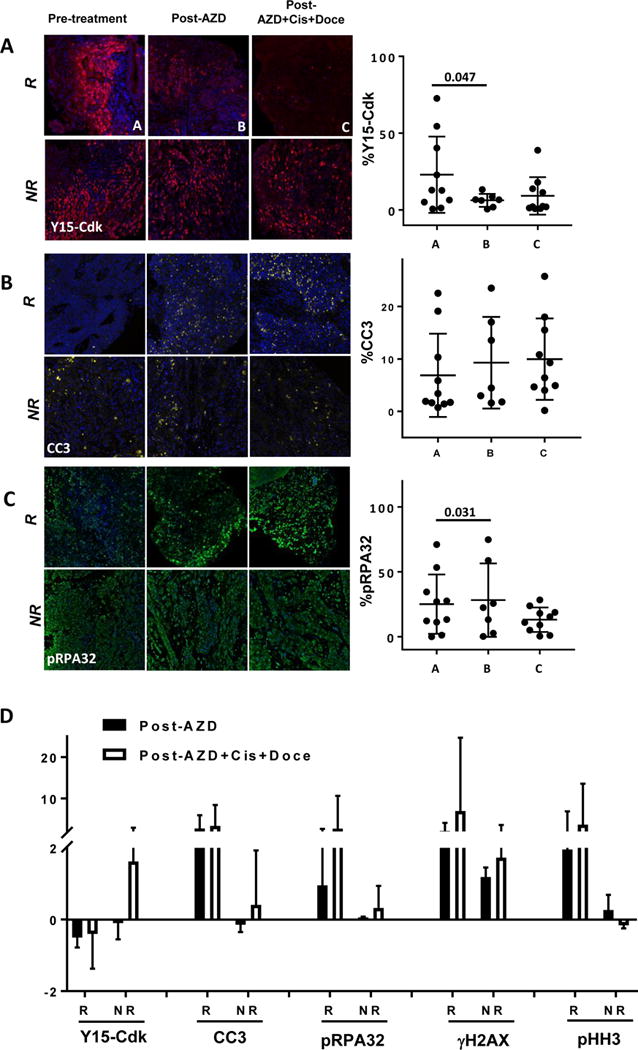
Biomarker response as demonstrated by IF (left panels) and quantified IHC data as % cells stained (right panels) for (A) Y15-Cdk, (B) CC3, and (C) pRPA32 in biopsies pre-treatment (sample “A”), post-AZD alone (sample “B”), and post-AZD plus cisplatin plus docetaxel (sample “C”). (D) Fold changes in biomarkers Y15-Cdk, CC3, pRPA32, γH2AX, and pHH3 post-AZD alone (black bars) and post-AZD plus cisplatin/docetaxel (white bars), after subdividing into clinical responders (“R”) and non-responders (“NR”).
Reductions in Y15-Cdk before and after therapy were significant (Fig. 3A p=0.047) and correlated with degree of response. These results suggest evidence of target engagement and a positive association between greater clinical response and more pronounced suppression of Y15-Cdk (Fig. 3A, 3D, eFig. 2A R2= 0.3461). In fact, patient #9, who progressed through treatment, showed an increase in Y15-Cdk (Fig. 3A). A trend towards increased apoptotic marker CC3 staining was also observed after therapy (Fig. 3B). Moreover, this gain was correlated with clinical response and an increase was noted in responders vs. non-responders when comparing biopsies before and after AZD1775/chemotherapy (Fig. 3B, 3D, eFig. 2B R2= 0.4073). Further evidence of effective WEE1 inhibition is seen with increased staining of the replication stress marker pRPA32, particularly in biopsies after AZD1775 treatment alone (Fig. 3C, P=0.031), and when comparing responders vs. non-responders (Fig. 3D). Finally, the DNA damage marker γH2AX showed near-significant increased staining after treatment with either AZD1775 or in combination (eFig. 2C). This trend remains when comparing responders vs. non-responders (Fig. 3D). In contrast, the mitotic marker HH3 showed overall more heterogeneous staining (eFig. 2D). When compared to non-responders, responders tended to have increased HH3 staining not reaching significance (Fig. 3D).
Genomic Analyses
Mutational analysis of the 262 cancer-relevant genes evaluated by the UW-OncoPlex test for all enrolled patients is shown in eTable4. In agreement with several studies on the mutational landscape of HNSCC, there was a high frequency of tumor suppressor loss, including TP53 mutations (9 of 12 patients), CDKN2A (4 of 12 patients), and FBXW7 in in one patient who was p16+ with a history of smoking (patient #2). NOTCH1 which is involved in differentiation, and thought to play a tumor suppressor role in HNSCC2 was mutated in 3 patients. Alterations common in HNSCC such as EGFR, FGFR3, EPHA3, CCND1 and HRAS were also noted. All p16+ OPSCC patients also carried PIK3CA mutations or copy number gains as previously shown in other studies1–3. A summary oncoplot of the most frequent mutations by type for all 12 patients is depicted in Figure 4A and a comparison between responders vs. non-responders (by RECIST adjusted by histological responses as explained above) in Figure 4B. Mutations in CDKN2A and NOTCH1 were noted in three of the clinically-responding patients. All enrolled p16+ patients had a response either by pathology (patient #2) or RECIST alone (patient #3, 7 and 11). Although three patients with TP53 mutations (patients #2, 3 and 6) showed minimal regression by RECIST alone (-3, -11 and -16%), all three had robust histologically confirmed responses, and patient #3 had a complete PET response (Fig. 1B).
Figure 4. Genetic landscape of tumors from enrolled patients.
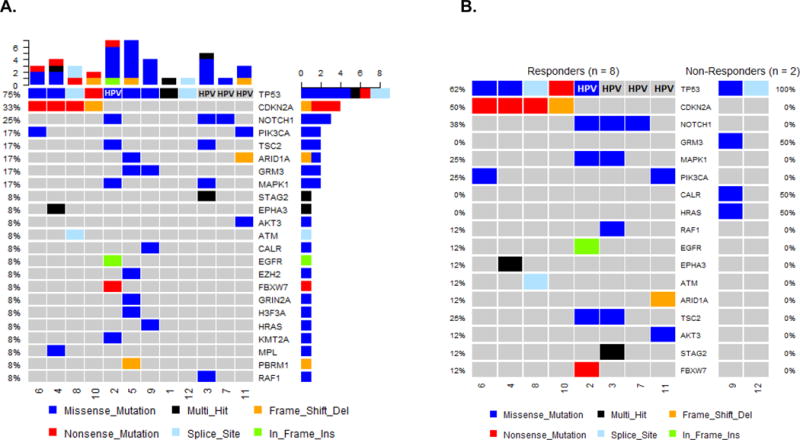
(A) Schematic representation (Oncoplot) of most commonly mutated genes and HPV status in all patients enrolled. Each column represents a patient. Colors depict the type of mutations for each gene. (B) Oncoplot comparison between responders vs. non-responders by RECIST criteria adjusted by histological responses as classified for the pharmakodynamic analysis.
DISCUSSION
This completed phase I clinical trial represents the first published clinical experience using the novel therapeutic combination of AZD1775 with cisplatin and docetaxel. Our goal was to leverage on the role of WEE1 as a G2 checkpoint regulator (through CDK1) - activated upon genotoxic therapy - and as guardian of genomic stability in S-phase (through CDK2) such that a synergistic interaction with a better tolerated DNA damaging regimen which omitted 5-FU would yield similar or higher rates of response but with significantly less toxicity. Towards this end, we observed low rates of Grade ≥ 3 toxicity; successful completion of the induction treatment regimen, and encouraging therapeutic responses. The PD biomarker data coupled with genomic characterization for all patients led to mechanistic insights that reflect our clinical observations. This novel therapeutic approach exploits the underlying biology of tumors with high frequency of TP53 inactivation and genomic instability as supported by PD data showing evidence of replication stress and to a lesser degree, G2/M override leading to apoptosis. The MTD was established at 150mg orally twice daily for 5 doses with the only major DLT at DL2 (175 mg) being diarrhea in two patients. Pharmacokinetic analyses were consistent with previously reported AZD1775 regimens in combination with chemotherapeutic agents, and the target concentration of 240 nM was achieved on Day 4 at all three doses tested.
Induction chemotherapy or neoadjuvant chemotherapy has been extensively studied in locally advanced HNSCC. This approach has the potential to reduce tumor volume to limit toxicity from definitive therapy, decrease micrometastatic disease, and improve surgical resectability23–25. This is of particular relevance for oral cavity tumors where upfront surgery is the preferred approach, followed by adjuvant therapy based on pathologic findings. Despite these advantages, the existing neoadjuvant regimens for HNSCC are rarely used due to exceptionally high rates of hematologic toxicity which limits the anti-tumor efficacy. Two multi-institutional trials, PARADIGM and DECIDE, compared induction chemotherapy followed by definitive chemoradiation compared to chemoradiation alone and found no differences in survival in the induction group26,27. Of note, in both these studies, there was a significantly high proportion of OPSCCs with unknown HPV status where high response rates could have biased results towards the null hypothesis. In addition, both trials utilized induction regimens associated with protracted delivery and significant toxicities that can hamper delivery of definitive chemoradiation. Both trials were prematurely closed and were underpowered to detect survival differences.
One challenging aspect of inclusion criteria in this study was the use of “borderline” resectability which can be subjective. We addressed this by obtaining three surgeons’ consensus in tumor board regarding resectability in a tertiary referral institution where all resources and expertise were available to perform complex resections and reconstructions. Notwithstanding the subjectivity of some of these measures, the success of this premise was illustrated well in patients #4, 6 and 8. In all three, it is clear that surgery would have incurred either significant if not unacceptable disfigurement (patient #4, Fig. 1C), or functional speech and swallowing impairment as a result of a total glossectomy (patients #6 and 8). Moreover, these responses have been durable almost 1.5-2 years out. This illustrates the notion that if a safe and effective short-course regimen of about 4 weeks for oral cavity tumors where upfront surgery is typically preferred, tumor cytoreduction could make surgery feasible. Longer follow up times are needed to ensure that PFS is not shortened via this approach, but we have not observed any recurrences in our cohort.
For the majority of markers, statistically significant changes were difficult to obtain given the small sample size of our Phase I trial. Despite this, our exploratory biomarker data revealed a statistically significant reduction in Y15-Cdk following treatment, which seemed to correlate with clinical response. This suggests that target engagement is a critical component of response in our cohort. Similar trends, though not significant, were observed with γH2AX and CC3 suggesting that AZD1775-mediated DNA damage could lead to apoptosis particularly in HPV+ tumors28. Interestingly, although the trends demonstrated that a higher proportion of responders had increases in pHH3, here the correlation between response and staining was much weaker. One potential implication is that an increase in pre-mature mitosis may not be required for AZD1775/chemotherapy cytotoxicity as suggested by Guertin el al14, though this will require further study. In fact, the statistically significant increase in pRPA32, a marker of replication stress, after AZD1775 alone points towards replication stress as a potentially more pronounced component of anti-tumor activity in this treatment regimen10. Albeit not conclusive, these biomarker findings provide a foundation for further scientific investigation and clinical trials.
The patients enrolled carried a similar pattern of mutations and copy number aberrations typical for HNSCC – mainly, loss of tumor suppressor function such as TP53 and CDKN2A, few shared oncogenic mutations and PI3K, EGFR and CCND1 amplifications. These genomic aberrations are conducive to genomic instability and reliance on the G2 checkpoint. Given that all patients carried either TP53 mutations or a deficient p53 pathway via HPV E6-mediated p53 degradation, it is not possible to ascertain the role of TP53 in responses observed. It is clear that HPV+ patients with intact TP53 had substantial responses. This emphasizes the issue that it may not matter the manner in which TP53 is inactivated so long as the p53 pathway is deficient. Another possibility is that HPV E6/E7 oncoproteins may play a role in DNA damage response or other pathways and synergize with AZD1775 through a p53-independent mechanism. What is clear from preclinical studies is that in isogenic cell line models, knock-down of TP53 confers sensitivity to AZD1775 particularly in the presence of DNA damage5,6. In addition, there is evidence of replication stress upon single agent treatment in a p53-independent manner12. It is yet unclear how all these genomic aberrations might affect AZD1775 response in this trial schema, or if AZD1775 alters the mutational profile of treated cancers. These findings highlight the complexity of the mutational context in HNSCC and underscore the need to understand how different networks and pathways are disrupted rather than single mutations in isolation.
As PD-1 checkpoint blockade and immune modulatory approaches increasingly play a role in HNSCC therapy, the possibility of combinatorial therapeutic approaches is enticing. Future research on the impact of AZD1775 on the immune response, the possible exposure of neo-antigens in AZD1775 treated cancers, and combination AZD1775/PD-1 checkpoint blockade may be relevant to the treatment of HNSCC. The intriguing results described here suggest that the therapeutic combination of AZD1775 and platinum is a promising approach for HNSCC patients, particularly in difficult-to-treat TP53-mutated HNSCC patients. Additional scientific and therapeutic studies involving AZD1775 merit further scientific and clinical evaluation.
The PD biomarker results are very suggestive of the presence of replication stress and potentially G2/M override in certain cases. Our PK and PD analyses illustrate the successful translation of preclinical data into clinically safe and efficacious therapeutic strategies for HNSCC. This data augments the previously published prospectively collected information on the activity of AZD1775 in HNSCC5,6. The encouraging safety profile and response rates observed in our rather small phase I study merit further investigation of this neoadjuvant regimen prior to definitive therapy for locally advanced HNSCC.
Supplementary Material
Statement of significance.
The regimen of neoadjuvant AZD1775 in combination with cisplatin/docetaxel in advanced HNSCC seemed tolerable with encouraging anti-tumor activity. Given the favorable safety profile, the use of AZD1775 in this setting could reduce morbidity of definitive therapy in advanced HNSCC where there is a desperate need for targeted agents that leverage on the genomic profile of this disease.
STATEMENT OF TRANSLATIONAL RELEVANCE.
This completed phase I clinical trial represents the first published clinical experience using the WEE1 inhibitor, AZD1775, with cisplatin and docetaxel in the neoadjuvant setting. This novel therapeutic approach exploits the underlying biology of TP53 inactivated tumors, which are incapable of G1 arrest and reliant on the G2/M checkpoint for DNA repair following genotoxic therapy. Additionally, WEE1’s inhibition of CDK2 maintains genomic stability in S-phase, thereby protecting cells against replication stress and subsequent cell death. We tested if the synergistic interaction between AZD1775 and a less toxic DNA damaging regimen without 5-FU would yield similar or higher rates of response while reducing toxicity. Our pharmacodynamic data was consistent with evidence of replication stress and to a lesser degree, G2/M override leading to apoptosis. Given its favorable safety profile, AZD1775 in this setting could reduce morbidity of definitive therapy in borderline-resectable, advanced HNSCC, where there is desperate need for targeted agents that leverage the genomic profile of this disease.
Acknowledgments
Histology and immunohistochemistry was performed by Experimental Histopathology Shared Resources at Fred Hutchinson Cancer Research Center.
Funding Sources:
This work was funded by NCI (5 P30 CA015704-40, U01CA176303-01 and R01 CA215647-01A); the American Cancer Society (FDN-RSG-13-0661-01-TBG); NIDCD (T32DC000018); and internal funds from University of Washington/Seattle Cancer Care Alliance and Fred Hutchinson Cancer Center. AstraZeneca provided AZD1775 and funding for pharmacokinetic data and genomic analysis.
Footnotes
Disclosures:
Dr. Cristina P. Rodriguez and Dr. Eduardo Mendez receive institutional research funding from Merck, Bristol-Myers Squibb, Astra Zeneca, and Ignyta.
Dr. Chow reports Advisory Board participation personal fees and institutional grant funding from Seattle Genetics, Novartis, Genentech, AstraZeneca/Medimmune, Bristol Myers Squibb and Pfizer; only institutional grant funding from Lily/Imclone, Incyte and VentiRx; Advisory Board personal fees from Takeda and Sanofi-Genzyme; and Consultation personal fees from Amgen outside the submitted work, during the conduct of the study.
Dr. Mugundu is an employee of AstraZeneca and holds stocks of AstraZeneca & Pfizer.
Other than the disclosures noted above, none of the other authors have financial conflicts of interest to disclose.
Declarations:
Ethics approval and consent to participate: This study was approved by the institutional review offices of the University of Washington (UW) and the Fred Hutchinson Cancer Research Center (FHCRC).
Availability of data and material: All data generated or analyzed during this study are included in this published article and its supplementary information files.
Authors’ contributions -
Conceptualization: E.M., C.R., N.F., L.C., R.M., R.S.D.
Methodology: E.M., C.R., L.C.
Validation: E.M., C.R., M.K., S.R., A.D., A.H., E.K.
Formal Analysis: E.M., C.R., M.K., S.R., A.D., A.H., E.K.
Data Curation: E.M., C.R., M.K., S.R., A.D., G.M
Writing-Original Draft: E.M.
Writing-Review & Editing: All authors
Visualization- E.M., A.D., S.R., C.R., M.K.
Supervision, E.M., C.R., L.C., N.F., R.M.
Project Administration, E.M.
Funding Acquisition: E.M., L.C., C.R., N.F.
References
- 1.Stransky N, Egloff AM, Tward AD, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333(6046):1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agrawal N, Frederick MJ, Pickering CR, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333(6046):1154–1157. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517(7536):576–582. doi: 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poeta ML, Manola J, Goldwasser MA, et al. TP53 Mutations and Survival in Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med. 2007;357(25):2552–2561. doi: 10.1056/NEJMoa073770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moser R, Xu C, Kao M, et al. Functional Kinomics Identifies Candidate Therapeutic Targets in Head and Neck Cancer. Clin Cancer Res. 2014;20(16):4274. doi: 10.1158/1078-0432.CCR-13-2858. LP-4288. http://clincancerres.aacrjournals.org/content/20/16/4274.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Osman AA, Monroe MM, Ortega Alves MV, et al. Wee-1 kinase inhibition overcomes cisplatin resistance associated with high-risk TP53 mutations in head and neck cancer through mitotic arrest followed by senescence. Mol Cancer Ther. 2015;14(2):608–619. doi: 10.1158/1535-7163.MCT-14-0735-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aarts M, Sharpe R, Garcia-Murillas I, et al. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov. 2012;2(6):524–539. doi: 10.1158/2159-8290.CD-11-0320. [DOI] [PubMed] [Google Scholar]
- 8.Hirai H, Arai T, Okada M, et al. MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol Ther. 2010;9(7):514–522. doi: 10.4161/cbt.9.7.11115. [DOI] [PubMed] [Google Scholar]
- 9.Odajima J, Kalaszczynska I, Sicinski P. Cyclins A and E trigger DNA damage. Cell Cycle. 2010;9(7):1231–1232. doi: 10.4161/cc.9.7.11313. [DOI] [PubMed] [Google Scholar]
- 10.Beck H, Nahse-Kumpf V, Larsen MSY, et al. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol Cell Biol. 2012;32(20):4226–4236. doi: 10.1128/MCB.00412-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dominguez-Kelly R, Martin Y, Koundrioukoff S, et al. Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J Cell Biol. 2011;194(4):567–579. doi: 10.1083/jcb.201101047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kao M, Green C, Sidorova J, Mendez E. Strategies for Targeted Therapy in Head and Neck Squamous Cell Carcinoma Using WEE1 Inhibitor AZD1775. JAMA Otolaryngol Head Neck Surg. 2017;143(6):631–633. doi: 10.1001/jamaoto.2016.4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kreahling JM, Gemmer JY, Reed D, Letson D, Bui M, Altiok S. MK1775, a selective Wee1 inhibitor, shows single-agent antitumor activity against sarcoma cells. Mol Cancer Ther. 2012;11(1):174–182. doi: 10.1158/1535-7163.MCT-11-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guertin AD, Li J, Liu Y, et al. Preclinical evaluation of the WEE1 inhibitor MK-1775 as single-agent anticancer therapy. Mol Cancer Ther. 2013;12(8):1442–1452. doi: 10.1158/1535-7163.MCT-13-0025. [DOI] [PubMed] [Google Scholar]
- 15.Do K, Wilsker D, Ji J, et al. Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients With Refractory Solid Tumors. J Clin Oncol. 2015;33(30):3409–3415. doi: 10.1200/JCO.2014.60.4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leijen S, van Geel RMJM, Pavlick AC, et al. Phase I Study Evaluating WEE1 Inhibitor AZD1775 As Monotherapy and in Combination With Gemcitabine, Cisplatin, or Carboplatin in Patients With Advanced Solid Tumors. J Clin Oncol. 2016;34(36):4371–4380. doi: 10.1200/JCO.2016.67.5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu Y, Fang W, Zeng W, Leijen S, Woolf EJ. Evaluation of dried blood spot (DBS) technology versus plasma analysis for the determination of MK-1775 by HILIC-MS/MS in support of clinical studies. Anal Bioanal Chem. 2012;404(10):3037–3048. doi: 10.1007/s00216-012-6440-6. [DOI] [PubMed] [Google Scholar]
- 18.Sakurikar N, Eastman A. Critical reanalysis of the methods that discriminate the activity of CDK2 from CDK1. Cell Cycle. 2016;15(9):1184–1188. doi: 10.1080/15384101.2016.1160983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pritchard CC, Salipante SJ, Koehler K, et al. Validation and implementation of targeted capture and sequencing for the detection of actionable mutation, copy number variation, and gene rearrangement in clinical cancer specimens. J Mol Diagn. 2014;16(1):56–67. doi: 10.1016/j.jmoldx.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salipante SJ, Scroggins SM, Hampel HL, Turner EH, Pritchard CC. Microsatellite instability detection by next generation sequencing. Clin Chem. 2014;60(9):1192–1199. doi: 10.1373/clinchem.2014.223677. [DOI] [PubMed] [Google Scholar]
- 21.Mathias PC, Turner EH, Scroggins SM, et al. Applying Ancestry and Sex Computation as a Quality Control Tool in Targeted Next-Generation Sequencing. Am J Clin Pathol. 2016;145(3):308–315. doi: 10.1093/ajcp/aqv098. [DOI] [PubMed] [Google Scholar]
- 22.Awasthi P, Foiani M, Kumar A. ATM and ATR signaling at a glance. J Cell Sci. 2015;128(23):4255. doi: 10.1242/jcs.169730. LP-4262. http://jcs.biologists.org/content/128/23/4255.abstract. [DOI] [PubMed] [Google Scholar]
- 23.Zorat PL, Paccagnella A, Cavaniglia G, et al. Randomized Phase III Trial of Neoadjuvant Chemotherapy in Head and Neck Cancer: 10-Year Follow-Up. JNCI J Natl Cancer Inst. 2004;96(22):1714–1717. doi: 10.1093/jnci/djh306. http://dx.doi.org/10.1093/jnci/djh306. [DOI] [PubMed] [Google Scholar]
- 24.Forastiere AA. Is There a New Role for Induction Chemotherapy in the Treatment of Head and Neck Cancer? JNCI J Natl Cancer Inst. 2004;96(22):1647–1649. doi: 10.1093/jnci/djh339. http://dx.doi.org/10.1093/jnci/djh339. [DOI] [PubMed] [Google Scholar]
- 25.Pignon JP, Bourhis J, Domenge C, Designe L. Chemotherapy added to locoregional treatment for head and neck squamous-cell carcinoma: three meta-analyses of updated individual data. MACH-NC Collaborative Group. Meta-Analysis of Chemotherapy on Head and Neck Cancer. Lancet (London, England) 2000;355(9208):949–955. [PubMed] [Google Scholar]
- 26.Haddad R, O’Neill A, Rabinowits G, et al. Induction chemotherapy followed by concurrent chemoradiotherapy (sequential chemoradiotherapy) versus concurrent chemoradiotherapy alone in locally advanced head and neck cancer (PARADIGM): a randomised phase 3 trial. Lancet Oncol. 2013;14(3):257–264. doi: 10.1016/S1470-2045(13)70011-1. [DOI] [PubMed] [Google Scholar]
- 27.Cohen EEW, Karrison TG, Kocherginsky M, et al. Phase III randomized trial of induction chemotherapy in patients with N2 or N3 locally advanced head and neck cancer. J Clin Oncol. 2014;32(25):2735–2743. doi: 10.1200/JCO.2013.54.6309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanaka N, Patel AA, Wang J, et al. Wee-1 Kinase Inhibition Sensitizes High-Risk HPV+ HNSCC to Apoptosis Accompanied by Downregulation of MCl-1 and XIAP Antiapoptotic Proteins. Clin Cancer Res. 2015;21(21):4831–4844. doi: 10.1158/1078-0432.CCR-15-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.