

Abstract
Enzalutamide is a second-generation nonsteroidal antiandrogen clinically approved for the treatment of castration-resistant prostate cancer (CRPC), yet resistance to endocrine therapy has limited its success in this setting. Although the androgen receptor (AR) has been associated with therapy failure, the mechanisms underlying this failure have not been elucidated. Bioinformatics analysis predicted that activation of the Wnt/β-catenin pathway and its interaction with AR play a major role in acquisition of enzalutamide resistance. To validate the finding, we show upregulation of β-catenin and AR in enzalutamide-resistant cells, partially due to reduction of β-TrCP mediated-ubiquitination. While activation of the Wnt/β-catenin pathway in enzalutamide-sensitive cells led to drug resistance, combination of β-catenin inhibitor ICG001 with enzalutamide inhibited expression of stem-like markers, cell proliferation, and tumor growth synergistically in various models. Analysis of clinical datasets revealed a molecule pattern shift in different stages of PCa, where we detected a significant correlation between AR and β-catenin expression. These data identify activation of the Wnt/β-catenin pathway as a major mechanism contributing to enzalutamide resistance and demonstrate the potential to stratify patients with high risk of said resistance.
Keywords: enzalutamide resistance, androgen receptor, β-catenin, cancer stem cell, castration-resistant prostate cancer
Introduction
Prostate cancer (PCa) is the second most frequently diagnosed malignancies and the sixth leading cause of cancer-related death in men worldwide (1). Although PCa can often be cured at early stage (2), patients have to go through androgen deprivation therapy (ADT) (3). AR is necessary for the function, survival, and differentiation of prostatic tissue (4,5). In contrast, during carcinogenesis the function of AR signaling is altered from tumor suppressive to tumor promoting (6,7). The disease eventually progresses into a stage called castration-resistant prostate cancer (CRPC), when AR signaling is reactivated again (8). Enzalutamide, a second-generation AR antagonist, was approved in clinic to treat CRPC (9,10). Some mechanisms contributing to enzalutamide resistance have been reported (11,12). However, a more comprehensive approach is needed to further explore the underlying mechanisms for enzalutamide resistance.
β-catenin is a multifunctional protein. Numerous studies indicate that activation of β-catenin signaling contributes to human cancer (13–16). A high-level β-catenin expression always induces tumorigenic traits and promotes cancer cell proliferation and correlates with poor overall survival (16,17). Further, β-catenin signaling is crucial for the pluripotent phenotype and self-renewal of both normal and cancer stem cells (CSCs) (18,19). CSCs are resistant to classic chemotherapy and radiotherapy. In PCa, it is possible that CSCs survive after androgen ablation therapy, causing CRPC (20). Growing evidence shows that Wnt/β-catenin signaling is highly active in CSCs and may have a role in prostate stem cell self-renewal (21,22). In addition, AR and β-catenin may enforce each other to elicit target genes for promoting androgen-independent growth and progress (23). However, the role of Wnt/β-catenin signaling in enzalutamide resistance has not yet been reported, albeit it is urgently needed in clinic.
Herein, we aim to probe for mechanisms of enzalutamide resistance using an unbiased systems biology approach. To our knowledge, this is the first report to evaluate the association between AR, β-catenin, and CSCs in enzalutamide-resistant PCa. We show that inhibition of β-catenin overcomes enzalutamide resistance, thus enhancing its efficacy in CRPC.
Materials and Methods
RNA-seq datasets
PCa patient data
1) TCGA Prostate Adenocarcinoma Patient Data. 498 PCa RNA sequence and their-associated clinical information were obtained from Genomic Data Commons Data Portal (GDC, https://portal.gdc.cancer.gov/). All of these RNA sequence data were collected from Level 3 (for Segmented or Interpreted Data, IllumninaHiSeq_RNASeqV2 of TCGA. 2) Hormone sensitive and resistant for high Gleason score signature in TCGA PCas. Cancer samples were divided into low-grade (Gleason scores <8) and high-grade (Gleason scores ≥ 8). Samples with a high grade of Gleason score plus hormone treatment were considered to be potentially hormone resistance. To integrate patient demography and drug treatment annotation together, 140 out of 497 cases with clinical hormone treatment were selected as observed objectives. 140 tumors were classified into low Gleason score (<8) and high Gleason score (≥8) groups and identified differential gene expression based on RPKM value by standard microarray analysis techniques (quantile normalization and moderated t-statistics). 3) RNA-seq data of 7 patients with or without treatment of ADT for ~22 weeks was collected from Gene Expression Omnibus, GSE48403.
Segregation of clinic specimens
We used Gleason score of 8 as a stricter criterion to segregate samples into “potential hormone sensitive or resistant”. Patients response to hormone therapy should show less aggressive. If the patients received hormone-therapy but continuously progressed to more aggressive prostate cancer (Gleason score≥8), patients should be considered as no or weak response to hormone therapy which was termed as hormone-therapy resistance. Accordingly, we set patients with Gleason score <8 as sensitive but Gleason score ≥8 as resistant to hormone-therapy.
Immunofluorescence (IF) staining
IF staining was performed as described previously(24). Cells were incubated with antibodies against β-catenin (catalog no. 610153, BD Transduction Laboratory) for 1 h at room temperature, followed by incubation with secondary antibody and 4′,6-diamidino-2-phenylindole (DAPI; Sigma) for 1 h.
Proteasome activity assay
Prepare by homogenizing cells with 0.5 % NP-40 in dH2O or PBS. Add up to 50 μl of each cell extract or other proteasome sample to be tested to paired wells. Bring the volume of each well to 100 μl with Assay Buffer. Add 1 μl of the Proteasome inhibitor to one of the paired wells, 1 μl of Assay Buffer to the other well, mix. Add 1 μl of Proteasome Substrate to all wells, mix, protected from light, mix. Measure kinetics of fluorescence development at Ex/Em = 350/440 nm in a micro-plate reader at 37°C for 30 – 60 min (catalog no. K245, BIOVISION).
Luciferase assay
A T-cell factor/lymphoid enhancing factor (TCF/LEF) reporter kit was purchased from BPS Bioscience (catalog no. 60500), and luciferase assays were performed according to the manufacturer’s instructions.
Xenograft experiments
All the animal experiments described in this study were approved by the Purdue University Animal Care and Use Committee (PACUC). After 22RV1 cells (2.5 × 105 cells per mouse) were inoculated into nude mice (Harlan Laboratories) for two weeks, animals were randomized into treatment and control groups of 6 mice each, followed by injection of enzalutamide into the tail vein twice weekly. The ICG001 suspension was prepared in coin oil and administered into mice via oral gavage twice weekly. Tumor volumes, calculated from the formula V = L × W2/2 (where V is volume [cubic millimeters], L is length [millimeters], and W is width [millimeters]).
LuCaP35CR xenograft model
Mice carrying LuCaP35CR tumors were obtained from Robert Vessella at the University of Washington. Tumors were amplified and then implanted into pre-castrated nude mice. After enough tumors were amplified, tumors were harvested and cut into ~20- to 30-mm3 pieces before being implanted into 20 pre-castrated nude mice. When tumors reached 200 to 300 mm3, mice were randomly separated into 4 groups for different treatments.
Spheroid formation assay
Single cells were cultured in serum-free DMEM/F12 medium (Invitrogen) supplemented with 20 ng/ml human recombinant EGF (Sigma-Aldrich), 10 ng/ml human recombinant bFGF (Sigma-Aldrich), 4 μg/ml insulin (Sigma-Aldrich), 500 U/ml penicillin, 500 μg/ml streptomycin (Invitrogen) and 1% methylcellulose (Sigma-Aldrich). After cells were cultured in suspension in poly-HEMA-coated 24-well plates, cells were replenished with supplemented medium every other day. To propagate spheres in vitro, spheres were collected by gentle centrifugation and dissociated to single cells using TrypLE Express (Invitrogen). Following dissociation, trypsin inhibitor (Invitrogen) was used.
Results
Systematic detection of enzalutamide-resistant mechanisms in CRPC cells and tumors based on transcriptomic analysis
To identify novel targets or pathways that might be responsible for enzalutamide resistance in PCa, we analyzed expression levels of 514 human mRNAomes in three sample groups (4 PCa cell lines, 498 tumors from PCa patients treated with hormone therapy (sensitive vs resistant), and 52 pairs PCa specimens (Fig 1A). The three gene expression datasets were obtained from RNA-seq experiments of in-house cell lines, the Cancer Genome Atlas (TCGA) of prostatic adenocarcinoma patients, and Gene Expression Omnibus (GEO) GSE48403. Genome-wide transcriptome of all samples were profiled by high throughput RNA-seq technique and preprocessed aligning reads toward genome and transcriptome with NGSQC software (25) and its junction-mapping extension by STAR software (26) (see Material and Methods). After associated pathway enrichment of DEGs was detected by Gene Set Enrichment Analysis (GSEA), gene control network was constructed by ingenuity pathway analysis (IPA) software. Venn diagrams as shown in Fig. 1B reveal 226 genes that might be involved in enzalutamide resistance. The detailed gene list is shown in Supplementary Table S1 and Table S2.
Figure 1.

Systematic transcriptome comparison before and after enzalutamide resistance led to discovery of molecular pathways rewiring in PCa. A, Systematically dissecting enzalutamide-resistant mechanisms in PCa based on cell and tumor transcriptomics. B, Venn diagram showing the number of DEGs in PCa with different responses to enzalutamide. C, Gene signature with Wnt signaling pathway analysis by GSEA in normal adjacent tissues, primary PCa and enzalutamide-sensitive vs -resistant cell lines. D and E, Correlation between AR and β-catenin in PCa (D) and adjacent normal tissue (E). F, Wnt pathway rewiring upon enzalutamide resistance.
350 pathways were found to be activated in enzalutamide-resistant C4-2 cells by RNA-seq analysis, 24 pathways were activated based on comparison of 7 pairs of next-generation sequencing of advanced PCa tumors before and after treatment with androgen-deprivation therapy via Gene set enrichment analysis (GSEA) (Supplementary Table S3). The overlapping top six pathways enriched (p value <0.05) in both C4-2R cells and androgen-deprivation therapy-resistant tumors (GSE48403) are displayed in Supplementary Table S4. As indicated, the Wnt signaling pathway show strong activation upon enzalutamide resistance in both PCa cell lines and PCa tumors. However, we found it was detected only in tumor-adjacent normal tissue and enzalutamide-resistant PCa cells but not in primary PCa, i.e., the Wnt pathway is rewired from an active state in normal tissue, to an inactive state in primary cancer, then to an active state again upon acquisition of enzalutamide resistance (Fig. 1C).
Strong interaction between AR and β-catenin in CRPC based on Pearson correlation analysis
Next, Pearson correlation analysis was performed to detect the gene expression interaction of AR and β-catenin in 52 pairs of PCa tumors and tumor-adjacent normal tissues from TCGA. Strong gene co-expression between AR and β-catenin in CRPC was observed with a correlation coefficient r=0.6171 (Fig. 1D), whereas the co-expression was much weaker in tumor-adjacent normal tissue with r=0.4516 (Fig. 1E). Further, the correlation was also observed in 498 tumors with a correlation coefficiency r=0.5115 (Supplementary Fig. S1A). However, the correlation of AR and β-catenin is dramatically reduced in 62 hormone therapy-resistant tumors with r=0.3607 for unclear reasons (Supplementary Fig. S1B).
We then performed IPA analysis and found the interaction activity between AR and β-catenin is linked to the activity of important stem cell markers, such as OCT-4, SOX2, CD44, ALDH1A1, ALDH7A2, ABCB1 and TGFβ (Fig. 1F, Supplementary Fig. S1C, S1D and S1E). In summary, AR, β-catenin and stem cells show a strong correlation upon acquisition of enzalutamide resistance.
β-catenin was upregulated in enzalutamide-resistant PCa cells
To support the role of β-catenin in development of chemotherapy-resistance in CRPC, we evaluated the possible alteration of β-catenin level in PCa cells. As indicated, the levels of β-catenin were clearly elevated in MR49F and C4-2R than in LNCaP and C4-2 respectively (Fig. 2A, 2B, 2C and Supplementary Fig. S2A). Next, we investigated subcellular localization of β-catenin. Consistent with the immunofluorescence (IF) data, we also detected elevated levels of nuclear β-catenin in enzalutamide-resistant cells than their parental cells (Fig. 2D). To further confirm activation of the Wnt/β-catenin pathway, TCGA dataset (http://www.cbioportal.org/study?id=nepc_wcm_2016#summary) analysis was used to explore genomic and transcriptional alterations of relevant genes involved in the pathway. As shown in Supplementary Fig. S2B, amplifications of CTNNB1 (26%), c-myc (53%) and CCND1 (27%) were found in PCa specimens. Correspondingly, we found mRNA upregulation in 43% of CTNNB1, 43% of c-myc and 28% of CCND1 (Supplementary Fig. S2C). In addition, we also found that multiple components of the Wnt pathway were increased, supporting activation of this pathway (Supplementary Fig. S2B, S2C). These genomic and RNA-seq analyses in dataset from TCGA are consistent with our findings.
Figure 2.

β-catenin is upregulated in enzalutamide-resistant PCa. A, Cells were treated with 2 μM ICG001 for 48 h and harvested for IF staining. B, Cells were treated with ICG001, subjected for IF staining of β-catenin, and quantified. ***: p<0.01. C, Cells were subjected to immunoblotting (IB) to detect the protein levels of β-catenin. D, PCa cells were subjected to subcellular fractionation (Nuc: nuclear, Cyto: cytosolic), followed by IB. M: protein marker. E, mRNA levels of β-catenin were measured by Q-PCR in four PCa cell lines. F, PCa cells were subjected to anti-β-catenin immunoprecipitation (IP), followed by anti-ubiquitin IB to compare the polyubiquitination status of β-catenin. G, Cells were treated with 10 mM MG132 for 6 h and harvested for IB. H, Cells were treated with 10 μg/ml cycloheximide (CHX) for indicated time, harvested for IB, and quantified. I, Anti-β-TrCP IB of PCa cells. J, IB to compare protein levels of β-catenin and AR in PCa cells.
Suppression of the proteasome pathway contributes to upregulation of β-catenin
To distinguish the accumulation of β-catenin is due to increased transcription or altered protein modification that contributes to increased stabilization, we first compared mRNA levels of four PCa cell lines and found that there was only slight higher of β-catenin mRNA level in MR49F cells than in LNCaP cells. Interestingly, the level of β-catenin mRNA in C4-2R cells was actually lower than that of C4-2 cells (Fig. 2E), suggesting transcriptional alteration doesn’t contribute to the elevation of β-catenin in resistant cells. We then switched to evaluate the role of protein modification by ubiquitination assay. As shown in Fig. 2F, less polyubiquitination was detected in C4-2R and MR49F cells in comparison to their enzalutamide-sensitive counterparts. Furthermore, treatment with MG132, a proteasome inhibitor, clearly increased β-catenin levels in both LNCaP and MR49F cells. Of note, less increase of β-catenin in MR49F cells compared with that in LNCaP cells after MG132 treatment indicates more activation of proteasome pathway (Fig. 2G). To further rule out β-catenin regulation at the gene expression level, we treated LNCaP and MR49F cells with cycloheximide, an inhibitor of protein translation, and then directly measured turnover of β-catenin. As indicated, the levels of β-catenin protein were gradually decreased in LNCaP cells, but largely unchanged in MR49F cells (Fig. 2H). It was established that β-TrCP is an E3 ligase that is responsible for polyubiquitination of β-catenin, resulting in its degradation through the proteasome pathway (27,28). To support, we compared β-TrCP protein levels in sensitive versus resistant cells. We found less β-TrCP in MR49F cells compared with LNCaP cells, consistent with the data obtained from cells treated with cycloheximide and MG132. To support the conclusion that proteasome pathway was more activated in enzalutamide-sensitive than in -resistant cell lines, we performed proteasome activity assay and found less proteasome activity in 22RV1 and MR49F cells than that of C4-2 and LNCaP cells, respectively (Fig. S2D). In addition, ectopic expression of β-TrCP indeed reduced the levels of β-catenin in 22RV1 and MR49F cells, further supporting the notion that increased levels of β-catenin in resistant cells was at least partially due to loss of the E3 ligase β-TrCP (supplementary Fig. S2E). However, more β-TrCP was found in C4-2R than that in C4-2 cells (Fig. 2I). Heterogeneity of PCa may be one of the reasons for this phenomenon. The role of interaction between AR and β-catenin in enzalutamide-resistant PCa has not yet been elucidated. Upon acquisition of enzalutamide resistance, we found that AR was upregulated. In addition, the prostate-specific antigen (PSA) level was increased in MR49F cells but almost completely disappeared in C4-2R cells (Fig. 2J). Thus, the nuclear colocalization of β-catenin with AR helps to reactivate AR signaling and contribute to enzalutamide resistance.
Inhibition of the Wnt/β-catenin pathway overcomes enzalutamide resistance
To further explore the effects of β-catenin on acquisition of enzalutamide resistance, we treated C4-2R and MR49F cells with ICG001, an inhibitor of Wnt/β-catenin pathway, and/or enzalutamide and then performed cell viability and colony formation assays. As indicated, monotherapy with ICG001 or enzalutamide only exerted slight effect on cell growth compared to combination of ICG001 and enzalutamide (Fig. 3A, 3B, 3C, 3D). To address potential off-target effects associated with pharmacological inhibition of β-catenin, we depleted β-catenin with RNAi in C4-2R, MR49F and 22RV1 cells. We found siRNA-mediated β-catenin silencing suppressed the growth of these cells, which was more significant following combinatory treatment with enzalutamide (Fig. 3E, 3F and supplementary Fig. S3A). Treatment with enzalutamide also significantly inhibited colony formation (Fig. 3G, 3H). Furthermore, cell proliferation assay and colony formation assay showed that ICG001 had no effects on β-catenin-depleted cells, suggesting ICG001 specifically inhibited β-catenin (supplementary Fig. S3B). To test whether ICG001 can overcome resistance to enzalutamide in vivo, we treated mice carrying 22RV1 xenograft tumors with enzalutamide, ICG001 or both for 3 weeks. As shown in Fig. 3I, tumors were highly resistant to enzalutamide alone. ICG001 alone slightly decreased the tumor volume, whereas combination of ICG001 and enzalutamide synergistically decreased tumor volume, indicating that ICG001 overcomes enzalutamide resistance and restores sensitivity to enzalutamide. It is important to point out that the combination treatment did not affect animal weight (Fig. 3J). Although both wet weight and size of the tumors were reduced with monotherapy of ICG001, the effect was more significant with dual treatments (Fig. 3K and 3L). H&E staining of tumors with dual treatment showed more necrosis and increased numbers of apoptotic bodies in comparison with monotherapy-treated tumors. For the untreated group, we detected more mitotic cells, suggesting that cell division was active (Fig. 3M). To determine whether ICG001 alone or in combination with enzalutamide represses tumor proliferation and promotes apoptosis, tumor samples were analyzed by immunofluorescence (IFC) staining for Ki67 and cleaved caspase 3. As shown in Fig. 3N and 3O, ICG001 inhibited Ki67 expression while combination treatment further decreased the level of Ki67 but increased the level of cleaved caspase 3. Finally, to determine cell origin, we analyzed tumors by staining for CK8/18 (luminal cells) and P63 (basal cells) and found that CK8/18+ cell population was increased after treatment with ICG001 and more significant increase was observed with combination of enzalutamide. However, single treatment with enzalutamide had no effects on luminal cell population. We observed no expression of P63 in tumors derived from 22Rv1 cells, in agreement with previous reports (29) (Fig. 3P).
Figure 3.
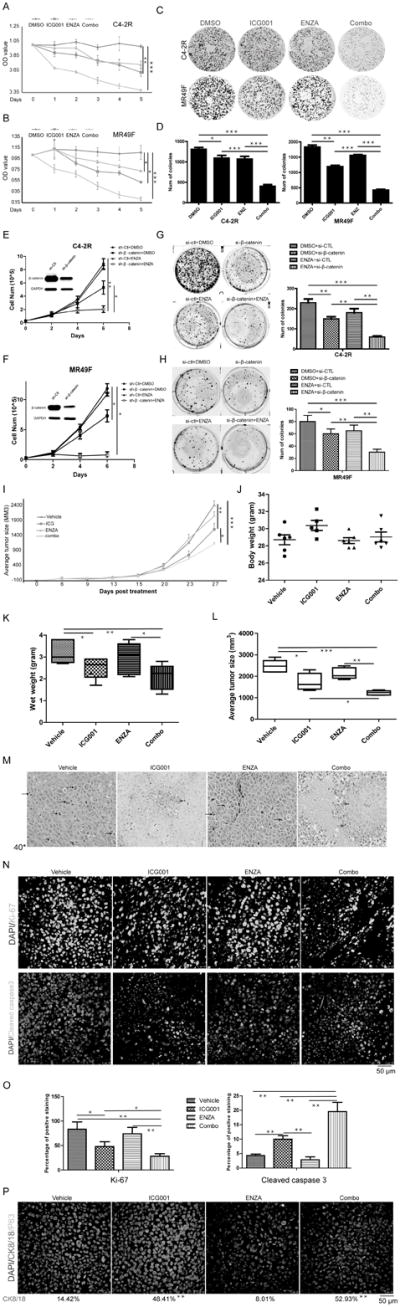
Suppression of β-catenin overcomes enzalutamide resistance. A and B, After 5 × 103 of C4-2R (A) and MR49F (B) cells were seeded onto 96-well plates overnight, cells were treated with ICG001 (2 μM), enzalutamide (10 μM for MR49F and 20 μM for C4-2R), or both, and harvested for MTT assays. C and D, 3 × 103 of C4-2R and MR49F cells were seeded onto 6-well plates, treated with indicated drugs, and cultured for two weeks with culture medium refresh every three days. E and F, 5 × 104 of MR49F (E) and C4-2R (F) cells were depleted of β-catenin with RNAi, treated with enzalutamide, and harvested for cell number counting at 6 d post-transfection. G and H, 3 × 103 of C4-2R (G) and MR49F (H) cells were depleted of β-catenin and treated with enzlutamide for two weeks. I, Nude mice were inoculated with CWR22RV1 cells (2.5×105) for 3 days and treated with ICG001 (5 mg/kg, intravenous injection), enzalutamide (20 mg/kg by oral gavage), or both. Tumor size were measured every 3 days (means±standard errors; n=6). J, Body weight were measured on day 27. K, Wet weight. L, Average tumor size on day 27. M, Representative H&E staining of tumor sections. (Black arrows: mitotic cells, red arrow: apoptotic cells). N, IHC staining for Ki67 and cleaved caspase 3 of tumor sections. O, Quantification of Ki67 and cleaved caspase 3 signals (means±standard errors). P, Representative images of IHC staining for P63 and CK8/18. *, p<0.05, **, 0.01<p<0.05, ***, p<0.01.
To rule out the potential off-target effects of ICG001 on tumor suppression in vivo, a more stable genetic approach (β-catenin shRNA) was used. As demonstrated in supplementary Fig. S3C–3G, β-catenin depletion slightly decreased tumor volume, whereas administration of enzalutamide showed synergistically effects, indicating that knockdown of β-catenin overcomes enzalutamide resistance and restores sensitivity to enzalutamide. Although both wet weight and size of the tumors were reduced with β-catenin knockdown alone, the effect was more significant with β-catenin depletion plus enzalutamide treatment. The levels of β-catenin in tissues were evaluated. As shown in supplementary Fig. S3H, only ICG001 but not enzalutamide treatment reduced β-catenin level. Combination treatment of enzalutamide and ICG001 did not show synergic effects on suppressing the level of β-catenin in tumors.
Activation of the Wnt/β-catenin pathway results in enzalutamide resistance in PCa
To validate the notion that the Wnt/β-catenin pathway is hyper-activated in enzalutamide-resistant PCa, a β-catenin-responsive TCF/LEF reporter assay was performed. As indicted, transcriptional activity of β-catenin was significantly elevated in C4-2R and MR49F cells in comparison to their enzalutamide-sensitive counterparts (Fig. 4A). In support, cyclin D1 and c-myc were also higher in enzalutamide-resistant cells than their enzalutamide-sensitive counterparts. We also detected reduced phosphorylation and inactivation of GSK3β in enzalutamide-resistant cells (Fig. 4B). As illustrated in Fig. 4C, several components of β-catenin pathway, such as WNT3, WNT5A, WNT7B, FZD2, FZD7, FZD9 and LRP6 were upregulated in C4-2R cells. When we investigated ligands of Wnt signaling pathway in public TCGA dataset, we found WNT5A (29%), FZD2 (21%), FZD9 (26%) were amplified in PCa and that 6% of WNT5A upregulated (Supplementary Fig. S2B, S2C). Further, GSEA supported the notion that Wnt signaling pathway is enriched in enzalutamide-resistant cells. Interestingly, we also found enrichment of Wnt pathway in normal adjacent prostate tissue compared with primary PCa (Fig. 1C). Collectively, our data implies a likely reactivation of Wnt pathway that may contribute to enzalutamide-resistance. To further investigate the role of Wnt/β-catenin pathway in enzalutamide resistance, we treated LNCaP and C4-2 cells with LiCl to activate β-catenin to mimic its upregulation in enzalutamide-resistant cells. As demonstrated in Fig. 4D, LiCl treatment-induced upregulation of β-catenin expression was accompanied by reduced enzalutamide-associated apoptosis, likely contributing to resistance to enzalutamide therapy in PCa. To support, LiCl treatment also antagonized enzalutamide-induced inhibition of cell proliferation and colony formation in LNCaP and C4-2 cells (Fig. 4E, 4F).
Figure 4.

Activation of the Wnt/β-catenin pathway contributes to enzalutamide resistance. A, Transcriptional activity assay. Luciferase activity is reported as relative activity (luciferase/Rellina) and presented as means±standard deviation of data from 3 replicates. B, Evaluation of transcription of target genes involved in the Wnt/β-catenin pathway by IB. C, Heat-map of genes involved in the Wnt/β-catenin pathway by comparing C4-2 and C4-2R cells with 3 replicates for each cell line. D-F, Cells were treated with 2.5 mM LiCl, 10 μM enzalutamide or both, followed by IB (D), MTT assay (E), and colony formation assay (F). G, Cells were ectopically expressed β-catenin, and then treated with 10 μM enzalutamide, followed by IB. H-I, C4-2 cells were infected with lentivirus expressing TOPOGFP, followed by isolation of GFP+ and GFP− cells. Then treated with indicated drugs, cells and followed viability (H) and colony formation assays (I). *, p<0.05, **, 0.01<p<0.05, ***, p<0.01.
To rule out off-target effects of LiCl on activation of β-catenin, we ectopically expressed β-catenin, and treated cells with or without enzalutamide to investigate the role of β-catenin in the acquisition of enzalutamide resistance. As demonstrated in Fig. 4G, β-catenin overexpression attenuated enzalutamide-induced apoptosis. Consistently, ectopic expression of β-catenin also antagonized enzalutamide-induced inhibition of cell proliferation and colony formation (supplementary Fig. S4A and S4B), similar to what we have observed with LiCl treatment.
To further confirm that enzalutamide resistance is mediated by Wnt signaling, we infected C4-2 cells with lentivirus expressing green fluorescent protein (GFP) from Wnt-responsive TCF/LEF promoter. Therapy with ICG001 and/or enzalutamide can suppress proliferation of GFP− C4-2 cells. Compared to GFP− cells, monotherapy with ICG001 has more significant effects on suppressing growth than in GFP+ cells. However, more cells were survived after only treated with enzalutamide in GFP+ cells, indicating that GFP+ cells (β-catenin active) were more resistant to enzalutamide than GFP− cells (β-catenin inactive). Interestingly, combinatory treatment has more dramatic effects on reducing cell survival in GFP+ cells but not in GFP− cells, supporting the notion that activation of β-catenin plays a critical role in development of enzalutamide resistance (Fig. 4H). We also found that GFP+ cells formed more and larger colonies than GFP− cells in the presence of enzalutamide (Fig. 4I), which means more resistant to enzalutamide treatment. Finally, we examined stem cell potential of GFP+ cells using xenograft mouse model. As indicated, GFP+ cells showed a stronger ability to form tumors than that of GFP− cells (Supplementary Table S5).
Pluripotency properties are enhanced upon acquisition of enzalutamide resistance
To support the role of β-catenin in CSCs, mRNA levels of stem cell markers OCT-4, SOX2, CD44, ALDH1A1, ALDH7A2 and transporter gene ABCB1 were found dramatically increased in C4-2R cells in comparison to C4-2 cells. In addition, a slight augment of transcription of NANOG and CD133 was also found in C4-2R cells (Fig. 5A), consistent with the results derived from published TCGA database (Supplementary Fig. S2B, S2C). Gene amplification was found in multiple stemness markers, including CD44 (17%), SOX2 (29%), PROM1 (10%), POU5F1 (19%) and ALDH1A1 (25%). In addition, transcription upregulation was found in 2.5% of CD44, 9% of SOX2, 28% of PROM1 and 16% of ALDH1A1. To validate the finding based on Q-PCR, we showed that the protein levels of CD44 and OCT-4 were significantly increased in C4-2R and MR49F cells compared with their enzalutamide-sensitive counterparts (Fig. 5B). Further, spheroid formation assay was used to investigate pluripotency of resistant cells. Monotherapy with ICG001 or enzalutamide alone can partially suppress spheroid formation while more significant inhibition was observed with combinatory treatment (Fig. 5C). Finally, reduction of protein levels of CD44 level but upregulation of cleaved-PARP, a marker for apoptosis, were detected in C4-2R and MR49F cells especially after treatment with ICG001 plus enzalutamide (Fig. 5D). Consistently, as shown in supplementary Fig. 4C, transcription of SOX2, CD44 and ALDH1A1 were suppressed especially upon combinatory treatment by real-time PCR. To rule out the potential off-target effects of ICG001 and further validate the molecular events in different cell derivatives, we investigated the associated molecular markers upon depletion of β-catenin in C4-2R, MR49F and 22RV1 cells. As indicated, reduction of CD44 and ALDH1A1 but increased levels of cleaved PARP were observed especially after treatment with enzalutamide in β-catenin-depleted cells (Fig 5E). Therefore, the stemness genes, upregulated in enzalutamide-resistant cells, can be suppressed by inhibition of Wnt/β-catenin pathway.
Figure 5.

Stem-cell-like properties were enhanced in enzalutamide-resistant cells. A, mRNA levels of stemness genes. B, Protein levels of three stemness genes. C, Spheroid formation of cells after drug treatment. D, Protein levels of stemness genes after drug treatment. Data was shown as means±standard errors from 3 replicates. E, Protein levels of stemness genes after drug treatment. Data was shown as means±standard errors from 3 replicates. *, p<0.05, **, 0.01<p<0.05, ***, p<0.01.
Combination therapy with ICG001 and enzalutamide blocks PCa growth in a patient-derived xenograft (PDX) model
To validate the synergic effect of ICG001 and enzalutamide in inhibition of tumor growth, we switched to an AR-positive but castration-resistant LuCaP35CR human PDX model (30,31). As shown in Fig. 6A, monotherapy with ICG001 or enzalutamide partially suppressed tumor growth, but the inhibitory effect was much stronger for ICG001 plus enzalutamide. No significant side effect was observed as body weight did not change much (Fig. 6B). Although both wet weight and size of the tumors were reduced with monotherapy of enzalutamide or ICG001, the effect was much more significant with combinatory treatment (Fig. 6C, 6D and 6E). Since H&E staining indicated necrosis at the interior of the tumors, we mainly analyzed the exterior of the tumors where cells were still actively growing. Tumors with combination therapy showed increased numbers of apoptotic bodies compared with monotherapy (Fig. 6F). Immunostaining for Ki67 and cleaved caspase-3 also confirmed that tumors after combination therapy had a significant reduction in overall proliferation and a dramatic increase in apoptosis (Fig. 6G, 6H). Similarly, although monotherapy with ICG001 or enzalutamide increased CK8/18+ cell population, a more significant increase was observed after dual treatment with both drugs. However, there was no dramatic change of P63 expression in different treatment groups except for slightly decreased with monotherapy of ICG001 (Fig. 6I). Finally, the PSA level in the serum was also decreased upon combination treatment compared with single-drug treatments, indicating activity of AR was inhibited (Fig. 6J). To determine the readout of efficacy of ICG001 in PDX tumors, we evaluated β-catenin in treated tissues. As demonstrated in Fig. 6K, only ICG001 reduced β-catenin level but not enzalutamide treatment. Combination treatment of enzalutamide and ICG001 did not show synergic effects on suppressing the level of β-catenin in tumors.
Figure 6.
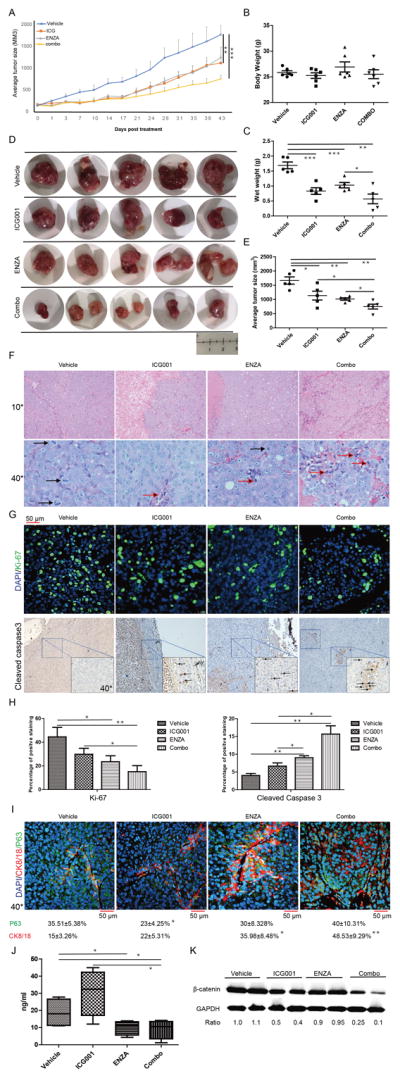
ICG001 and enzalutamide inhibit LuCaP35CR xenografts synergistically. A, Tumor growth curves (means±standard errors, n=5). B, Body weight on day 43. C, Wet weight of tumors. D, Tumors at the end of the study. E, Average tumor sizes on day 43. F, H&E staining of tumors. (Black arrows: mitotic cells, red arrow: apoptotic cells). G, IHC staining for Ki67 and cleaved caspase 3. H, Quantification of G. I, IHC staining for P63 and CK8/18. J, Inhibition of PSA levels by ICG001 and enzalutamide. Serum PSA levels were measured on day 43. K, Protein level of β-catenin in PDX tumors. *, p<0.05, **, 0.01<p<0.05, ***, p<0.01.
To examine treatment-associated toxicity within each group, we performed histopathologic examination of vital organs. Overall, no significant pathological changes were noted in any group. As shown in Supplementary Fig. S5A, all the vital organs were normal after 3 weeks of treatment; the livers did not show any vacuolar changes and there was no sign of inflammation at the renal pelvis in single treatment or combination treatment group. We concluded that both ICG001 alone and combination treatment were well tolerated.
Activation pattern of Wnt/β-catenin pathway switches during PCa progression
To explore the possibility of translating our findings into clinic, we stratified 498 PCa specimens into hormone therapy-sensitive (Gleason score <8) and -resistant groups (Gleason score ≥8) and performed GSEA. As indicated, the cell adhesion and focal adhesion function-related β-catenin show strong activation upon β-catenin upregulation in hormone therapy-resistance PCa tumors (Fig 7A and 7B). Several members of Wnt pathway were upregulated in therapy-resistant individuals compared to therapy-sensitive patients (Fig. 7C). In agreement, upregulation of stemness genes including NANOG, CD44, SOX10 and CD9 was also observed in hormone therapy-resistant patients (Fig. 7D). More interestingly, we found components of Wnt pathway to be activated in all the normal adjacent tissue, primary PCa and hormone therapy-resistant specimens PCa. To dissect the different functions of Wnt/β-catenin pathway in these three types of samples, we analyzed specific components involved in activation of the pathway and found significantly diverse alterations of molecules within each group. The data suggest that significant molecule shifts in different disease stages likely contribute to PCa progression (Fig. 7E), indicating that activation of Wnt/β-catenin pathway, especially different activation patterns, likely reflects various disease stages of PCa.
Figure 7.

Dataset analysis of 498 human PCa specimens from TCGA. A and B, Cell adhesion and focal adhesion pathways were activated in chemotherapy-resistant PCa revealed by GSEA. C and D, Heat maps to compare gene expression data from human specimens. E, Heat map of gene expression pattern compared in normal prostate tissues, primary PCa and therapy-resistant PCa. F and G, Gene expression of AR and β-catenin shown as RPKM in PCa samples. H, A predictive model for chemotherapy-resistant PCa via correlation of gene expression levels between AR and β-catenin.
Correlation between β-catenin and AR can be used as a predictable biomarker for chemotherapy-resistant PCa
To test whether the correlation between AR and β-catenin has any prognostic value in clinic, Activation of AR-associated transcription factor signaling pathway occurred in hormone therapy-resistant PCa was observed via GESA (supplementary Fig S5B). Further, we found a strong correlation between stem-like genes and β-catenin transcription level in therapy-resistant samples (supplementary Fig S5C), consistent with our cell culture-based findings (Fig. 5). In addition, increased levels of AR (p=0.00255) and downstream factor c-myc (p=0.02931) correlated with poor survival probability in PCa (supplementary Fig S5D and 5E). Kaplan-Meier curves shown higher β-catenin levels correlated with worse overall survival in both neuroblastoma and lung tumor (Supplementary Fig. S5F, S5G, S5H). To identify correlation of AR and β-catenin with disease progression, we explored mRNA levels of AR and β-catenin in adjacent normal prostate tissues, primary tumors and therapy-resistant tumors, respectively. As shown in Fig. 7F, we found low AR expression but high β-catenin expression in adjacent normal tissues compared with primary tumors. Of note, we observed high levels of both AR and β-catenin in resistant tumors compared to sensitive tumors (Fig. 7G). The collective data implies a low level of AR but a high level of β-catenin in normal tissue, and a high level of AR but a low level of β-catenin in primary PCa. Finally, upon progression into therapy-resistant PCa, the molecule expression pattern switches to high levels of both AR and β-catenin (Fig. 7H). These findings have important clinic implications. Because of high expression of AR, primary PCa usually responds well to androgen-deprivation therapy by suppressing AR function. Upon relapse of the disease, reactivation of AR signaling pathway and β-catenin pathway eventually drives tumors to be chemotherapy resistant. In support, expression of both AR and β-catenin exhibits a strong correlation with clinical parameters (Supplementary Table S6, Supplementary Fig. S5I). Specifically, p-values for correlations between lymph node metastasis and β-catenin and AR are 0.008 and 0.038, respectively. In addition, depth of tumor invasion is strongly correlated to β-catenin with p-value of 0.016. The correlations between follow-up days (survival) and the levels of β-catenin (p-value = 0.001) and AR (p-value = 0.007) are significant as well. Finally, the expression level of c-myc is also correlated to the overall survival (Supplementary Table S6). In conclusion, correlation between AR and β-catenin can be used to stratify patients and guide clinical practice in future PCa therapy.
Discussion
20% to 40% of PCa patients eventually end up with biochemical recurrence (32,33), when the patients are mainly treated with ADT, including surgical or medical castration methods (34). However, almost all of them eventually develop CRPC (35). Despite the fact that enzalutamide can effectively block AR signaling (9), it only improves the overall survival for several months. As expected, multiple mechanisms were reported as possible reasons for enzalutamide resistance (36–39). However, an unbiased approach is urgently needed to address this question systematically.
In this study, we employed RNA-seq in enzalutamide-resistant cell lines and patient specimens to characterize the cellular and molecular features of enzalutamide resistance in CRPC. We identified activation of Wnt and AR signaling pathways and the interaction of β-catenin and AR involved in enzalutamide resistance (Fig. 1). Wnt signaling was activated in normal adjacent prostate tissues but not in primary PCa. More significantly, this pathway was reactivated in enzalutamide-resistant cells, suggesting that reactivation of Wnt signaling may play an important role in acquisition of enzalutamide resistance (Fig. 1C). In addition, the correlation between AR and β-catenin of tumor (r=0.6171) is stronger than that of normal tissues (r=0.4516). Furthermore, there is strong correlation in chemotherapy-resistant PCa (r=0.3607). These data demonstrate that correlation between AR and β-catenin plays an important role not only in PCa initiation but also in progression to chemotherapy resistance.
A hallmark of Wnt pathway is stabilization and nuclear localization of β-catenin. Here, we showed β-catenin upregulated and mainly localized in the nucleus of enzalutamide-resistant cells, partially due to inhibition of β-TrCP-mediated polyubiquitination of β-catenin (Fig. 2). While inhibition of Wnt signaling improved sensitivity to enzalutamide in enzalutamide-resistant PCa, reactivation of Wnt pathway resulted in drug resistance in enzalutamide-sensitive cells (Fig. 3, 4). Of significance, the inhibitor of Wnt pathway ICG001 overcame enzalutamide resistance in two xenograft models (Fig. 3, 6). Such findings suggest an immediate clinical trial for combining Wnt signaling inhibitor with enzalutamide in CRPC. To explore underlying mechanisms, we then identified stem-like gene signature which was induced by activation of Wnt signaling upon development of drug resistance (Fig. 5).
Aberrant activation of Wnt signaling contributes to the progression of several major human cancers, including colorectal, liver and prostate cancer (16). To validate clinical impact of our findings, we showed activation of some components of Wnt pathway in normal prostate tissue, primary PCa and chemotherapy-resistant PCa. However, when we analyzed the molecules involved, we found that the molecular patterns were totally different at each stage of disease (Fig. 7), arguing that Wnt pathway is multifaceted in different stages of PCa. We showed here that the molecule pattern shift occurred during different disease stages was followed by reactivation of AR pathway, which may be one of the reasons for failure of ADT in clinic. As such, upregulation of stem-like genes contributes to drug resistance. In addition, expression levels of AR and c-myc, the target of Wnt signaling, were positively correlated with poor survival (supplementary Fig. S5D and S5E), suggesting that the correlation between AR and β-catenin might be a useful biomarker to stratify different disease stages. Of note, elevated β-catenin levels were correlated with poor prognosis in several cancer types (Supplementary Fig. S5F, S5G, S5H).
We divided specimens into three groups: normal tissues, primary PCa, chemotherapy-resistant PCa. We found a relatively high level of β-catenin but a low level of AR in normal tissues, a low level of β-catenin but a high level of AR in primary PCa, and high levels of both β-catenin and AR in chemotherapy-resistant PCa (Fig. 7H). A high level of AR in primary PCa is consistent with the fact that PCa at this stage is sensitive to ADT. After a period of ADT treatment, relapse, which is characterized with a higher level of β-catenin but a lower level of AR compared with normal prostate, happens. Eventually, a higher level of β-catenin leads to reactivation of AR, thus chemotherapy resistance. We acknowledge that we likely need more normal, chemotherapy-sensitive and -resistant tumors to determine the cutoff of AR and β-catenin levels to translate the finding into clinic. We also acknowledge that the underlying mechanisms for β-catenin activation during acquisition of enzalutamide resistance remain unclear. Ideally, it’s best to obtain enough enzalutamide-resistant specimens to perform bioinformatic and biological study to explore the underlying mechanisms.
In summary, we showed that activation of Wnt pathway induces expression of stem-like genes, eventually contributing to enzalutamide resistance by reactivating AR signaling. Furthermore, the correlation of AR and β-catenin levels with PCa stages has the potential to classify patients who will likely resist to chemotherapy during treatment, thus helping to improve clinical practice in future (supplementary Fig S5J).
Supplementary Material
Acknowledgments
This work was supported by NIH grants R01 CA157429 (X. Liu), R01 CA192894 (X. Liu), R01 CA196835 (X. Liu), and R01 CA196634 (X. Liu). The work was also supported by Purdue University Center for Cancer Research (P30 CA023168).
Footnotes
Statement of significance
Wnt/β-catenin inhibition resensitizes prostate cancer cells to enzalutamide.
Disclosures of Potential Conflicts of Interest: We declare no conflict of interest.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Kohli M, Tindall DJ. New developments in the medical management of prostate cancer. Mayo Clin Proc. 2010;85:77–86. doi: 10.4065/mcp.2009.0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.James ND, Sydes MR, Clarke NW, Mason MD, Dearnaley DP, Spears MR, et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet. 2016;387:1163–77. doi: 10.1016/S0140-6736(15)01037-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lonergan PE, Tindall DJ. Androgen receptor signaling in prostate cancer development and progression. J Carcinog. 2011;10:20. doi: 10.4103/1477-3163.83937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Isaacs JT, Furuya Y, Berges R. The role of androgen in the regulation of programmed cell death/apoptosis in normal and malignant prostatic tissue. Semin Cancer Biol. 1994;5:391–400. [PubMed] [Google Scholar]
- 6.Chmelar R, Buchanan G, Need EF, Tilley W, Greenberg NM. Androgen receptor coregulators and their involvement in the development and progression of prostate cancer. Int J Cancer. 2007;120:719–33. doi: 10.1002/ijc.22365. [DOI] [PubMed] [Google Scholar]
- 7.Niu Y, Altuwaijri S, Lai KP, Wu CT, Ricke WA, Messing EM, et al. Androgen receptor is a tumor suppressor and proliferator in prostate cancer. Proc Natl Acad Sci U S A. 2008;105:12182–7. doi: 10.1073/pnas.0804700105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schally AV, Block NL, Rick FG. New therapies for relapsed castration-resistant prostate cancer based on peptide analogs of hypothalamic hormones. Asian J Androl. 2015;17:925–8. doi: 10.4103/1008-682X.152819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–97. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 10.Froehner M, Wirth MP. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. 2014;371:1755. doi: 10.1056/NEJMc1410239. [DOI] [PubMed] [Google Scholar]
- 11.Claessens F, Helsen C, Prekovic S, Van den Broeck T, Spans L, Van Poppel H, et al. Emerging mechanisms of enzalutamide resistance in prostate cancer. Nat Rev Urol. 2014;11:712–6. doi: 10.1038/nrurol.2014.243. [DOI] [PubMed] [Google Scholar]
- 12.Liu C, Lou W, Zhu Y, Yang JC, Nadiminty N, Gaikwad NW, et al. Intracrine Androgens and AKR1C3 Activation Confer Resistance to Enzalutamide in Prostate Cancer. Cancer Res. 2015;75:1413–22. doi: 10.1158/0008-5472.CAN-14-3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khramtsov AI, Khramtsova GF, Tretiakova M, Huo D, Olopade OI, Goss KH. Wnt/beta-catenin pathway activation is enriched in basal-like breast cancers and predicts poor outcome. Am J Pathol. 2010;176:2911–20. doi: 10.2353/ajpath.2010.091125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tao J, Calvisi DF, Ranganathan S, Cigliano A, Zhou L, Singh S, et al. Activation of beta-catenin and Yap1 in human hepatoblastoma and induction of hepatocarcinogenesis in mice. Gastroenterology. 2014;147:690–701. doi: 10.1053/j.gastro.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Damsky WE, Curley DP, Santhanakrishnan M, Rosenbaum LE, Platt JT, Gould Rothberg BE, et al. beta-catenin signaling controls metastasis in Braf-activated Pten-deficient melanomas. Cancer Cell. 2011;20:741–54. doi: 10.1016/j.ccr.2011.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Polakis P. Wnt signaling in cancer. Cold Spring Harb Perspect Biol. 2012:4. doi: 10.1101/cshperspect.a008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Valkenburg KC, Graveel CR, Zylstra-Diegel CR, Zhong Z, Williams BO. Wnt/beta-catenin Signaling in Normal and Cancer Stem Cells. Cancers (Basel) 2011;3:2050–79. doi: 10.3390/cancers3022050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miki T, Yasuda SY, Kahn M. Wnt/beta-catenin signaling in embryonic stem cell self-renewal and somatic cell reprogramming. Stem Cell Rev. 2011;7:836–46. doi: 10.1007/s12015-011-9275-1. [DOI] [PubMed] [Google Scholar]
- 19.Yang K, Wang X, Zhang H, Wang Z, Nan G, Li Y, et al. The evolving roles of canonical WNT signaling in stem cells and tumorigenesis: implications in targeted cancer therapies. Lab Invest. 2016;96:116–36. doi: 10.1038/labinvest.2015.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lawson DA, Witte ON. Stem cells in prostate cancer initiation and progression. J Clin Invest. 2007;117:2044–50. doi: 10.1172/JCI32810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bisson I, Prowse DM. WNT signaling regulates self-renewal and differentiation of prostate cancer cells with stem cell characteristics. Cell Res. 2009;19:683–97. doi: 10.1038/cr.2009.43. [DOI] [PubMed] [Google Scholar]
- 22.Korkaya H, Paulson A, Charafe-Jauffret E, Ginestier C, Brown M, Dutcher J, et al. Regulation of mammary stem/progenitor cells by PTEN/Akt/beta-catenin signaling. PLoS Biol. 2009;7:e1000121. doi: 10.1371/journal.pbio.1000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yokoyama NN, Shao S, Hoang BH, Mercola D, Zi X. Wnt signaling in castration-resistant prostate cancer: implications for therapy. Am J Clin Exp Urol. 2014;2:27–44. [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Z, Hou X, Shao C, Li J, Cheng JX, Kuang S, et al. Plk1 inhibition enhances the efficacy of androgen signaling blockade in castration-resistant prostate cancer. Cancer Res. 2014;74:6635–47. doi: 10.1158/0008-5472.CAN-14-1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dai M, Thompson RC, Maher C, Contreras-Galindo R, Kaplan MH, Markovitz DM, et al. NGSQC: cross-platform quality analysis pipeline for deep sequencing data. BMC Genomics. 2010;11(Suppl 4):S7. doi: 10.1186/1471-2164-11-S4-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu C, Kato Y, Zhang Z, Do VM, Yankner BA, He X. beta-Trcp couples beta-catenin phosphorylation-degradation and regulates Xenopus axis formation. Proc Natl Acad Sci U S A. 1999;96:6273–8. doi: 10.1073/pnas.96.11.6273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao C, Chen G, Romero G, Moschos S, Xu X, Hu J. Induction of Gsk3beta-beta-TrCP interaction is required for late phase stabilization of beta-catenin in canonical Wnt signaling. J Biol Chem. 2014;289:7099–108. doi: 10.1074/jbc.M113.532606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olsen JR, Azeem W, Hellem MR, Marvyin K, Hua Y, Qu Y, et al. Context dependent regulatory patterns of the androgen receptor and androgen receptor target genes. BMC Cancer. 2016;16:377. doi: 10.1186/s12885-016-2453-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Corey E, Quinn JE, Buhler KR, Nelson PS, Macoska JA, True LD, et al. LuCaP 35: a new model of prostate cancer progression to androgen independence. Prostate. 2003;55:239–46. doi: 10.1002/pros.10198. [DOI] [PubMed] [Google Scholar]
- 31.Mostaghel EA, Marck BT, Plymate SR, Vessella RL, Balk S, Matsumoto AM, et al. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res. 2011;17:5913–25. doi: 10.1158/1078-0432.CCR-11-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lennernas B, Edgren M, Haggman M, Norlen BJ, Nilsson S. Postoperative radiotherapy after prostatectomy--a review. Scand J Urol Nephrol. 2003;37:10–5. doi: 10.1080/00365590310008613. [DOI] [PubMed] [Google Scholar]
- 33.Ahlering TE, Skarecky DW. Long-term outcome of detectable PSA levels after radical prostatectomy. Prostate Cancer Prostatic Dis. 2005;8:163–6. doi: 10.1038/sj.pcan.4500788. [DOI] [PubMed] [Google Scholar]
- 34.Kozlowski JM, Ellis WJ, Grayhack JT. Advanced prostatic carcinoma. Early versus late endocrine therapy. Urol Clin North Am. 1991;18:15–24. [PubMed] [Google Scholar]
- 35.Karantanos T, Corn PG, Thompson TC. Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene. 2013;32:5501–11. doi: 10.1038/onc.2013.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy DA, Doshi S, et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide) Cancer Discov. 2013;3:1030–43. doi: 10.1158/2159-8290.CD-13-0142. [DOI] [PubMed] [Google Scholar]
- 37.Yuan X, Cai C, Chen S, Chen S, Yu Z, Balk SP. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene. 2014;33:2815–25. doi: 10.1038/onc.2013.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D, et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013;3:1020–9. doi: 10.1158/2159-8290.CD-13-0226. [DOI] [PubMed] [Google Scholar]
- 39.Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013;73:483–9. doi: 10.1158/0008-5472.CAN-12-3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.