

Abstract
Activation of the Wnt/β-catenin signaling pathway drives colorectal cancer (CRC) growth by deregulating expression of downstream target genes including the c-Myc proto-oncogene. The critical targets that mediate the functions of oncogenic c-Myc in CRC have yet to be fully elucidated. Previously, we showed that activation of PI3K/Akt/mTOR contributes to CRC growth and metastasis. Here we show that Deptor, a suppressor of mTOR, is a direct target of Wnt/β-catenin/c-Myc signaling in CRC cells. Inhibition of Wnt/β-catenin or knockdown of c-Myc decreased, while activation of Wnt/β-catenin or overexpression of c-Myc increased, the expression of Deptor. c-Myc bound the promoter of Deptor and transcriptionally regulated Deptor expression. Inhibition of Wnt/β-catenin/c-Myc signaling increased mTOR activation, and the combination of Wnt and Akt/mTOR inhibitors enhanced inhibition of CRC cell growth in vitro and in vivo. Deptor expression was increased in CRC cells; knockdown of Deptor induced differentiation, decreased expression of B lymphoma Mo-MLV insertion region 1 (Bmi1), and decreased proliferation in CRC cell lines and primary human CRC cells. Importantly, our work identifies Deptor as a downstream target of the Wnt/β-catenin/c-Myc signaling pathway, acting as a tumor promoter in CRC cells. Moreover, we provide a molecular basis for the synergistic combination of Wnt and mTOR inhibitors in treating CRC with elevated c-Myc.
Keywords: Wnt, c-Myc, Deptor, mTOR, CRC
INTRODUCTION
Wnt/β-catenin signaling plays an important role in normal tissue development; conversely, deregulation of Wnt signaling contributes to tumorigenesis.(1, 2) Wnt binding stabilizes the transcription factor β-catenin, which enters the nucleus to regulate Wnt pathway target genes such as Axin2, c-Myc, and cyclin D1.(1) More than 90% of colorectal cancers (CRCs) contain a mutation in APC or β-catenin, and these mutations stabilize β-catenin and activate Wnt signaling.(3) Intercepting and blocking the Wnt pathway at various points in the signaling cascade offers an attractive approach for CRC chemoprevention and therapeutics.
mTOR is a serine/threonine kinase that exists in two complexes(4): mTORC1 (containing mTOR, Raptor, etc.) and mTORC2 (containing mTOR, Rictor, etc.). The phosphatidylinositol 3-kinase (PI3K)/Akt pathway is an important upstream mediator of mTORC1, which exerts its effects through growth factor-mediated activation of Akt via PI3K.(5) mTORC1 influences cell growth and proliferation by promoting biosynthesis of proteins, lipids, and organelles, and by limiting catabolic processes. Regulation of protein biosynthesis by mTORC1 occurs via phosphorylation of ribosomal protein S6 kinase 1 (S6K1) and eukaryotic initiation factor 4E-binding protein 1 (4E-BP1).(5) mTORC2 is a major hydrophobic kinase that plays important roles in Akt phosphorylation and in the regulation of actin cytoskeleton.(5) Activation of PI3K, by activating mutations or loss of the tumor suppressor protein PTEN, is associated with growth and progression of a number of cancers including CRC. Activation of PI3K/Akt/mTOR increases normal intestinal proliferation.(6) Conversely, inhibition of PI3K/Akt/mTOR decreases viability and growth, and increases differentiation and apoptosis of human CRC cells.(7) Although Wnt activation in mice with APC mutation results in the activation of mTORC1 signaling in intestinal polyps,(8–10) the interaction between Wnt signaling and mTOR signaling in CRC cells is not well delineated.
The DEP domain containing mTOR interacting (Deptor) protein binds and inhibits mTOR, whose activity is mostly hyperactivated in the majority of human tumors.(4) Conversely, the potential role of Deptor as an oncogene has been described in certain tumor types.(11) Deptor is overexpressed in many tumors including breast, prostate, and lung cancers, and chronic myeloid leukemia.(11–14)Although Deptor acts as a suppressor of mTOR, Deptor has been shown to contribute to cancer cell proliferation and survival of various cancer types.(15–19) Deptor has emerged as an attractive pharmacological target for novel cancer therapy.(11, 20) In our current study, we identify Deptor as a direct target of Wnt/β-catenin/c-Myc signaling in CRCs. Inhibition of mTOR enhances the anticancer effect of Wnt inhibition. Moreover, Deptor is required for CRC cell proliferation, and its expression is elevated in human CRCs.
MATERIALS AND METHODS
Materials
FH535 and β-actin were from Sigma Chemical Co. (St. Louis, MO). ICG001 and MK2206 were from Selleckchem (Radnor, PA). AZD8055 was from Apexbio (Houston, TX). Rabbit Monoclonal anti-c-Myc antibody was from Epitomics Inc. (Burlingame, CA). Rabbit anti-Deptor, β-catenin, cyclin D1, phospho-S6 (pS235/236), S6, phospho-Akt (S473), Akt, and Axin2 antibodies were from Cell Signaling (Beverly, MA). Rabbit anti-HMGCS2 was from Abcam (Cambridge, MA). Adenovirus vectors encoding GFP (Ad-GFP) and human c-Myc (Ad-c-Myc) were from Vector BioLabs (Malvern, PA). Human c-Myc, β-catenin and non-targeting control siRNA SMARTpool were from Dharmacon, Inc. (Lafayette, CO). MISSION control shRNA and shRNAs to human Deptor shRNAs constructed in pLKO.1-puro vector were from Sigma-Aldrich. Human tissue collection was performed in accordance with the US Common Rule. Collections utilized an IRB approved protocol, with patient informed consent. Excess surgical tissue was collected by an honest broker service de-identified; tissue and associated clinical data was dispensed to the researchers.
Animal studies
All animal procedures were conducted with approval and in compliance with University of Kentucky Institutional Animal Care and Use Committee.
ApcMin mice
C57BL/6J-ApcMin mice (stock number 002020) were from the Jackson Laboratory (Sacramento, CA). The presence and morphology of intestinal adenomas were confirmed by H&E staining and by immunohistochemical (IHC) analysis for either Deptor, β-catenin or c-Myc.
HT29 and MC38 xenograft model
HT29 cells (2 × 106 cells in PBS, 200 μl/mouse) were subcutaneously injected into athymic nude mice (male, 6-week-old), from Jackson Laboratory. MC38 cells (1 × 106 cells in PBS, 200 μl/mouse) were subcutaneously injected into C57BL/6J mice (female, 7-week-old), from Jackson Laboratory. Prior to initiation of treatment, mice were randomized among control and treated groups and treated with AZD8055, ICG001, AZD8055 plus ICG001 and vehicle alone when the subcutaneous tumors grew 8 days (HT29) or 5 days (MC38) after tumor cell injections. ICG001 was formulated in 3% DMSO, 50% PEG300 and 0.5% Tween 80 as suggested by Selleckchem, and administered intraperitoneally at a dose of 100 mg/kg daily. AZD8055 was formulated in 30% capsitol as described (21) and administered orally at a dose of 30 mg/kg daily. For combination treatment, both drugs were given concurrently. Control mice received vehicle alone for both drugs. The average tumor diameter (two perpendicular axes of the tumor) was measured in control and treated groups using a caliper. The data are expressed as the increase or decrease in tumor volume in mm3 (mm3 = π/6 × (larger diameter) × (smaller diameter)2). Tumors were excised for IHC.
Cell culture and transfection
Human CRC cell lines, HT29 and DLD1, were maintained in McCoy’s 5A supplemented with 10% fetal calf serum (FCS), and DMEM supplemented with 10% FCS, respectively. HT29 and DLD1 cells were tested for authentication via STR profiling in February 2016 by Genetica DNA Laboratories (LabCorp Specialty Testing Group; Burlington, NC). Authentications were confirmed by a 100% match in comparison to the reference STR profiles from ATCC. In addition, both cell lines were tested for mycoplasma contamination (Genetica DNA Laboratories) and were found to be negative. The human CRC cell line, LS174T, purchased in February 2016 from ATCC, was maintained in MEM supplemented with 10% FCS. The mouse CRC cell line MC38 was purchased in November 2017 from Kerafast (Boston, MA) and was maintained in DMEM supplemented with 10% FCS, 2mM glutamine, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, and 10 mM Hepe. Cells were transfected with the siRNA duplexes (100 nM) by electroporation (Gene Pulser, Bio-Rad). Cells were infected with lentiviral vectors containing control shRNA or shRNA to human Deptor and stably expressing cells were selected with puromycin at a concentration of 5 μg/ml.
Primary human CRC cells
Patient-derived xenografts (PDXs) were established in NOD-SCID-IL2rg-/- (NSG) mice using freshly resected CRC specimens from patients treated at UK Chandler Medical Center. Primary CRC Pt93 and Pt130 cells were isolated and established from PDX tumors and cultured in DMEM supplemented with 10% FBS and 1% penicillin–streptomycin as previously described.(22) These cell lines were authenticated as unique human cell lines and tested for mycoplasma contamination and found to be negative.
Cell proliferation and colony formation assays
Cell proliferation was analyzed by counting the number of viable cells in response to the knockdown of Deptor or treatment with inhibitors. Colony formation assays were performed as previously described.(23) Briefly, HT29 cells were seeded in 12-well plates at approximately 250 cells/well. Cells were treated with inhibitors 24 h after seeding. During colony growth, the culture medium was replaced every 3 days. On the 10th day after seeding, the cells were fixed and then stained with crystal violet.
Western blot analysis
Total protein was resolved on a 10% polyacrylamide gel and transferred to PVDF membranes. Membranes were incubated for 1 h at room temperature in blotting solution. Deptor, c-Myc, β-catenin, Axin2, phospho-Akt (S473), Akt, phospho-S6 (pS235/236), S6, cyclin D1, HMGCS2 and β-actin were detected as we have described previously.(24)
Quantitative real time RT-PCR analysis
Total RNA was extracted and treated with DNase (RQ1, Promega, Madison, WI). Synthesis of cDNA was performed with 1 μg of total RNA using reagents in the TaqMan® Reverse Transcription Reagents Kit (ABI #N8080234). TaqMan® probe and primers were purchased from Applied Biosystems (Foster City, CA). Quantitative real time RT-PCR analysis was performed as we have described previously.(24)
Electrophoretic mobility shift assays (EMSAs)
EMSAs were performed with 32P-labeled consensus oligonucleotides (wide type [WT]: 5′-TCCTGACCTCAGGTGATCTGCCCGCC-3′; mutant [MUT]: 5′TCCTGACCTCAGGGAATCTGCCCGCC-3′) using the Gel Shift Assay Systems (Promega) according to the manufacturer’s protocol as we have described previously.(25)
Chromatin immunoprecipitation (ChIP) assay
The ChIP assay was performed using the ChIP-IT Express Enzymatic Kit (Active Motif) according to the manufacturer’s protocol as we have described previously.(25) PCR of the human Deptor promoter containing the c-Myc binding sites was performed using total (input) or immunoprecipitated chromatin with the following pair of oligonucleotide primers: 5′- AGTCTCGCTCTTGTTGC-3′ and 5′-AGAATGCAGGCTTGGAG-3′.
Immunohistochemical analysis
Immunohistochemistry staining was performed as we have described previously.(24) Tissue was processed for routine IHC staining using the antibodies against Deptor and c-Myc (Santa Cruz Biotechnology, Santa Cruz, CA), phospho-S6 (pS235/236), phospho-Akt (S473), β-catenin (Cell Signaling, Beverly, MA) and Cd8α (Thermo Fisher Scientific, San Diego, CA). Negative controls (including no primary antibody or isotype-matched mouse immunoglobulin G) were used in each assessment. Fifty-six clinical specimens of primary CRC with matching normal colonic mucosa were analyzed for expression of Deptor (Cell Signaling, Beverly, MA). Blind scoring performed by a pathologist used a semi-quantitative method. The extent of expression score was assessed on a scale of 0 to 3 (no positive cells = 0, <10% = 1, 10%–50% = 2, positive staining of >50% = 3); the intensity score was also measured on a scale of 0 to 3 (negative = 0, weak = 1, moderate = 2, strong = 3). Multiplication of the values for intensity and extent of expression provided a score for immunoreactivity as we have described previously.(26)
Statistical analysis
Pairwise comparisons for two groups were performed using two-sample t-test or analysis of variance for multiple groups with contrast statements. Adjustments for multiple testing between groups within an experiment were performed using Holm’s p-value adjustment method. Comparisons were performed for control versus ICG001, MK2206 and AZD8055 alone or with combination treatment, control shRNA versus Deptor shRNA, or GFP versus c-Myc. Tumor volume growth curves were compared across treatment groups using linear mixed models with linear and quadratic terms for time and random effects for the intercept and time factors. Contrast statements were used to assess differences in tumor growth curves between groups. RNA-Seq data for 456 primary tumor and 41 matched normal samples from The Cancer Genome Atlas (TCGA) colorectal cancer study were downloaded from the Genomic Data Commons (GDC) database (Access date June, 2017). The expression of Deptor, MYC, CTNNB1 and HMGCS2 in tumor vs. normal samples was compared by the linear mixed model. The correlation between expressions of each pair of genes in tumor samples was quantified by Spearman’s correlation coefficient. The analyses were performed by SAS (version 9.4) and R (version 3.2.3).
RESULTS
Regulation of Deptor expression by Wnt/β-catenin signaling in CRC cells
Wnt/β-catenin and mTOR signaling are two critical pathways controlling CRC cell proliferation.(2, 5) Our previous studies have shown that inhibition of either of these pathways significantly inhibits CRC growth.(5, 27, 28) To better delineate the interaction between these two pathways in CRC, LS174T, HT29 and DLD1 cells were treated with the Wnt small molecule inhibitor ICG001, which selectively binds to CBP and prevents its interaction with β-catenin, resulting in the suppression of Wnt/β-catenin-driven gene expression.(29) As shown in Fig. 1A, treatment with ICG001 induced mTOR activation in LS174T and HT29 cells as noted by the increased expression of phospho-Akt and phospho-S6. Deptor protein, which binds mTOR through a PDZ domain, associates with both mTORC1 and mTORC2 and inhibits their activity.(11) Consistent with the inhibition of mTOR signaling, treatment with ICG001 resulted in a dose-dependent inhibition of Deptor protein expression in LS174 and HT29 cells. Similarly, treatment with ICG001 increased Akt phosphorylation and decreased Deptor expression in DLD cells (Fig. 1A). In contrast, ICG001 treatment did not affect the phospho-S6 levels in DLD1 cells, suggesting a differential effect on mTORC1 activity (Fig. 1A). Consistently, treatment with FH535, which inhibits Wnt and peroxisome proliferator-activated receptor (PPAR) signaling,(30) also decreased Deptor protein expression and increased Akt phosphorylation (Supplemental figure 1A). Interestingly, treatment with FH535 decreased S6 phosphorylation which may be through the inhibition of PPAR signaling since PPAR activation is known to increase mTORC1 activity.(31)
Figure 1. Regulation of Deptor expression by Wnt/β-catenin signaling.
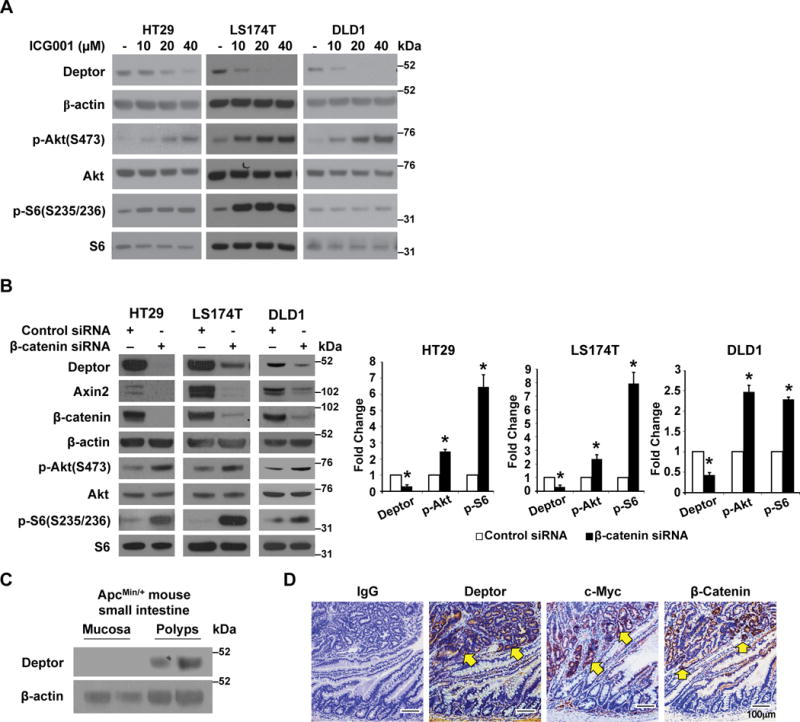
A. Inhibition of Wnt/β-catenin signaling decreases Deptor protein expression. Treatment with Wnt inhibitor ICG001 decreased Deptor expression. LS174T, HT29, and DLD1 cells were treated with Wnt inhibitors ICG001 for 24 h. Cells were lysed and western blot analysis was performed using antibodies against Deptor, p-Akt, Akt, p-S6, S6, and β-actin. The images are representative of three independent experiments. B. Knockdown of β-catenin results in the induction of mTOR activation and a decrease of Deptor protein expression. HT29, LS174T and DLD1 cells were transfected with β-catenin or non-targeting control siRNA. After 48 h incubation, transfected cells were lysed and western blot analysis was performed using antibodies against Deptor, Axin2, β-catenin, β-actin, p-Akt, Akt, p-S6 and S6. The images are representative of three independent experiments. Deptor, p-Akt and p-S6 signals from three separate experiments were quantitated densitometrically and expressed as fold change with respect to β-actin, total Akt or total S6, respectively. (n= 3, data represent mean±SEM; *P<0.01 versus control siRNA). C&D. Activation of Wnt signaling increases Deptor expression. Tumors of ApcMin/+ mice show increased expression of Deptor (C). Western blots of extracts from either normal intestine mucosa or a pool of polyps from the small intestine of ApcMin/+ mice (n=2). Representative IHC staining of Deptor, c-Myc and β-catenin in polyp and adjacent normal ileum (D). Scale bars, 100 μm.
To further demonstrate the regulation of Wnt/β-catenin on Deptor expression and mTOR activation, LS174T, HT29 and DLD1 cells were transiently transfected with control siRNA or siRNA targeting β-catenin. As shown in Fig. 1B and Supplemental figure 1B, knockdown of β-catenin resulted in the activation of mTORC1 and mTORC2 as noted by increased expression of phospho-Akt and phospho-S6. Consistent with mTOR activation, knockdown of β-catenin significantly decreased Deptor protein expression. As expected, knockdown of Deptor in these cell lines resulted in mTOR activation (Supplemental figure 1C). Together, these results demonstrate that inhibition of Wnt/ β-catenin results in decreased Deptor expression and mTORC activation in CRC cells.
To further determine the regulation of Deptor expression by Wnt activation, we treated 293T and HCT116 cells with Wnt conditioned medium as we have described previously.(32) Activation of Wnt signaling increased Deptor protein expression (Supplemental figure 1D) and mRNA levels (Supplemental figure 1E). Next, we examined the expression of Deptor protein in the intestinal polyps and the normal mucosa of ApcMin+ mice by western blotting and immunostaining. Western blot analysis showed a significant increase of Deptor protein expression in the polyps as compared with the normal mucosa, which demonstrated minimal to no Deptor expression (Fig. 1C). Deptor protein was expressed strongly in the adenoma epithelium where Wnt signaling was activated as noted by the similar pattern of staining for β-catenin and its target c-Myc (Fig. 1D), which suggests induction of Deptor by Wnt activation.
c-Myc directly binds the Deptor promoter and regulates Deptor expression in CRC cells
T-cell factor/Lymphoid enhancer factor (TCF/LEF) transcription factors bind Wnt responsive DNA elements (WREs) and recruit the β-catenin transcriptional co-activator to activate target gene expression.(33) However, we could not identify a consensus TCF/LEF binding site in the promoter region of Deptor, suggesting that Wnt/β-catenin signaling may regulate Deptor indirectly, probably through other β-catenin target genes. Increased expression of c-Myc by aberrant Wnt/β-catenin signaling drives colorectal carcinogenesis.(34) To test whether c-Myc regulates Deptor expression, HT29, LS174T, and DLD1 cells were infected with an adenovirus construct encoding GFP control or human c-Myc. Overexpression of c-Myc increased Deptor protein expression (Fig. 2A) along with the inhibition of mTOR activation as shown by the decreased expression of phospho-Akt and phospho-S6. To further demonstrate the role of c-Myc in the regulation of Deptor expression, HT29, LS174T and DLD1 cells were transiently transfected with control siRNA or siRNA targeting c-Myc. As shown in Fig. 2B, knockdown of c-Myc resulted in the activation of mTOR as shown by the increased expression of phospho-Akt and phospho-S6. Consistent with the mTOR activation, knockdown of c-Myc significantly decreased Deptor protein expression. Importantly, these results further demonstrate that Wnt/β-catenin/c-Myc regulates Deptor expression, which affects mTOR activation in CRC cells.
Figure 2. c-Myc controls Deptor expression and mTOR activation in CRC cells.

A. HT29, LS174T and DLD1 cells were infected with a recombinant adenovirus encoding the human c-Myc or vector control encoding GFP. After 48 h, cells were lysed and protein extracted. Deptor, c-Myc, p-Akt, Akt, p-S6, S6 and β-actin were determined by western blotting. The images are representative of three independent experiments. Deptor, p-Akt and p-S6 signals from three separate experiments were quantitated densitometrically and expressed as fold change with respect to β-actin, total Akt or total S6, respectively. (n= 3, data represent mean±SEM; *P<0.01 versus control GFP). B. HT29, LS174T and DLD1 cells were transfected with c-Myc or non-targeting control siRNA. After 48 h incubation, transfected cells were lysed and western blot analysis was performed using antibodies against Deptor, c-Myc, p-Akt, Akt, p-S6, S6 and β-actin. The images are representative of three independent experiments. Deptor, p-Akt and p-S6 signals from three separate experiments were quantitated densitometrically and expressed as fold change with respect to β-actin, total Akt or total S6, respectively. (n= 3, data represent mean±SEM; *P<0.01 versus control siRNA). C. HT29 cells were infected with a recombinant adenovirus encoding human c-Myc or vector control encoding GFP. After 48 h, cells were lysed and extracted for RNA. Deptor mRNA expression was assessed by real-time RT-PCR. (n=3, data represent mean ± SEM; *P<0.01 versus GFP). D. Promoter region of the Deptor gene with a putative c-Myc binding site. E. ChIP assays assessing the binding of c-Myc to the Deptor promoter. Normal rabbit IgG and anti-c-Myc antibody were used to precipitate chromatin DNA fragments as indicated. F. EMSA assays. Nuclear protein extracted from HT29 cells was incubated with 32P-labeled wild type (WT) or mutant (MUT) oligonucleotide and DNA binding was analyzed. Unlabeled WT or MUT oligonucleotide were added in molar excess to confirm binding specificity (competitor). G. Nuclear extracts were incubated with 32P-labeled WT oligonucleotide probe alone or in the presence of specific antibody to c-Myc. Normal rabbit IgG was used as negative control. The images are representative of three independent experiments.
To determine whether c-Myc also increases Deptor mRNA expression, RNA from HT29 cells infected with Ad-c-Myc was analyzed. Overexpression of c-Myc increased Deptor mRNA expression, suggesting a transcriptional regulation of Deptor by c-Myc (Fig. 2C). c-Myc binds to E-box sequences near the core promoter elements of actively transcribed genes and causes transcriptional amplification of its target genes.(35) An essential binding site for c-Myc is 5′-CAGGTG-3′,(35) which we identified in the Deptor promoter (Fig. 2D). To determine whether c-Myc binds to the Deptor promoter in CRC cells, we performed a ChIP assay using an anti-c-Myc antibody in HT29 cells. Cross-linked chromatin was prepared from HT29 cells and immunoprecipitation was performed using either the anti-c-Myc antibody or IgG, and the sequence (-1131/-779 bp) containing the putative c-Myc binding site, was amplified. As shown in Fig. 2E, PCR analysis demonstrated that c-Myc binds to the Deptor promoter. To further confirm c-Myc binding, EMSAs were performed. As shown in Fig. 2F, three major binding bands were found in the complex with wild type (WT) probe. Binding specificity was confirmed by competition studies using cold WT (5′-CAGGTG-3′) and mutant (MUT, 5′-CAGGGA-3′) competitor probes. Addition of cold WT probe, but not cold MUT probe, abolished bands a and b. Moreover, the binding specificity was further demonstrated by the dimunition of binding of bands a and b that was noted in the complex using the MUT probe. Furthermore, addition of anti-c-Myc antibody to the mixture abolished bands a and b (Fig. 2G). Together, these complementary EMSA results further demonstrated c-Myc binding to the Deptor promoter, suggesting that c-Myc regulates Deptor expression through recruitment to the promoter.
Co-targeting Wnt/β-catenin and mTOR signaling pathways leads to augmented anticancer effects
Inhibition of Wnt/β-catenin signaling increases sensitivity to chemotherapeutic agents in cancers.(29, 36) Our data show that suppression of Wnt/β-catenin activates mTOR signaling in CRC cells. As a result, we speculated that mTOR activation would counteract the anticancer effects of Wnt/β-catenin inhibition. If this hypothesis is correct, then the concurrent suppression of the Wnt/β-catenin axis and blockage of mTOR activation could be predicted to augment the growth inhibitory effects. We examined the effects of either a pan-Akt inhibitor MK2206(37) or mTOR inhibitor AZD8055(21) combined with the Wnt inhibitor, ICG001, on the growth of HT29 cells. Importantly, we found that the combination of ICG001 with either MK2206 or AZD8055 synergistically inhibited HT29 cell growth compared with single agent treatment (Fig. 3A). Consistently, treatment with ICG001 increased phosphorylation of Akt and S6; this induction was blocked by combination treatment with either MK2206 or AZD8055 (Supplemental figure 2A). Similar results on growth inhibition were noted in 10-day colony formation assays; the combination of the two agents significantly decreased colony size and number compared with either single agent alone (Fig. 3B). Similar effects were also observed in LS174T (Fig. 3C) and primary human pt130 CRCs (Fig. 3D), derived from one of our PDX tumors, after combination treatment with ICG001 with either MK2206 or AZD8055. Collectively, our results demonstrate that the combination of a Wnt/β-catenin inhibitor and either an Akt or mTOR inhibitor results in an augmented growth inhibition of CRC cells.
Figure 3. Combined inhibition of Wnt and Akt/mTOR results in synergistic inhibition of CRC cell proliferation.
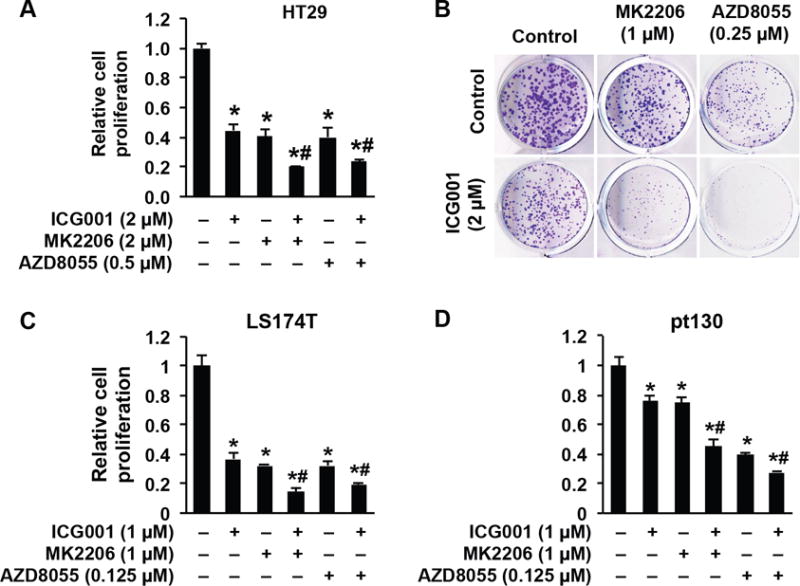
A. HT29 cells were treated with the indicated inhibitors for 3 d. Cell proliferation was determined using the Vi-CELL XR 2.03 cell viability analyzer. B. HT29 cells were treated with the indicated inhibitors for 10 d. Colony formation was analyzed. C&D. LS174T (C) and primary human CRC cells (pt130) (D) were treated with the indicated inhibitors for 3 d. Cell proliferation was determined using the Vi-CELL XR 2.03 cell viability analyzer. (n=3, data represent mean ± SEM; *p<0.01 vs control; #p<0.01 vs single inhibitor). Data are from one of three independent experiments with similar results.
mTOR inhibition enhances the antitumor activity of ICG001 in vivo
The profound anti-proliferative effects of the combination of Wnt and mTOR inhibitors in vitro suggest that targeting both Wnt and mTOR signaling may be a rational strategy for the treatment of CRC. To explore the feasibility of this therapeutic strategy, we tested the efficacy of inhibiting mTOR and Wnt in HT29 tumor xenografts in vivo. The Wnt inhibitor ICG001, at a dosage of 100 mg/kg, has shown to be an effective inhibitor of tumor xenograft growth.(38) Although the mTOR inhibitor AZD8055, at 10 mg/kg, was shown to be an effective inhibitor of tumor xenograft growth,(21) our preliminary study found that AZD8055, at 15 mg/kg, exhibited only a minor effect on HT29 xenograft growth. Therefore, we treated athymic nude mice bearing established HT29 xenografts with AZD8055 (30 mg/kg), ICG001 (100 mg/kg), a combination of both drugs, or vehicle control daily for 27 days. Administration of AZD8055 or ICG001 alone slowed the growth of tumors. In contrast, treatment with the combination of ICG001 and AZD8055 strongly suppressed tumor growth (Fig. 4A&B). In addition, chronic administration of both drugs, at the indicated dose and schedule, was well tolerated with no weight loss in the animals (Supplemental figure 2B). Consistent with the in vitro findings, IHC staining of tumor tissues demonstrated that ICG001 potently induced phosphorylation of Akt and S6 (Fig. 4C); the induction was effectively blocked by combination treatment with mTOR inhibitor AZD8055. Together, these data highlight the effectiveness of concomitant inhibition of mTOR and Wnt.
Figure 4. Combined inhibition of Wnt and mTOR significantly reduces tumor growth in vivo.
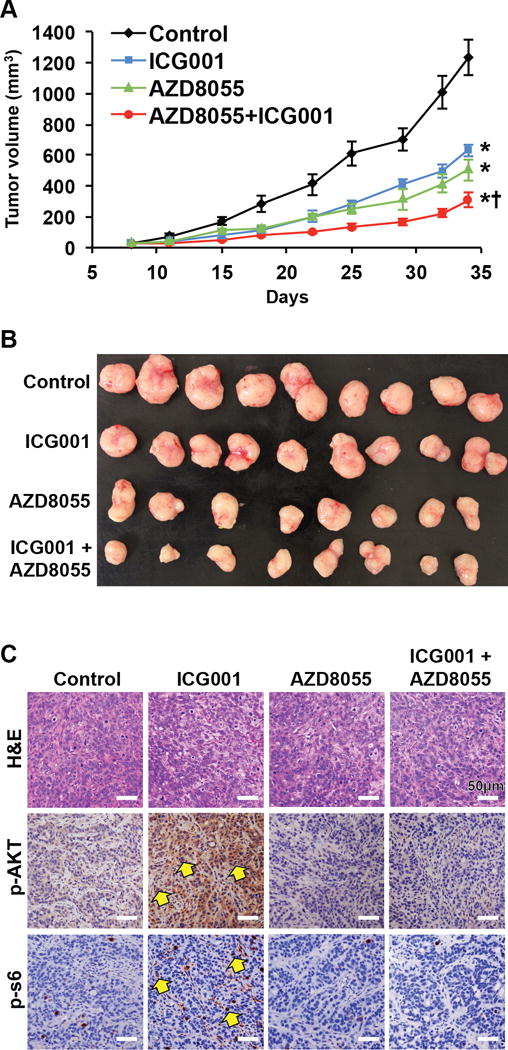
A, B. Athymic nude mice bearing established HT29 xenografts were treated with control vehicle (n=9), ICG001 (100 mg/kg, n=9), AZD8055 (30 mg/kg, n=8), or combination of both drugs (n=8) once daily and tumor size was measured by caliper two times per week. The results are presented as the mean tumor volume ± SEM. *P < 0.05 for either ICG001, AZD8055 or combination of ICG001 and AZD8055 versus vehicle control. +P < 0.05 for combination of ICG001 and AZD8055 versus either ICG001 or AZD8055 alone. C. Tumor sections from xenografts were prepared for IHC. Representative IHC staining of the tumor tissues for p-Akt(S473) and p-S6(S235/236) (arrows).
To determine if this tumor inhibition occurs in immune competent mice, we used the mouse CRC cell line MC38. Similar to human CRC cells, treatment of MC38 cells with ICG001 decreased Deptor expression along with increased mTOR activation (Fig. 5A), and the combination of ICG001 and AZD8055 significantly inhibited tumor cell growth compared with single agent treatment (Fig. 5B; Supplemental figure 2C). We next tested the efficacy of inhibiting mTOR and Wnt in MC38 xenografts in immune competent mice. Administration of AZD8055 or ICG001 alone slowed the growth of tumors while treatment with the combination of ICG001 and AZD8055 strongly suppressed tumor growth (Fig. 5C&D). As expected, ICG001 potently induced phosphorylation of Akt and S6; the induction was effectively blocked by combination treatment with AZD8055 (Supplemental figure 2D). Inhibition of mTOR by AZD8055 enhances antitumor immunity.(39) Moreover, Wnt/β-catenin signaling blocks CD8α T cell activity; loss of β-catenin or Wnt signaling reverses this effect and enhances the anti-tumor immune response.(40) Therefore, we determined the recruitment of CD8α T cells by IHC staining. Increased infiltration of CD8α T cells was found in tumors treated with ICG001 and the combination of ICG001 and AZD8055 but not by AZD8055 alone (Supplemental figure 2E). These results further confirm the effectiveness of concomitant inhibition of mTOR and Wnt in immune competent mice, and the enhanced anti-tumor immune response contributes to this growth inhibition.
Figure 5. Inhibition of Wnt and mTOR significantly reduces tumor growth in immune competent mice.
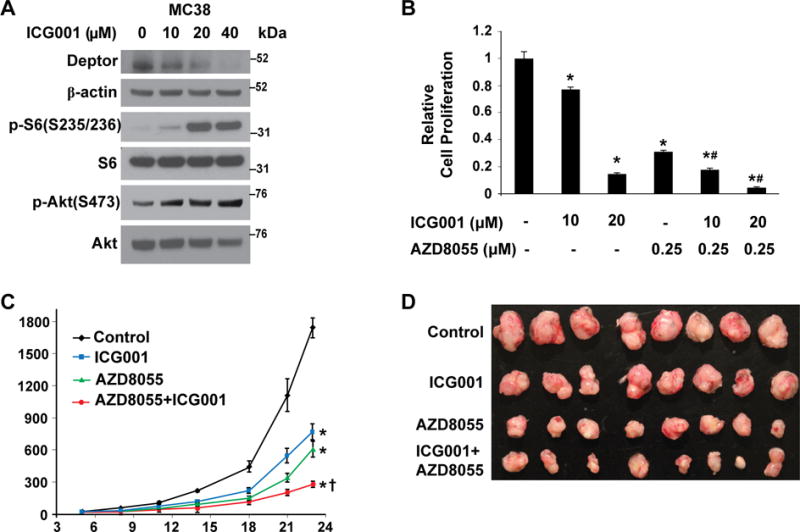
A. MC38 cells were treated with Wnt inhibitors ICG001 for 24 h. Cells were lysed and western blot analysis was performed using antibodies against Deptor, p-Akt, Akt, p-S6, S6, and β-actin. The images are representative of three independent experiments. B. MC38 cells were treated with the indicated inhibitors for 3 d. Cell proliferation was determined using the Vi-CELL XR 2.03 cell viability analyzer. (n=3, data represent mean ± SEM; *p<0.01 vs control; #p<0.01 vs single inhibitor). Data are from one of three independent experiments with similar results. C, D. C57BL/6J Mice bearing established MC38 xenografts were treated with control vehicle (n=8), ICG001 (100 mg/kg, n=8), AZD8055 (30 mg/kg, n=8), or combination of both drugs (n=8) once daily and tumor size was measured by caliper two times per week. The results are presented as the mean tumor volume ± SEM. *P < 0.05 for either ICG001, AZD8055 or combination of ICG001 and AZD8055 versus vehicle control. +P < 0.05 for combination of ICG001 and AZD8055 versus either ICG001 or AZD8055 alone.
Deptor is overexpressed in CRC Cells
Because Deptor is overexpressed in many tumor types including breast, prostate, and lung cancers, and chronic myeloid leukemia,(11, 13, 14) we next assessed Deptor expression in human CRC specimens and matched normal colonic mucosa from our surgical patients. Deptor protein expression was uniformly increased in the 8 CRCs compared to paired normal mucosa (Fig. 6A). As a further confirmation, Deptor expression was determined in CRCs and matched normal colonic mucosa from 56 patients. Consistent with our initial findings, increased expression of Deptor was noted in CRCs as compared with normal colon mucosa. (Fig. 6B). Using the TCGA CRC dataset, we determined the expression of Deptor mRNA. As shown in Fig. 6C, in agreement with the increased protein expression determined by IHC stating (Fig. 6B), elevated Deptor mRNA levels were found in cancer samples compared with normal mucosa. Increased β-catenin (CTNNB1) and MYC in cancer samples was also confirmed (Supplemental figure 3A&B). Importantly, Deptor mRNA levels were significantly correlated with the expression of β-catenin (CTNNB1) (Fig. 6D) and c-Myc (MYC) (Fig. 6E) in human CRC tissues. These correlations further suggest the regulation of Deptor by Wnt/β-catenin/c-Myc and a functional connection between Wnt/β-catenin/c-Myc and Deptor protein in CRCs.
Figure 6. Deptor is overexpressed in CRC cells.
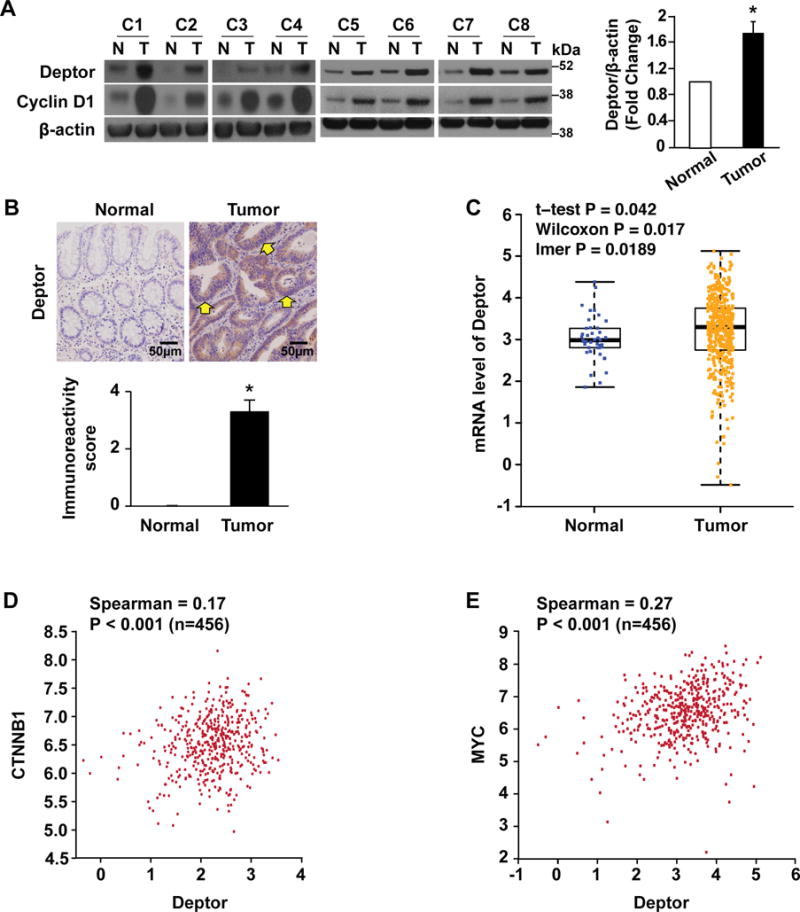
A. Deptor expression is significantly upregulated in CRC specimens. Tissue extracts from eight pairs of human specimens from primary colon tumor (T) and its surrounding normal mucosa (N) were prepared for western blot. Cyclin D1 was used as a proliferation marker. Deptor signals from eight pairs of human specimens were quantitated densitometrically and expressed as fold change with respect to β-actin. (n= 8, data represent mean±SEM; *P<0.01 versus normal mucosa). B. Expression of Deptor was analyzed in human tissues (matching primary CRC and normal colon mucosa), n = 56, using IHC. Expression of Deptor in representative colorectal adenocarcinomas and matched adjacent normal mucosa (upper panel). Elevated expression of Deptor protein was found in CRC cells (arrows). Scale bars=50 μm. Immunoreactivity score was determined by multiplication of the values for staining intensity (lower panel). *p < 0.01 as compared to normal mucosa tissue. C. Box plots show the mRNA levels of Deptor in CRC tissues (n.456) and normal tissues (n.41). Data were obtained from the TCGA dataset. D&E. The scatter plot of correlated mRNA levels between Deptor and CTNNB1 (D) and Deptor and MYC (E) in CRC tissues (n .456). Expression data were obtained from the TCGA dataset.
Deptor contributes to CRC cell growth
As Deptor acts as an oncogene contributing to cancer cell proliferation and survival of various types of cancers,(15–19, 41) we next determined whether Deptor also regulates proliferation of CRC cells. CRC cell lines or primary CRCs were infected with lentiviral vectors containing either Deptor shRNA or control. Deptor down-regulation inhibited the growth of CRC cell lines HT29 and LS174T (Fig. 7A) and primary human pt130 and pt93 CRC cells derived from PDX tumors (Fig. 7B). Moreover, knockdown of Deptor enhanced the inhibitory effect of either MK2206 or AZD8055 on LS174T cell proliferation (Supplemental figure 4A).
Figure 7. Deptor contributes to CRC cell growth.
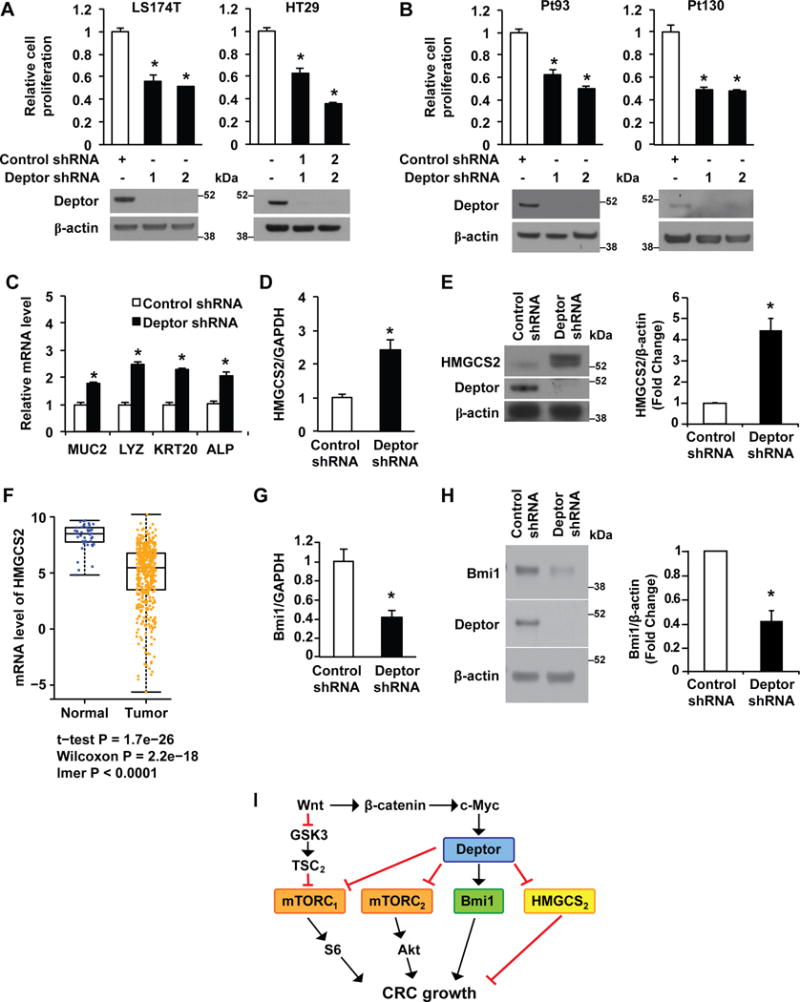
A&B. Knockdown of Deptor decreases the proliferation of CRC cells. LS174T, HT29 (A) and primary human CRC cells (pt93 and pt130) (B), stably transfected with control shRNA or Deptor shRNA, were seeded into 6-well plates. After incubation for 72 h, cell numbers were counted. (n=3, data represent mean ± SEM; *P<0.01 vs control shRNA). Data are from one of three independent experiments with similar results. C-E. Knockdown of Deptor induces differentiation in LS174T cells. Total RNA (C&D) or protein (E) was extracted from LS174T cells, stably transfected with control or Deptor shRNA. mRNA levels of differentiation markers MUC2, LYZ, KRT20 and ALP (C), and HMGCS2 (D) were determined by real time RT-PCR. (n=3, data represent mean ± SEM; *P<0.01 versus control shRNA). Data are from one of three independent experiments with similar results. HMGCS2 protein levels were determined by western blot analysis (E). The images are representative of three independent experiments. HMGCS2 signals from three separate experiments were quantitated densitometrically and expressed as fold change with respect to β-actin. (n= 3, data represent mean± SEM; *P<0.01 versus control shRNA). F. Box plots show the mRNA levels of HMGCS2 in CRC tissues (n.456) and normal tissues (n.41). Data were obtained from the TCGA dataset. G&H. Knockdown of Deptor inhibits Bmi1 expression in LS174T cells. Total RNA or protein was extracted from LS174T cells, stably transfected with control or Deptor shRNA. mRNA levels of Bmi1 (G) were determined by real time RT-PCR. (n=3, data represent mean ± SEM; *P<0.01 versus control shRNA). Data are from one of three independent experiments with similar results. Bmi1 protein levels (H) were determined by western blot analysis (left panels). The images are representative of three independent experiments. Bmi1 signals from three separate experiments were quantitated densitometrically and expressed as fold change with respect to β-actin (right panels). (n= 3, data represent mean±S.D.; *P<0.01 versus control shRNA). I. Model proposed to explain the regulation and the role of Deptor in CRC cell growth.
Wnt inhibition by APC restoration promotes cellular differentiation and reestablishes crypt homeostasis in CRCs.(3) We have shown the importance of the Wnt pathway in the regulation of intestinal cell proliferation and differentiation.(32) To determine whether Deptor knockdown contributes to the differentiation of CRC cells, mRNA levels of mucin2 (MUC2, a goblet cell marker), lysozyme (LYZ, paneth cell marker), keratin 20 (KRT20) and alkaline phosphatase (ALP) (both enterocyte markers) were determined by real time RT-PCR.(24) As shown in Fig. 7C, knockdown of Deptor increases expression of MUC2, LYZ, KRT20 and ALP in LS174T cells. In addition, knockdown of Deptor increased the expression of HMGCS2 (Fig. 7D&E), which we have recently found contributes to CRC cell differentiation.(24) Moreover, using the TCGA CRC dataset, we found that HMGCS2 mRNA levels were decreased in cancer samples compared with normal mucosa tissues (Fig. 7F). Knockdown of Deptor also decreased the expression of Polycomb complex protein (Bmi1) (Fig. 7G&H), a CRC stem cell marker which we have recently found is essential for colon cancer cell proliferation and tumor formation.(42) Knockdown of Deptor did not affect the expression of leucine-rich-repeat containing G-protein-coupled receptor 5 (Lgr5) (Supplemental figure 4B), another CRC stem cell marker. Taken together, although Deptor is a suppressor of mTOR, these novel findings suggest that Deptor acts as a downstream effector of Wnt/β-catenin/c-Myc signaling contributing to CRC growth, at least in part, through the dedifferentiation of CRC cells and regulation of Bmi1 in CRC cells (Fig. 7I).
DISCUSSION
Wnt/β-catenin signaling plays a critical role for CRC growth. Studies from our laboratory and others have shown that targeted inhibition of Wnt/β-catenin significantly inhibits CRC growth.(1, 28) Here, we report that Deptor is a downstream target of Wnt/β-catenin/c-Myc signaling in CRC cells. Inhibition of Akt or mTOR enhances the anticancer effect of Wnt inhibition. Moreover, we found that Deptor is overexpressed in CRC cells; knockdown of Deptor increases CRC cell differentiation, decreases Bmi1 expression and inhibits CRC cell proliferation.
Wnt activation in APC-mutated mice increases mTORC1 activity.(8–10) APC loss also triggers mTORC1 activation through a Wnt/β-catenin independent pathway.(43) Wnt activates mTOR by inhibiting GSK3 without involving β-catenin-dependent transcription. GSK3 inhibits the mTOR pathway by phosphorylating TSC2.(44) Moreover, Wnt signaling contributes to mTORC1 activation in the intestinal polyps also through the increased level of mTOR mediated by APC mutation.(8) Our results show that inhibition of Wnt decreases Deptor expression and increases mTOR activation. Thus, Wnt signaling contributes to mTORC1 activation through the GSK3/TSC2 pathway or through the increased expression of mTOR protein. Conversely, activation of Wnt/β-catenin/c-Myc signaling pathway increases Deptor expression and, as a result, inhibits mTORC1 and mTORC2 activation (Fig. 7I).
We show that Wnt/β-catenin/c-Myc inhibition results in the activation of mTOR signaling. This suggests a potential strategy of combining Akt/mTOR and Wnt/β-catenin inhibitory agents to enhance the growth inhibitory effects of either agent alone. Indeed, our results showed the enhanced inhibition of CRC cell growth in vitro and in vivo by combining Akt/mTOR and Wnt/β-catenin inhibitory agents. The benefit of Wnt inhibitors is currently being tested in clinical trials in CRC patients with advanced metastatic disease(45); these inhibitors may be tested as part of adjuvant therapy in the future. Moreover, our results further indicate that treatment of tumors with elevated expression of Deptor results in the activation of mTOR which may attenuate the therapeutic efficacy for using Wnt/β-catenin/c-Myc inhibitory agents. Furthermore, Deptor expression may be used as a biomarker to predict growth inhibition using a combination of Akt/mTOR and Wnt/β-catenin inhibitory drugs. This would be advisable for Wnt/β-catenin inhibitors and may be extended to other drugs that target c-Myc in CRC.
Although Deptor appears to act as a suppressor of mTOR in certain cell types, the potential role of Deptor as an oncogene has also been reported. Increased expression of Deptor has been noted in numerous tumor types,(12–14) and, moreover, Deptor contributes to cancer cell proliferation and survival.(15–19, 41) Our results demonstrate that activation of the Wnt/β-catenin/c-Myc signaling pathway increases Deptor expression. There is now strong evidence showing that increased c-Myc expression is a key component of Wnt signaling downstream of APC loss.(46) c-Myc overexpression is commonly observed in human CRC samples indicating that overexpression of this Wnt target gene, due to transcriptional deregulation, may play a central role in intestinal transformation.(47) In our current study, we demonstrated elevated expression of Deptor protein in CRC cells compared with the adjacent normal mucosa. Knockdown of Deptor inhibits CRC cell proliferation. Thus, our findings support an oncogene function of Deptor in CRC cells.
Deptor is upregulated in numerous cancers, however, very little is known regarding the mechanisms for this increase.(12–14) Our data identified Deptor as a direct target of Wnt/β-catenin/c-Myc signaling and that c-Myc binds to the Deptor promoter. In addition, our finding that c-Myc levels control Deptor expression provides an important step in unraveling the precise molecular regulators of Deptor expression in cancer. As activation of Wnt signaling and overexpression of c-Myc are hallmarks of various tumor types,(34, 48) elevated expression of Deptor in these cancers may also result from deregulated Wnt/c-Myc signaling, suggesting broader implications of our findings. For example, in multiple myeloma cell lines with high expression of Deptor, inhibition of Deptor has been shown to be an effective therapeutic strategy.(41) Our findings identifying Deptor as a tumor promoter in CRCs suggest that Deptor may also represent an attractive pharmacological target for the development of novel cancer therapies targeting CRC.
Knockdown of Deptor results in a feedback inhibition of the PI3K/AKT/SGK cascade, thus contributing to the inhibition of cell proliferation and survival.(18, 41) However, this is not the case in CRC cells as we and others have shown that knockdown increases Akt phosphorylation.(41) In this study, we found that knockdown of Deptor induces differentiation of CRC cells. Differentiation therapy, which results in induction of terminal differentiation or apoptosis, is a valid option for the treatment of recurrent CRCs.(49) Wnt inhibition by APC restoration promotes cellular differentiation and reestablishes crypt homeostasis in CRC cells.(3) Previously, we showed the importance of the Wnt pathway in the regulation of intestinal cell proliferation and differentiation. (32) Our current study demonstrate that Deptor is a Wnt/c-Myc target gene and the knockdown of Deptor increased the expression of the ketogenic enzyme HMGCS2. In agreement with these findings, ketogenic HMGCS2 has been shown to function as a c-Myc target gene that is down-regulated in CRC.(50) Recently, we have shown that HMGCS2 and its product β-hydroxybutyrate play an important role in the induction of differentiation in normal intestinal epithelium and CRC cells.(24) Moreover, we have shown that treatment with β-hydroxybutyrate inhibits CRC cell growth.(24) In this study, we also found that knockdown of Deptor decreases the expression of Bmi1 which contributes to tumor propagation.(42) Taken together, our results suggest that knockdown of Deptor may induce differentiation and inhibition of CRC cells through increased ketogenesis and decreased Bmi1 expression. Our future studies will delineate how Deptor regulates HMGCS2 and Bmi1expression and the role of HMGCS2 and Bmi1 in the effects of Deptor on the regulation of CRC cell proliferation and differentiation.
In conclusion, our results demonstrate that Deptor, which is deregulated in CRCs, is a direct target gene of Wnt/β-catenin/c-Myc signaling. Moreover, knockdown of Deptor inhibits CRC cell proliferation. In CRC, overexpression of c-Myc due to hyperactive Wnt/β-catenin signaling is a key driver of tumor progression; however, effective strategies to target this oncogene remain elusive. Importantly, our data provide a novel mechanism for the promotion of CRC growth through Deptor, an important downstream target of Wnt/β-catenin/c-Myc signaling that promotes CRC cell growth, and suggests that Deptor could be a potential therapeutic target for human CRCs.
Supplementary Material
SIGNIFICANCE.
The mTOR inhibitor DEPTOR acts as a tumor promoter and could be a potential therapeutic target in colorectal cancer
Acknowledgments
The authors thank Donna Gilbreath and Heather N. Russell-Simmons for manuscript preparation; D. Napier for tissue sectioning and staining; Tianxin Yu for tissue collection from ApcMin mice. We also acknowledge support from the University of Kentucky Markey Cancer Center’s Biostatistics and Bioinformatics Shared Resource Facility and the Biospecimen Procurement and Translational Pathology Shared Resource Facility. This work was supported by NIH grants R01 DK48498, R01 CA172379, T32 CA160003, and P30 CA177558.
Footnotes
Conflict of Interest: The authors declare no potential conflicts of interest.
References
- 1.Bahrami A, Amerizadeh F, ShahidSales S, et al. Therapeutic Potential of Targeting Wnt/beta-Catenin Pathway in Treatment of Colorectal Cancer: Rational and Progress. J Cell Biochem. 2017 doi: 10.1002/jcb.25903. [DOI] [PubMed] [Google Scholar]
- 2.Schatoff EM, Leach BI, Dow LE. Wnt Signaling and Colorectal Cancer. Current colorectal cancer reports. 2017;13:101–10. doi: 10.1007/s11888-017-0354-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dow LE, O’Rourke KP, Simon J, et al. Apc Restoration Promotes Cellular Differentiation and Reestablishes Crypt Homeostasis in Colorectal Cancer. Cell. 2015;161:1539–52. doi: 10.1016/j.cell.2015.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 5.Zaytseva YY, Valentino JD, Gulhati P, Evers BM. mTOR inhibitors in cancer therapy. Cancer Lett. 2012;319:1–7. doi: 10.1016/j.canlet.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 6.Sheng H, Shao J, Townsend CM, Jr, Evers BM. Phosphatidylinositol 3-kinase mediates proliferative signals in intestinal epithelial cells. Gut. 2003;52:1472–8. doi: 10.1136/gut.52.10.1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Q, Wang X, Hernandez A, Kim S, Evers BM. Inhibition of the phosphatidylinositol 3-kinase pathway contributes to HT29 and Caco-2 intestinal cell differentiation. Gastroenterology. 2001;120:1381–92. doi: 10.1053/gast.2001.24044. [DOI] [PubMed] [Google Scholar]
- 8.Fujishita T, Aoki K, Lane HA, Aoki M, Taketo MM. Inhibition of the mTORC1 pathway suppresses intestinal polyp formation and reduces mortality in ApcDelta716 mice. Proc Natl Acad Sci U S A. 2008;105:13544–9. doi: 10.1073/pnas.0800041105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Faller WJ, Jackson TJ, Knight JR, et al. mTORC1-mediated translational elongation limits intestinal tumour initiation and growth. Nature. 2015;517:497–500. doi: 10.1038/nature13896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hardiman KM, Liu J, Feng Y, Greenson JK, Fearon ER. Rapamycin inhibition of polyposis and progression to dysplasia in a mouse model. PLoS One. 2014;9:e96023. doi: 10.1371/journal.pone.0096023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Z, Zhong J, Inuzuka H, et al. An evolving role for DEPTOR in tumor development and progression. Neoplasia. 2012;14:368–75. doi: 10.1593/neo.12542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chin SF, Wang Y, Thorne NP, et al. Using array-comparative genomic hybridization to define molecular portraits of primary breast cancers. Oncogene. 2007;26:1959–70. doi: 10.1038/sj.onc.1209985. [DOI] [PubMed] [Google Scholar]
- 13.Duan S, Skaar JR, Kuchay S, et al. mTOR generates an auto-amplification loop by triggering the betaTrCP- and CK1alpha-dependent degradation of DEPTOR. Mol Cell. 2011;44:317–24. doi: 10.1016/j.molcel.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pei L, Xie P, Zhou E, Yang Q, Luo Y, Tang Z. Overexpression of DEP domain containing mTOR-interacting protein correlates with poor prognosis in differentiated thyroid carcinoma. Mol Med Rep. 2011;4:817–23. doi: 10.3892/mmr.2011.503. [DOI] [PubMed] [Google Scholar]
- 15.Zhang HR, Chen JM, Zeng ZY, Que WZ. Knockdown of DEPTOR inhibits cell proliferation and increases chemosensitivity to melphalan in human multiple myeloma RPMI-8226 cells via inhibiting PI3K/AKT activity. J Int Med Res. 2013;41:584–95. doi: 10.1177/0300060513480920. [DOI] [PubMed] [Google Scholar]
- 16.Srinivas KP, Viji R, Dan VM, et al. DEPTOR promotes survival of cervical squamous cell carcinoma cells and its silencing induces apoptosis through downregulating PI3K/AKT and by up-regulating p38 MAP kinase. Oncotarget. 2016;17:24154–71. doi: 10.18632/oncotarget.8131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang H, Chen J, Zeng Z, Que W, Zhou L. Knockdown of DEPTOR induces apoptosis, increases chemosensitivity to doxorubicin and suppresses autophagy in RPMI-8226 human multiple myeloma cells in vitro. Int J Mol Med. 2013;31:1127–34. doi: 10.3892/ijmm.2013.1299. [DOI] [PubMed] [Google Scholar]
- 18.Yang Y, Bardeleben C, Frost P, et al. DEPTOR is linked to a TORC1-p21 survival proliferation pathway in multiple myeloma cells. Genes Cancer. 2014;5:407–19. doi: 10.18632/genesandcancer.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parvani JG, Davuluri G, Wendt MK, et al. Deptor enhances triple-negative breast cancer metastasis and chemoresistance through coupling to survivin expression. Neoplasia. 2015;17:317–28. doi: 10.1016/j.neo.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee J, Shi Y, Vega M, et al. Structure-activity relationship study of small molecule inhibitors of the DEPTOR-mTOR interaction. Bioorg Med Chem Lett. 2017;27:4714–24. doi: 10.1016/j.bmcl.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 21.Chresta CM, Davies BR, Hickson I, et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010;70:288–98. doi: 10.1158/0008-5472.CAN-09-1751. [DOI] [PubMed] [Google Scholar]
- 22.Kondo J, Endo H, Okuyama H, et al. Retaining cell-cell contact enables preparation and culture of spheroids composed of pure primary cancer cells from colorectal cancer. Proc Natl Acad Sci U S A. 2011;108:6235–40. doi: 10.1073/pnas.1015938108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun SY, Rosenberg LM, Wang X, et al. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005;65:7052–8. doi: 10.1158/0008-5472.CAN-05-0917. [DOI] [PubMed] [Google Scholar]
- 24.Wang Q, Zhou Y, Rychahou P, et al. Ketogenesis contributes to intestinal cell differentiation. Cell Death Differ. 2017;24:458–68. doi: 10.1038/cdd.2016.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Q, Zhou Y, Weiss HL, Chow CW, Evers BM. NFATc1 regulation of TRAIL expression in human intestinal cells. PLoS One. 2011;6:e19882. doi: 10.1371/journal.pone.0019882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zaytseva YY, Harris JW, Mitov MI, et al. Increased expression of fatty acid synthase provides a survival advantage to colorectal cancer cells via upregulation of cellular respiration. Oncotarget. 2015;6:18891–904. doi: 10.18632/oncotarget.3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gulhati P, Cai Q, Li J, et al. Targeted inhibition of mammalian target of rapamycin signaling inhibits tumorigenesis of colorectal cancer. Clin Cancer Res. 2009;15:7207–16. doi: 10.1158/1078-0432.CCR-09-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang W, Sviripa V, Kril LM, et al. Fluorinated N,N-dialkylaminostilbenes for Wnt pathway inhibition and colon cancer repression. Journal of medicinal chemistry. 2011;54:1288–97. doi: 10.1021/jm101248v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takahashi-Yanaga F, Kahn M. Targeting Wnt signaling: can we safely eradicate cancer stem cells? Clin Cancer Res. 2010;16:3153–62. doi: 10.1158/1078-0432.CCR-09-2943. [DOI] [PubMed] [Google Scholar]
- 30.Handeli S, Simon JA. A small-molecule inhibitor of Tcf/beta-catenin signaling down-regulates PPARgamma and PPARdelta activities. Mol Cancer Ther. 2008;7:521–9. doi: 10.1158/1535-7163.MCT-07-2063. [DOI] [PubMed] [Google Scholar]
- 31.Blanchard PG, Festuccia WT, Houde VP, et al. Major involvement of mTOR in the PPARgamma-induced stimulation of adipose tissue lipid uptake and fat accretion. J Lipid Res. 2012;53:1117–25. doi: 10.1194/jlr.M021485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Q, Zhou Y, Rychahou P, Liu C, Weiss HL, Evers BM. NFAT5 represses canonical Wnt signaling via inhibition of beta-catenin acetylation and participates in regulating intestinal cell differentiation. Cell Death Dis. 2013;4:e671. doi: 10.1038/cddis.2013.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–80. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 34.Gabay M, Li Y, Felsher DW. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harbor perspectives in medicine. 2014;4:a014241. doi: 10.1101/cshperspect.a014241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin CY, Loven J, Rahl PB, et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012;151:56–67. doi: 10.1016/j.cell.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heidel FH, Bullinger L, Feng Z, et al. Genetic and pharmacologic inhibition of beta-catenin targets imatinib-resistant leukemia stem cells in CML. Cell Stem Cell. 2012;10:412–24. doi: 10.1016/j.stem.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hanrahan AJ, Schultz N, Westfal ML, et al. Genomic complexity and AKT dependence in serous ovarian cancer. Cancer Discov. 2012;2:56–67. doi: 10.1158/2159-8290.CD-11-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holland JD, Gyorffy B, Vogel R, et al. Combined Wnt/beta-catenin, Met, and CXCL12/CXCR4 signals characterize basal breast cancer and predict disease outcome. Cell Rep. 2013;5:1214–27. doi: 10.1016/j.celrep.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 39.Jiang Q, Weiss JM, Back T, et al. mTOR kinase inhibitor AZD8055 enhances the immunotherapeutic activity of an agonist CD40 antibody in cancer treatment. Cancer Res. 2011;71:4074–84. doi: 10.1158/0008-5472.CAN-10-3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spranger S, Dai D, Horton B, Gajewski TF. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell. 2017;31:711–23.e4. doi: 10.1016/j.ccell.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peterson TR, Laplante M, Thoreen CC, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137:873–86. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu T, Chen X, Zhang W, et al. Regulation of the potential marker for intestinal cells, Bmi1, by beta-catenin and the zinc finger protein KLF4: implications for colon cancer. J Biol Chem. 2012;287:3760–8. doi: 10.1074/jbc.M111.316349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang S, Li N, Yousefi M, et al. Transformation of the intestinal epithelium by the MSI2 RNA-binding protein. Nat Commun. 2015;6:6517. doi: 10.1038/ncomms7517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Inoki K, Ouyang H, Zhu T, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126:955–68. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- 45.Sebio A, Kahn M, Lenz HJ. The potential of targeting Wnt/beta-catenin in colon cancer. Expert Opin Ther Targets. 2014;18:611–5. doi: 10.1517/14728222.2014.906580. [DOI] [PubMed] [Google Scholar]
- 46.Myant K, Sansom OJ. Wnt/Myc interactions in intestinal cancer: partners in crime. Exp Cell Res. 2011;317:2725–31. doi: 10.1016/j.yexcr.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 47.Erisman MD, Rothberg PG, Diehl RE, Morse CC, Spandorfer JM, Astrin SM. Deregulation of c-myc gene expression in human colon carcinoma is not accompanied by amplification or rearrangement of the gene. Mol Cell Biol. 1985;5:1969–76. doi: 10.1128/mcb.5.8.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36:1461–73. doi: 10.1038/onc.2016.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cruz FD, Matushansky I. Solid tumor differentiation therapy – is it possible? Oncotarget. 2012;3:559–67. doi: 10.18632/oncotarget.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Camarero N, Mascaro C, Mayordomo C, Vilardell F, Haro D, Marrero PF. Ketogenic HMGCS2 Is a c-Myc target gene expressed in differentiated cells of human colonic epithelium and down-regulated in colon cancer. Mol Cancer Res. 2006;4:645–53. doi: 10.1158/1541-7786.MCR-05-0267. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.