

Abstract
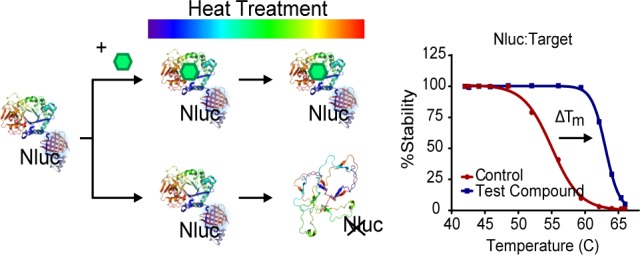
Protein thermal shift assays (TSAs) provide a means for characterizing target engagement through ligand-induced thermal stabilization. Although these assays are widely utilized for screening libraries and validating hits in drug discovery programs, they can impose encumbering operational requirements, such as the availability of purified proteins or selective antibodies. Appending the target protein with a small luciferase (NanoLuc) allows coupling of thermal denaturation with luminescent output, providing a rapid and sensitive means for assessing target engagement in compositionally complex environments such as permeabilized cells. The intrinsic thermal stability of NanoLuc is greater than mammalian proteins, and our results indicate that the appended luciferase does not alter thermal denaturation of the target protein. We have successfully applied the NanoLuc luciferase thermal shift assay (NaLTSA) to several clinically relevant protein families, including kinases, bromodomains, and histone deacetylases. We have also demonstrated the suitability of this assay method for library screening and compound profiling.
Keywords: Thermal shift assay, target engagement, NanoLuc luciferase, NaLTSA, ligand binding assay
As the therapeutic effect of most drugs is mediated through their binding interactions with target protein, characterization of target engagement is a standard experimental objective in preclinical drug development. Of the several approaches developed for this purpose (for reviews see refs (1) and (2)), thermal shift assays (TSA)3,4 have become among the most widely used. Conventional TSAs operate in a biochemical format, where purified target proteins in the presence of interacting ligands are subjected to a thermal challenge under defined conditions. In addition to the inherently artificial nature of this approach, it is also dependent on the ability to purify relatively large amounts of the target protein. More recently, a cell-based TSA (CETSA)5 has become popular, which arguably provides a more native environment for ascertaining engagement. However, it has a manually intensive workflow that can be difficult to perform reproducibly and is reliant upon antibodies necessary for selectively detecting the target protein. To offer a more robust and efficient means for assessing target engagement and to provide a method suitable for high throughput screening, we explored coupling the thermal denaturation to the luminescent output of a luciferase.
TSAs are based on the thermodynamic coupling of ligand binding with the temperature dependent denaturation of a target protein.4,6 At elevated temperatures, the loss of higher order protein structure leads to an unfolded state, which subsequently progresses toward protein aggregation and precipitation. Ligand binding modifies the denaturation profile by increasing resistance to unfolding. This results in a measurable shift in the protein’s melting temperature (Tm), which generally correlates with ligand concentration and affinity. As such, TSAs have become one of the preferred methods for verifying the hits from compound screening, and for rank ordering compound affinity.
The most commonly employed TSA for analyzing purified proteins is differential scanning fluorimetry (DSF), or thermofluor, which utilizes specialized fluorogenic dyes. The dyes are environmentally sensitive, exhibiting a significant increase in quantum yield when they interact with hydrophobic molecules.7 DSF takes advantage of this property to detect changes in protein structure at elevated temperatures. Hydrophobic residues are disproportionately sequestered in the core of soluble proteins and become exposed as the protein unfolds due to thermal denaturation, allowing the dye to bind and generate an increase in fluorescence. The method is relatively simple and compatible with high-throughput screening (HTS). Nonetheless, DSF is limited by its demand for large amounts of purified proteins, which can be difficult and expensive to obtain, and is known to suffer from high background originating from fluorescent compounds or hydrophobic proteins.3,7 To compensate for problems associated with the purification of full length proteins, DSF assays will frequently utilize protein domains that may not accurately reflect the target engagement attributes of the full length, native target.
The CETSA methodology was developed more recently to expand TSAs into a more physiologically relevant cellular environment.5 It employs the same thermodynamic principle of ligand-induced target stabilization but is applied within complex environments such as cell lysates, living cells, and tissues.5,8 Heat denaturation of proteins is measured through the resultant aggregation and determined by quantifying the remaining soluble fraction of the target protein using selective immunodetection (e.g., Western blot or AlphaScreen).5,8 CETSA has proven to be a very useful tool to provide an assessment of target engagement of endogenous proteins in their appropriate biological context. The technology has also been adapted to target identification using multiplexed quantitative mass spectrometry.9,10
However, CETSA relies on the availability of high quality antibodies, which limits the number of proteins that can be analyzed in this assay format. Other limitations include ligand-induced suppression of antibody recognition, a manually intensive workflow, and low throughput in the absence of homogeneous immunoassays. Recent reports also indicate that CETSA frequently fails when applied to large multidomain proteins.8,11 In addition, CETSA potentially suffers from reproducibility and sensitivity issues, particularly with low abundance proteins or when ligand stabilization results in relatively small shifts in Tm measurements.11,12 Lastly, separation methods for removing the aggregated proteins are biased against membrane and nuclear proteins.
Coupling the folded state of the target protein to a luminescent signal could avoid the need for specific antibodies or a separation step, as well as protein purification. We investigated this possibility by genetically fusing the small (19 kDa) NanoLuc (Nluc) luciferase to the target protein (Figure 1). Nluc was selected owing to its high thermostability and extremely bright luminescence providing the assay with a large dynamic range and assay sensitivity.13 Tagging the target protein with Nluc provides assay specificity in complex environments such as cellular lysates. In addition to removing the need for selective immune-detection, analysis of full-length proteins can be done without protein purification. These procedural simplifications aid in providing the assay with robustness and reproducibility, as well as improve suitability for high-throughput screening. We are referring to this method as the NanoLuc luciferase TSA, or NaLTSA.
Figure 1.
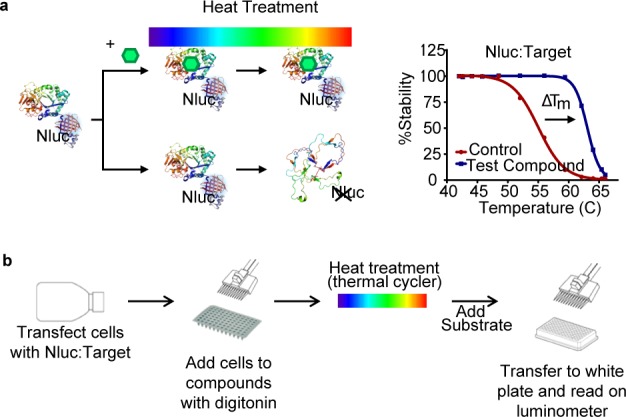
NaLTSA principle and workflow. (a) Compounds that bind Nluc-POI fusion will lead to stabilization compared to controls resulting in differences in Tm as detected by luminescence. (b) Assay workflow.
In the simplest iteration of NaLTSA, we introduced the protein of interest (POI) tagged with Nluc into cells by bulk transfection of the genetic fusion. After 24 h, the cells were harvested and dispensed into PCR plates preloaded with compounds in the presence of protease inhibitors and digitonin to permeabilize the cells (Figure 1). Following a short incubation, samples were subjected to a 3 min heat treatment using a thermal cycler. Luminescence was subsequently measured by adding substrate and transferring samples into a white 96-well plate for analysis on a luminometer. We found in general that the POI-Nluc fusion is sensitive to the heat treatment revealing reduced luminescent intensity as a function of temperature. Importantly, in the presence of a compound that specifically binds to the POI-Nluc fusion, stabilization is observed, which manifests as the loss of luminescence shifting toward higher temperatures (Figure 1).
We first assessed the validity of this approach using the target protein mitogen-activated protein kinase 14 (MAPK14 or p38α). MAPK14 is a small (41 kDa), cytoplasmic stress-activated serine/threonine-specific kinase involved in autoimmunity and other inflammatory diseases, as well as cancers.14,15 Digitonin permeabilized HeLa cells transiently expressing Nluc-MAPK14 were treated with either vehicle (DMSO) or kinase inhibitors AMG-548, SB 203580, and AZD 5438 for 1 h at 37 °C. The samples were then exposed to a thermal gradient followed by measurement of bioluminescence. The RLU values obtained at each temperature within the temperature gradient were used to calculate apparent thermal melting curves. The apparent Tm for Nluc-MAPK14 was 50.7 °C in the presence of vehicle but increased to 59.0 and 56.8 °C in the presence of known MAPK14 inhibitors AMG-548 and SB 203580, respectively (Figure 2a). In contrast, AZD 5438, an inhibitor selective to the kinase CDK2 and presumed not to bind to MAPK14, did not produce a significant shift in Tm (ΔTm) (Figure 2a). We found the assay to be robust, producing excellent curve fitting (R2 values ≥ 0.98, Figure 2a) and reproducibility across independent experiments (CVs < 15%, Figure 2b).
Figure 2.
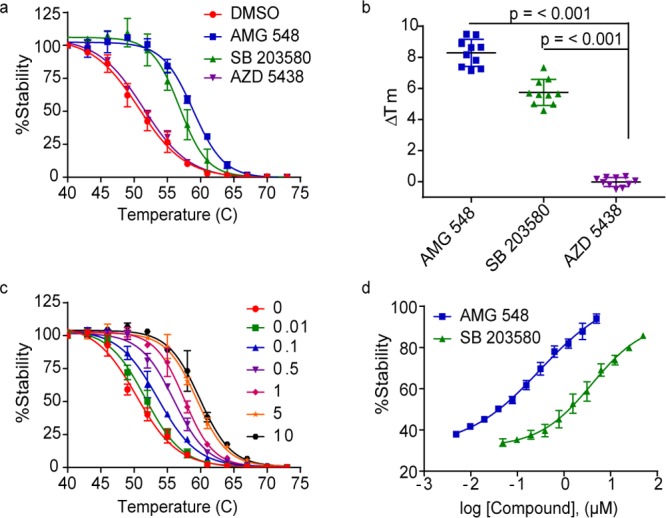
NaLTSA validation in permeabilized HeLa cells transiently expressing Nluc-MAPK14. (a) Samples were exposed to compounds [50 μM] and subjected to NaLTSA. (b) ΔTm results obtained from n = 10 independent NaLTSAs to demonstrate reproducibility. (c) Melting curves for each [AMG 548]. (d) Isothermal dose response curves were generated by exposing duplicate sample plates to increasing [compound] at isothermal temperatures of 40 or 54 °C. (a,c,d) Shown are mean ± SEM, n = 4 independent experiments.
For the assay to perform properly, the derived apparent Tm must be driven by the POI and not the reporter Nluc. Under these assay conditions, unfused Nluc expressed in HeLa cells had a Tm of 69.6 °C (Supporting Figure S1), indicating that the Tm of the fusion protein was determined by the fusion partner. Moreover, the Tm of unfused Nluc assayed was not altered by the test compounds, confirming that the ΔTm of Nluc-MAPK14 resulted from ligand stabilization specific to the POI (Supporting Figure S2). Considering that most mammalian proteins (>90%) have a reported melting point within the 37–67 °C range,9,10,16 the NaLTSA is expected to be broadly applicable across diverse target classes.
Results are displayed as percent stability to control confounding influences on light emission. This was done by normalizing the luminescence data for each compound against data obtained at the lowest temperature (i.e., RLU at X°C for compound A/RLU at 40 °C for compound A). For instance, compound SB 203580 was found to cause a 2-fold decrease in luminescence at low temperatures (Supporting Figures S2 and S3). Calculating relative stabilization in this manner rectifies unspecified signal changes caused by compound effects unrelated to target engagement without altering the relative Tm. Analogous normalization procedures are used in other TSA methods.8,12,17 Additionally, including an unfused Nluc control aids in revealing the presence of nonspecific effects on the luminescence.
To determine whether target engagement is influenced by the Nluc tag, we compared the thermal shift profiles of Nluc-MAPK14 to the endogenous MAPK14. Identical assay conditions for both configurations were ensured by applying CETSA (via Western blotting) to HeLa cells expressing Nluc-MAPK14 at a level comparable to the endogenous kinase. Prior to analysis, the cells were permeabilized using digitonin and treated with inhibitor (AMG-548). Apparent Tms for Nluc-MAPK14 and endogenous MAPK14 were found to be 49.7 and 50.3 °C, respectively, with similar Tm shifts induced by the presence of inhibitor (Supporting Figure S4). These data are in agreement with CETSA results previously reported for this target8 and demonstrate that target engagement as revealed by ΔTm does not appear altered by the Nluc tag.
To extend our evaluation to other clinically relevant protein classes, we applied the method to representative histone deacetylases (HDACs) and full-length bromodomain proteins of the BET family (Supporting Figure S5). These targets, tagged with Nluc and incubated in digitonin permeabilized cells with their corresponding high affinity inhibitors, produced ΔTms ranging between 2.2 to 6.3 °C (R2 ≥ 0.98 for each target). These results also indicate that NaLTSA is suitable to proteins located in the cytosol or nucleus.
TSA in living cells implies a physiological relevant cellular context for the target, including an intact cellular membrane and metabolism. When we applied NaLTSA to living cells, we found occurrences of false positives for direct target engagement, possibly explained as pathway effects, which are known to occur in live cell CETSA, but also difficulties in achieving assay reproducibility as demonstrated with Nluc-MAPK14 (Supporting Figure S6a,b). As expected from previously published reports,5,8 we found cellular membranes appeared largely intact following thermal challenge as determined by using standard dye exclusion assays (Supporting Figure S6c). However, similar experiments utilizing a cell impermeable Nluc inhibitor18 indicates that selective loss of membrane integrity might occur at temperatures as low as 58 °C (Supporting Figure S6d). Furthermore, a precipitous drop in cellular adenosine triphosphate (ATP) indicated a substantial metabolic impairment at temperatures above 56 °C, which overlaps with the Tm range for many proteins9,10,16 (Supporting Figure S6c). These data suggest that variability in membrane integrity and cell metabolism when cells are stressed by the thermal challenge may impact assay reliability in the live cell format. We have found that permeabilizing cells with digitonin substantially improves assay reproducibility and robustness, possibly by neutralizing these confounding cellular effects, and is consistent with previous studies that have shown mild detergents are compatible in CETSA applications.10
One of the draws to using TSAs is that relative binding affinity can be estimated by the dependence of ΔTm on compound concentration. As demonstrated using Nluc-MAPK14, while full thermal profiles may be determined over a range of compound concentrations (Figure 2c), it may be sufficient to determine relative compound EC50 for apparent stability at a single temperature (Figure 2d). For this isothermal mode, the optimal temperature may be determined from the thermal profile of the unbound target. Increasing ligand concentrations are known to be required for protein stabilization as the temperature is progressively raised above Tm(5,8,12,17) (Supporting Figure S7). Accordingly, EC50 values evident in the isothermal NaLTSA for inhibitors AMG-548 and SB 203580 are considerably higher than the reported IC50 values of 0.5 and 50–300 nM,19,20 which were generated using kinase activity assays as expected (Figure 2d). Nonetheless, the affinity rank ordering of these inhibitors is preserved in NaLTSA.
The simplicity of NaLTSA is compatible with workflow requirements typical for the high-throughput automation employed in compound screening and hit validation. We confirmed the suitability of isothermal NaLTSA for hit discovery by screening a small library comprising 80 kinase inhibitors for compounds selective to Nluc-MAPK14 (Figure 3). TSAs are orthogonal to kinase activity assays by having the ability to detect type I, type II, and allosteric inhibitors with equal proficiency5 (Supporting Figure S8). The library was screened at 10 μM concentration, and hits were scored by having relative stability greater than 3× the SD of the DMSO control.21 Unfused Nluc was also included as a control of nonspecific compound effects (Figure 3a,b).
Figure 3.

Primary hit screening, hit validation, and relative rank affinity for inhibitors of MAPK14. (a,b) Primary hit screening using HeLa cells transfected with Nluc-MAPK14 or unfused Nluc control using the ScreenWell Kinase Inhibitor library at [10 μM] in isothermal NaLTSA of 40 or 54 °C. Red lines denotes >40% stability hit identification. Dots: red, DMSO control; blue, AMG 548; green, hits >40% stabilization. Representative data shown. (c,d) Hit validation using NaLTSA in temperature gradient or isothermal dose response mode. Shown are mean ± SEM, n = 3 independent experiments. (e) Biochemical MAPK14 ADP-Glo kinase activity assay. Shown are mean RLU values, n = 3, of a representative experiment, with variability expressed as SD.
Considering that the ΔTm from compound binding may result in relatively small changes (1–2 °C),9,10,22 the high assay reproducibility of NaLTSA is critical for discriminating true hits within a relatively narrow screening window (Z′ = 0.80 as determined with positive and negative controls, Figure 3a). The relative stability of Nluc-MAPK14 was 32 ± 2.1% (DMSO control; 54 °C), allowing hits to be specified as >40% stability (absent anomalous effect on the unfused Nluc control) (Figure 3a,b). Lavendustin, an EGFR specific inhibitor, showed an apparent stabilization of both the Nluc-MAPK14 and the unfused Nluc control in the primary screen and was thus eliminated from the hit list. The positive control compound, AMG 548, produced the greatest stabilization of 90%. Seven additional hits were identified by the specified criteria, including two MAPK14-specific inhibitors SB 203580 and SB 202190 with 75% and 79% stability, respectively. Other hits included PP 1, PP 2, ZM 336372, LFM-A13, and Tyrphostin 9, exhibiting stabilization values ranging from 41–51%, indicating possibly lower affinity to MAPK14.
Hits from this primary screen were further qualified by determining their thermal profiles against the unfused Nluc control.
We found that the inhibitor Tyrphostin 9 had significant effects on the unfused Nluc bioluminescence and apparent Tm; consequently, this compound was eliminated from further consideration (Supporting Figure S9). The remaining hits had no effect on the Tm of the Nluc control. These were analyzed for rank order binding affinity to Nluc-MAPK14.
As expected, the three MAPK14-specific inhibitors, AMG-548, SB 203580, and SB 202190, produced the largest ΔTms (mean ± SEM) of 9.1 °C ± 0.35, 6.8 °C ± 0.24, and 7.8 °C ± 0.20, respectively. ZM 336372 produced a ΔTm of 4.9 °C ± 0.20 and appeared as the next ranked binder by isothermal dose response (Figure 3c). ZM 336372 has been shown to weakly inhibit SAPK2/p38α with an IC50 of 2 μM, indicating that the NaLTSA screen properly identified this low affinity inhibitor.23 PP 1 had a ΔTm of 3.05 °C ± 0.14, followed by LFM-A13 with a ΔTm of 3.43 °C ± 0.5, and lastly, PP 2 with a small ΔTm of 1.17 °C ± 0.34 (Figure 3c). PP 2 did not show any concentration-dependent increase in stability under isothermal dose–response conditions indicating that this compound either does not bind to MAPK14 or has extremely low affinity in the isothermal dose–response conditions (Figure 3d; Supporting Table S1). Interestingly, neither PP 1 nor LFM-A13 have previously been reported to bind MAPK14, indicating that MAPK14 may be a low affinity off-target for these inhibitors.
The hits identified by NaLTSA were validated as inhibitors of MAPK14 using a biochemical kinase activity assay. The results are largely consistent with the binding interactions and affinity ranking obtained by NaLTSA (Figure 3c,d; Supporting Table S1). Nonetheless, because the TSA assay relies on irreversible denaturation occurring at elevated temperatures, the results cannot be directly equated to a thermodynamic Kd obtained through enzymological analysis. Although Tm shifts generally correlate with ligand concentration and affinity, one should be aware that results are not always straightforward given the mechanistic complexity of structural disruption. The ΔTm may be influenced by a number of thermodynamic and ligand binding parameters, such as enthalpy, entropy, heat capacity, hydrogen bonds, ionic bonds, hydrophobic interactions, and binding kinetics.4,6
In contrast to interrogating a selected target with multiple compounds, NaLTSA may also be configured for evaluating a particular compound against a collection of targets. This provides a means for profiling compound binding without requiring functional assays of target activity. To investigate the utility of NaLTSA for compound profiling, we tested the effect of ponatinib, a clinically used type II kinase inhibitor,24 on a representative panel of kinases. A total of 38 Nluc kinase fusions were individually expressed in HeLa cells and assayed for increased protein stability upon treatment with ponatinib. The results show that NaLTSA accurately detected 19 of 20 known targets for ponatinib, with ΔTm values ranging from 2.05 to 9.06 °C (full melt curves for each kinase are shown in Supporting Figure S10). Notably, many of the biologically relevant targets are membrane proteins (PDGFRα, EPHA1, FGR1, FLT1, DDR1, IGF1R, and LTK) (Table 1).
Table 1. Ponatinib Kinase Profiling Using NaLTSA.
| tyrosine kinase | MW (kDa) | reported IC50 (nM)24,27 | NaLTSA ΔTm |
|---|---|---|---|
| PDGFRa | 124 | 1.1 | 6.31 |
| EPHA1 | 108 | 143 | 4.71 |
| FGFR1 | 92 | 2.2 | 4.24 |
| FLT1 | 151 | 3.7 | 2.35 |
| DDR1 | 101 | 9.4 | 5.60 |
| IGF1R | 155 | >1uM | 2.05 |
| LTK | 92 | >1uM | 2.59 |
| ABL1 | 123 | 0.37 | 3.11 |
| FYN | 61 | 0.36 | 6.04 |
| JAK1 | 133 | 32.2 | 4.90 |
| FRK | 58 | 1.3 | 4.07 |
| LYN | 59 | 0.16 | 5.25 |
| YES | 61 | 0.89 | 3.65 |
| HCK | 60 | 0.11 | 7.43 |
| SRC | 60 | 5.4 | 4.10 |
| LCK | 58 | 0.28 | 8.23 |
| CSK | 51 | 12.7 | 3.45 |
| JAK2 | 131 | 169 | –0.98 |
| JAK2–JH1 | 0.25 | ||
| JAK2–JH2 | 2.82 |
| serine/threonine kinase | MW (kDa) | reported IC50 (nM)24,27 | NaLTSA ΔTm |
|---|---|---|---|
| MAPK14 | 41 | <10 | 9.06 |
| PRKACA | 40 | 613 | 3.06 |
| MAP3K5 | 155 | 2.90 | |
| MAP4K1 | 91 | 2.40 | |
| MAP4K5 | 95 | 2.51 | |
| STK4 | 56 | 2.45 | |
| STK16 | 35 | 2.45 | |
| CAMK2G | 63 | 2.60 | |
| PIM1 | 45 | 2.57 | |
| AAK1 | 95 | 2.43 | |
| YSK4 | 151 | 2.34 | |
| PKN1 | 104 | 1.96 | |
| SIK1 | 85 | 1.36 | |
| SLK | 95 | 1.23 | |
| IRAK4 | 52 | 1.20 | |
| RIPK2 | 61 | 0.91 | |
| MEK5 | 49 | 0.80 | |
| GRK1 | 64 | –0.02 |
Full-length JAK2 reported as a false negative with a ΔTm of −0.98 °C, whereas reported biochemical data using isolated JAK2 domains revealed ponatinib binding with an IC50 = 169 nM.24 We considered the possibility that ligand binding to one domain of JAK2 may not protect the full-length protein from thermal denaturation resulting in a false negative for compound binding.9,25 To test this hypothesis, we fused Nluc to the segregated JH1 and JH2 domain of JAK2. Results showed that ponatinib stabilized the JH2 domain of JAK2 (ΔTm = 2.82 °C). This is consistent with a report showing thermal stabilization of the JH2 domain through binding ATP.26 Our results suggest that the JH2 domain is similarly stabilized through binding of ponatinib but that this binding event does not confer stability to the full-length protein (Table 1, Supporting Figure S10).
In addition to the known ponatinib targets, our results showed ponatinib induced thermal stabilization for several kinases not previously shown to bind this inhibitor (Table 1). Like many promiscuous drugs, ponatinib is associated with numerous and serious adverse side effects including cardiovascular, peripheral vascular, and cerebrovascular events.26 Profiling based on thermal shifts may indicate ponatinib binding to previously unknown targets, providing an alternative approach for identifying off-target influences. The relevance of these putative ponatinib off targets identified in this NaLTSA profiling experiment may warrant additional investigation.
While performing a similar profiling experiment using the type I kinase inhibitor staurosporine, we found that some known staurosporine targets showed no apparent shift in Tm (Supporting Table S2 and Figure S11). Similar results were reported for CETSA profiling of staurosporine in K562 cell extracts, where several kinases identified to bind to staurosporine by Kinobeads did not produce a Tm shift in CETSA.9 While false positives are considered rare for biochemical ligand-binding assays based on thermostability,11,28 false negatives are known to occur.9,11,25 Nonetheless, to ensure that these false negatives were not a result of the Nluc tag interfering with the ability for staurosporine to bind and stabilize the protein target, we performed standard DSF with purified, untagged, full-length kinases for comparison. The results obtained by DSF corroborated the NaLTSA results both for the positive control compounds and the false negatives produced by staurosporine, indicating that the observed false negative is not due to the fusion to Nluc but rather an inherent limitation of TSA (Supporting Table S2 and Figure S11).
It is not uncommon to find discrepancies in reported affinities between methods of analysis, particularly where experimental conditions vary. For example, substantial differences may result from choosing a catalytic domain over full-length protein or when incorporating differing post-translational modifications. It may also be possible that staurosporine engages some kinases with predominantly enthalpic contributions. This is a weakness of TSAs in general, as there is a bias toward recording entropically driven interactions, as these become more dominant with increasing temperatures, while enthalpically driven interactions move in the opposite direction.12,17 Ultimately, it is important to be aware that not every protein–ligand pairing will be suitable to study using TSAs. Interestingly, BTK produced high background in the DSF making full-length BTK incompatible with DSF analysis in general, whereas NaLTSA was able to report on ligand stabilization for this protein (Supporting Table S2 and Figure S11). It is estimated that up to 25% of recombinant proteins give nonideal denaturation curves that include high fluorescence at baseline or lack a clear transition to the unfolded state in DSF, highlighting the need for multiple, orthogonal approaches to studying small molecule–protein interactions.29
NaLTSA provides a homogeneous assay method that presents broad suitability for full-length proteins spanning various sizes and subcellular locations, including nuclear and membrane proteins. We have demonstrated its versatility using diverse clinical target classes, including kinases, bromodomains, and histone deacetylases. NaLTSA may be preferred under circumstances where suitable antibodies or functional assays are not available, where purification of the target protein is difficult in adequate quantities and proper configuration, or where simplified workflows are desired for increased throughput. Future applications include miniaturization into 384- and 1536-well plates to allow for automated HTS applications as well as the use of CRISPR to tag endogenous proteins with Nluc for analysis of ligand binding in lysates of disease-relevant cell types.
In conclusion, NaLTSA provides a simple and robust approach to verification of direct target engagement that should prove to be useful in addressing drug discovery challenges and bottlenecks found with similar assays.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00081.
Abbreviations, all experimental materials and methods, and supporting data figures (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All author have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Schurmann M.; Janning P.; Ziegler S.; Waldmann H. Small-Molecule Target Engagement in Cells. Cell chemical biology 2016, 23 (4), 435–41. 10.1016/j.chembiol.2016.03.008. [DOI] [PubMed] [Google Scholar]
- Fang Y. Ligand-receptor interaction platforms and their applications for drug discovery. Expert Opin. Drug Discovery 2012, 7 (10), 969–88. 10.1517/17460441.2012.715631. [DOI] [PubMed] [Google Scholar]
- Lo M. C.; Aulabaugh A.; Jin G.; Cowling R.; Bard J.; Malamas M.; Ellestad G. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal. Biochem. 2004, 332 (1), 153–9. 10.1016/j.ab.2004.04.031. [DOI] [PubMed] [Google Scholar]
- Vedadi M.; Niesen F. H.; Allali-Hassani A.; Fedorov O. Y.; Finerty P. J.; Wasney G. A.; Yeung R.; Arrowsmith C.; Ball L. J.; Berglund H. Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 15835. 10.1073/pnas.0605224103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina D. M.; Jafari R.; Ignatushchenko M.; Seki T.; Larsson E. A.; Dan C.; Sreekumar L.; Cao Y.; Nordlund P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 2013, 341 (6141), 84–7. 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- Straume M.; Freire E. Two-dimensional differential scanning calorimetry: simultaneous resolution of intrinsic protein structural energetics and ligand binding interactions by global linkage analysis. Anal. Biochem. 1992, 203 (2), 259–68. 10.1016/0003-2697(92)90311-T. [DOI] [PubMed] [Google Scholar]
- Zhang R.; Monsma F. Fluorescence-based thermal shift assays. Curr. Opin. Drug Discovery Dev. 2010, 13 (4), 389–402. [PubMed] [Google Scholar]
- Jafari R.; Almqvist H.; Axelsson H.; Ignatushchenko M.; Lundback T.; Nordlund P.; Martinez Molina D. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 2014, 9 (9), 2100–22. 10.1038/nprot.2014.138. [DOI] [PubMed] [Google Scholar]
- Savitski M. M.; Reinhard F. B.; Franken H.; Werner T.; Savitski M. F.; Eberhard D.; Martinez Molina D.; Jafari R.; Dovega R. B.; Klaeger S. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 2014, 346, 1255784. 10.1126/science.1255784. [DOI] [PubMed] [Google Scholar]
- Reinhard F. B.; Eberhard D.; Werner T.; Franken H.; Childs D.; Doce C.; Savitski M. F.; Huber W.; Bantscheff M.; Savitski M. M.; Drewes G. Thermal proteome profiling monitors ligand interactions with cellular membrane proteins. Nat. Methods 2015, 12 (12), 1129–31. 10.1038/nmeth.3652. [DOI] [PubMed] [Google Scholar]
- Martinez Molina D.; Nordlund P. The Cellular Thermal Shift Assay: A Novel Biophysical Assay for In Situ Drug Target Engagement and Mechanistic Biomarker Studies. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 141–61. 10.1146/annurev-pharmtox-010715-103715. [DOI] [PubMed] [Google Scholar]
- Seashore-Ludlow B.; Lundback T. Early Perspective: Microplate Applications of the Cellular Thermal Shift Assay (CETSA). J. Biomol. Screening 2016, 21 (10), 1019–1033. 10.1177/1087057116659256. [DOI] [PubMed] [Google Scholar]
- Hall M. P.; Unch J.; Binkowski B. F.; Valley M. P.; Butler B. L.; Wood M. G.; Otto P.; Zimmerman K.; Vidugiris G.; Machleidt T.; Robers M. B.; Benink H. A.; Eggers C. T.; Slater M. R.; Meisenheimer P. L.; Klaubert D. H.; Fan F.; Encell L. P.; Wood K. V. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem. Biol. 2012, 7 (11), 1848–57. 10.1021/cb3002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur J. S. C.; Ley S. C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013, 13 (9), 679–692. 10.1038/nri3495. [DOI] [PubMed] [Google Scholar]
- Igea A.; Nebreda A. R. The Stress Kinase p38α as a Target for Cancer Therapy. Cancer Res. 2015, 75 (19), 3997–4002. 10.1158/0008-5472.CAN-15-0173. [DOI] [PubMed] [Google Scholar]
- Franken H.; Mathieson T.; Childs D.; Sweetman G. M.; Werner T.; Togel I.; Doce C.; Gade S.; Bantscheff M.; Drewes G. Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry. Nat. Protoc. 2015, 10, 1567. 10.1038/nprot.2015.101. [DOI] [PubMed] [Google Scholar]
- Niesen F. H.; Berglund H.; Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 2 (9), 2212–21. 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- Walker J. R.; Hall M. P.; Zimprich C. A.; Robers M. B.; Duellman S. J.; Machleidt T.; Rodriguez J.; Zhou W. Highly Potent Cell-Permeable and Impermeable NanoLuc Luciferase Inhibitors. ACS Chem. Biol. 2017, 12 (4), 1028–1037. 10.1021/acschembio.6b01129. [DOI] [PubMed] [Google Scholar]
- Dominguez C.; Powers D. A.; Tamayo N. p38 MAP kinase inhibitors: many are made, but few are chosen. Curr. Opin. Drug Discovery Dev. 2005, 8 (4), 421–30. [PubMed] [Google Scholar]
- Swann S. L.; Merta P. J.; Kifle L.; Groebe D.; Sarris K.; Hajduk P. J.; Sun C. Biochemical and biophysical characterization of unique switch pocket inhibitors of p38α. Bioorg. Med. Chem. Lett. 2010, 20 (19), 5787–5792. 10.1016/j.bmcl.2010.04.097. [DOI] [PubMed] [Google Scholar]
- Sorrell F. J.; Greenwood G. K.; Birchall K.; Chen B. Development of a differential scanning fluorimetry based high throughput screening assay for the discovery of affinity binders against an anthrax protein. J. Pharm. Biomed. Anal. 2010, 52 (5), 802–808. 10.1016/j.jpba.2010.02.024. [DOI] [PubMed] [Google Scholar]
- McMahon R. M.; Scanlon M. J.; Martin J. L. Interrogating Fragments Using a Protein Thermal Shift Assay. Aust. J. Chem. 2013, 66 (12), 1502–1506. 10.1071/CH13279. [DOI] [Google Scholar]
- Hall-Jackson C. A.; Eyers P. A.; Cohen P.; Goedert M.; Tom Boyle F.; Hewitt N.; Plant H.; Hedge P. Paradoxical activation of Raf by a novel Raf inhibitor. Chem. Biol. 1999, 6 (8), 559–568. 10.1016/S1074-5521(99)80088-X. [DOI] [PubMed] [Google Scholar]
- O’Hare T.; Shakespeare W. C.; Zhu X.; Eide C. A.; Rivera V. M.; Wang F.; Adrian L. T.; Zhou T.; Huang W.-S.; Xu Q.; Metcalf C. A.; Tyner J. W.; Loriaux M. M.; Corbin A. S.; Wardwell S.; Ning Y.; Keats J. A.; Wang Y.; Sundaramoorthi R.; Thomas M.; Zhou D.; Snodgrass J.; Commodore L.; Sawyer T. K.; Dalgarno D. C.; Deininger M. W. N.; Druker B. J.; Clackson T. AP24534, a Pan-BCR-ABL Inhibitor for Chronic Myeloid Leukemia, Potently Inhibits the T315I Mutant and Overcomes Mutation-Based Resistance. Cancer Cell 2009, 16 (5), 401–412. 10.1016/j.ccr.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateus A.; Maatta T. A.; Savitski M. M. Thermal proteome profiling: unbiased assessment of protein state through heat-induced stability changes. Proteome Sci. 2016, 15, 13. 10.1186/s12953-017-0122-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasvolsky O.; Leader A.; Iakobishvili Z.; Wasserstrum Y.; Kornowski R.; Raanani P. Tyrosine kinase inhibitor associated vascular toxicity in chronic myeloid leukemia. Cardio-Oncology 2015, 1 (1), 5. 10.1186/s40959-015-0008-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de las Heras R.; Fry S. R.; Li J.; Arel E.; Kachab E. H.; Hazell S. L.; Huang C. Y. Development of homogeneous immunoassays based on protein fragment complementation. Biochem. Biophys. Res. Commun. 2008, 370 (1), 164–8. 10.1016/j.bbrc.2008.03.057. [DOI] [PubMed] [Google Scholar]
- Walters W. P.; Namchuk M. Designing screens: how to make your hits a hit. Nat. Rev. Drug Discovery 2003, 2 (4), 259–266. 10.1038/nrd1063. [DOI] [PubMed] [Google Scholar]
- Huynh K.; Partch C. L. Analysis of protein stability and ligand interactions by thermal shift assay. Current protocols in protein science 2015, 79, 28.9.1–28.9.14. 10.1002/0471140864.ps2809s79. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.