

Abstract
The optimization of the pharmacokinetic profile of a drug is one of the crucial aspects of medicinal chemistry campaigns. When efficacy is driven by a continuous coverage of the minimum efficacious plasma concentration, half-life must be optimized to achieve the optimal pharmacokinetic profile. The consensus in the field is that decreasing clearance, as opposed to increasing volume of distribution, is a better strategy to prolong half-life. While both the pharmacokinetic theory and the need for an optimal safety profile support this approach, this needs to be integrated with practical indications concerning the strategy to optimize clearance. This work presents an extensive analysis of Genentech’s in vitro and in vivo rat pharmacokinetic data, which highlights how half-life optimization through simple modulation of lipophilicity is generally not a successful strategy. Decreasing lipophilicity without addressing a metabolic soft-spot will often lead to both lower clearance and lower volume of distribution without extending half-life.
Keywords: Half-life, clearance, bioavailability, LogD, matched molecular pairs
Clearance (CL) and bioavailability (F) are the pharmacokinetic (PK) parameters that determine the exposure, which is area under the curve (AUC), of oral drugs. When the efficacy of a drug is driven by exposure (AUC driven efficacy), CL and F become the focus of PK optimization. However, when efficacy is dependent on a drug plasma concentration that is higher than the minimum efficacious concentration for the entire dosing interval (Cmin driven efficacy), improving F and CL might not suffice; in vivo drug half-life (T1/2) also needs to be optimized. Extending T1/2 involves balancing the interplay of CL and volume of distribution (Vd,ss). During the early drug discovery stage, the connection between PK and the pharmacodynamic (PD) response is often hard to establish. In the absence of a well-validated, in vivo proof-of-concept study, scientists working on novel biological targets have only a limited understanding of a drug’s PK–PD relationship. In these cases, the safest assumption to make is that efficacy is Cmin driven, and thus, T1/2 optimization should be pursued. This assumption is often a good reflection of the typical in vitro biological assays setup, which assumes reversible, direct target interaction. Furthermore, even in cases in which efficacy is AUC driven, prolonging T1/2 can be an effective strategy to lower the maximum drug concentration (Cmax) and decrease the risk of adverse toxicological outcomes. In a one-compartment PK model, T1/2 is defined as
where Vd,ss and Vd,ss,u (Vd,ss/fraction unbound in plasma) are the total and unbound volume of distribution, respectively, at steady state, while CLu (CL/fraction unbound in plasma) is the unbound CL. Vd,ss is the theoretical volume of fluid needed to contain the amount of drug present in the body at a concentration equal to the one observed in blood. This can largely exceed the volume of body fluids for compounds extensively distributed in tissues. Because only the free drug available in blood is readily eliminated from the body, a large volume of distribution is associated with prolonged half-life. CL is a proportionality constant relating the rate of elimination and the total amount of drug in the systemic circulation. High CL is therefore a synonym of fast elimination, which in turn leads to a short half-life.
Since CL is a component of both AUC and T1/2, and because rational approaches to increase Vd,ss,u (increasing lipophilicity and adding a positive charge) tend to introduce safety-related liabilities (e.g., promiscuity, hERG inhibition, lysosomal accumulation), the optimization of PK usually focuses on the lowering of CLu.1−3 Unlike Vd,ss,u, which is mainly a function of bulk physicochemical properties, the intrinsic CL of a drug is the result of the complex interplay between the topology of a molecule and the reactivity of potential metabolic soft-spots.
Higher lipophilicity increases drug promiscuity and clearance;4,5 thus, lowering the lipophilicity is a common drug design strategy for improving PK properties and safety profile. This approach offers more options than reducing the metabolic rate of elimination by addressing a metabolic soft-spot, a strategy that forces chemists to focus on the modification of a small portion of the molecule that is often also crucial for potency. This strategy may fail to improve T1/2 due to the interplay between CLu and Vd,ss,u, which are often affected by lipophilicity in an opposite fashion. Furthermore, a high degree of polarity may result in additional ADME related liabilities (e.g., low permeability, additional routes of elimination). If the importance of T1/2 is neglected and medicinal chemistry transformations are judged by their ability to improve PK solely on the basis of CLu, lipophilicity-based strategies to optimize dose could become counterproductive. Figure 1 shows that projected human dose increases drastically when T1/2 is considerably shorter than the dosing interval. The projected daily dose is calculated assuming a one compartment PK model including the kinetics of absorption. The following parameters are kept constant for calculation: minimum efficacious concentration (10 nM), molecular weight (450 g/mol), absorption rate constant (1/h), bioavailability (1).
Figure 1.
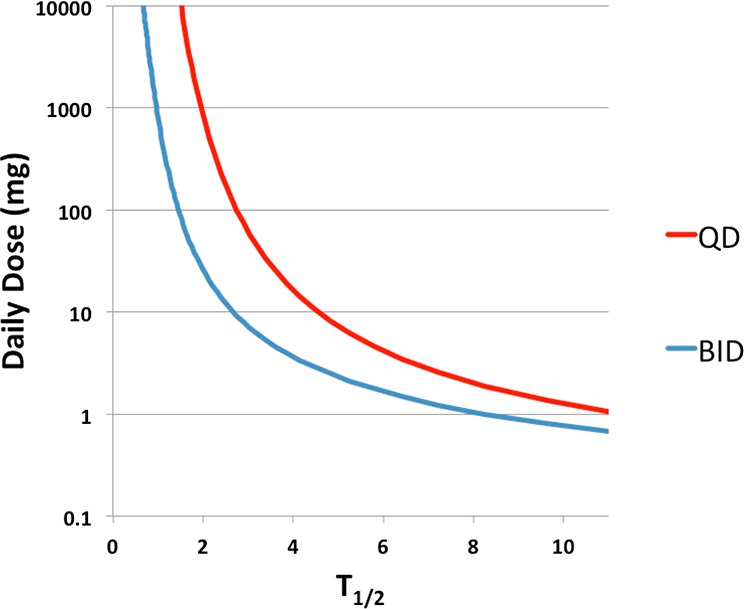
Human dose projection based on a one-compartment PK model including kinetics of absorption.
In this work, we leverage the Genentech internal data set of rat intravenous (IV) PK, in vitro metabolic stability in rat hepatocytes (RH CLint), and the log of the measured octanol–water partition coefficient at pH 7.4 (LogD7.4) to characterize the relationship between T1/2, lipophilicity, and metabolic stability. Experimental procedures to derive these data points have been described elsewhere.7−12 It is reasonable to extend the conclusion arising from this analysis to other mammal species under the assumptions that the passive biological phenomena underlying exposure are the same. This assumption is the basis of most of the modern PK science. Figure 2 depicts the distribution of T1/2, Vd,ss,u, and CLu with respect to measured LogD7.4. This set of 4767 includes all the small molecules with rat PK data available at Genentech that are unionized at pH 7.4 (based on calculated pKa13) since the presence of a charged species could potentially confound the relationship between T1/2 and lipophilicity.14 While data described in Figure 2A,B do not allow statistically significant conclusions, these trends highlight a tendency for both Vd,ss,u and CLu to increase with lipophilicity. Figure 2C clearly invalidates the hypothesis that lowering lipophilicity is likely to prolong in vivo T1/2. Figure 2D summarizes the relationships seen in Figures 2A–C. Vd,ss,u and CLu are highly correlated properties and are similarly affected by lipophilicity. Taken together, these observations suggest that T1/2 optimization via lipophilicity reduction without addressing a metabolic soft-spot is unlikely to work.
Figure 2.

Box-and-whisker plots representing the distribution of (A) rat IV Vd,ss,u, (B) rat IV CLu, and (C) rat IV T1/2 at different ranges of LogD7.4 for 4767 compounds. The lower and upper limits of the whiskers represent the minimum and maximum values, respectively, within the distributions (excluding outliers). The lower and upper edges of each box represent the 25th and 75th percentiles, respectively, while the line inside the box is the median value. (D) Scatter plot representing the relationship between Vd,ss,u and CLu in rat IV PK experiments; dots are color-coded by LogD7.4 value (the spectrum of colors from white to black represents LogD7.4 values ranging from <0 to >4, respectively).
The trends shown in Figure 2 may support the problem statement of this work, but offer little guidance for drug design. Matched molecular pairs analysis (MMPA) provides a systematic method to correlate and summarize property changes with structural modifications,15,16 thus addressing this concern. In our work, a matched molecular pair (MMP) analysis was applied to rat IV PK data, in vitro RH CLint, and LogD7.4. Our limited set of internal measurements for microsomal binding (fumic) suggests that a high level of binding in the in vitro incubation is likely to confound the interpretation of the assay only for compounds with a LogD7.4 higher than 2.5 (see Figure 3), in agreement with previous reports.17 Benet, Broccatelli, and Oprea showed that, when LogP is greater than 1, the route of elimination is most likely hepatic metabolism, suggesting that RH CLint may not be a good representation of in vivo CL for compounds with LogP below 1.18 When MMP analysis was only applied to neutral compounds with LogD7.4 1–2.5, this resulted in 9480 MMPs. It is important to mention that the RH CLint measurements below 14 mL/min/kg carry more uncertainty as a result of the in vitro T1/2 extrapolation that is over three times longer than the total incubation time. Similarly, other in vitro and in vivo measurements used in this study are not exempt from experimental error; consequently, a qualitative 2-fold change (0.3 for log values) was employed in this Letter to qualify changes in properties (e.g., T1/2) that are not just a result of experimental variability. While this may lead to a reduction of the sample analyzed, it allows drawing conclusions concerning the trends observed with a higher degree of confidence.
Figure 3.

Scatter plot characterizing the relationship between binding in the microsomal incubation and lipophilicity.
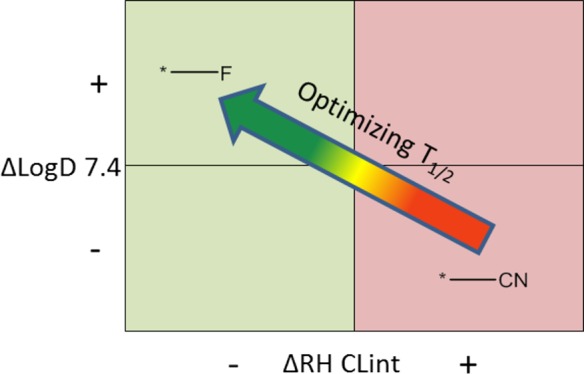
The MMP transformations were calculated by using KNIME19 and Vernalis MMP nodes.20 Changing fragments with less than 12 heavy atoms and a ratio of heavy atom counts of constant fragments to changing fragments of more than two were kept for the MMP calculation. Results from the MMP analysis are summarized in Figure 4, which illustrates the probability of various MMP classes resulting in a prolonged T1/2. The figure shows transformations for which the parameters of D7.4, T1/2, and RH CLint change by 2-fold or more. Overall, the data support the notion that improving RH CLint is a good strategy for optimizing T1/2 (67% probability), while decreasing lipophilicity is not (30% probability). Transformations in which RH CLint improvement can be achieved without decreasing lipophilicity are much more likely to result in better T1/2 (82% probability).
Figure 4.

Results of the MMP analysis described in this work. The plot includes only compounds for which at least a 2-fold variation in T1/2, RH CLint, and octanol–water partition at pH 7.4 (LogD7.4) was observed. Arrows represent decrease/increase in property.
Table 1 summarizes a selection of the half-life efficient transformations that were identified in the MMPs data set explored in this study. Inclusion criteria were a minimum of 10 examples spanning through at least three scaffolds, probability of improving T1/2 > 75%. and average T1/2 improvement of at least 2-fold. Only 0.4% of the transformations analyzed satisfied these criteria, highlighting that strategies for prolonging T1/2 are largely context dependent. Most of these transformations are characterized by the introduction of a chemical group with comparable or lower potential for CYP mediated metabolism and higher lipophilicity (e.g., H to halogen). A noticeable exception is the introduction of fluorine in place of a methyl, which is generally associated with reduction in lipophilicity and a considerably higher metabolic stability. Penning et al. reported a series of 1,5-diarylpyrazole cyclooxygenase-2 inhibitors and found substitution of the fluorine on the benzene ring with a metabolically labile methyl group reduced the half-life in the rat from 220 to 3.5 h21 (Figure 5). Many of these strategies are well-known to medicinal chemists and should be used judiciously; for example, the introduction of multiple halogen atoms might improve T1/2 but will most likely deteriorate water solubility and/or safety profile. While Table 1 may have anecdotal value to demonstrate the learning from this study, it clearly suggests that medicinal chemistry efforts striving to achieve T1/2 improvement through subtle point modifications of the metabolic soft-spot are more likely to be rewarding. Such approach is perfectly exemplified by deutetrabenazine, the deuterated analog of tetrabenazine, with improved safety profile in human; in this example, the introduction of a deuterium addressed the known metabolic soft-spot, leading to a significant improvement in oral half-life and consequent reduction in dose and Cmax(22) (Figure 5).
Table 1. Selection of the Half-life Efficient Transformations Identified in This Study.

Figure 5.

Examples of half-life efficient transformations.
Taken together, the results discussed in this work suggest that optimizing for minimum lipophilicity is not necessarily a PK-friendly strategy with only benefits and no risks. The need for practical dosing intervals in animal studies and human therapeutics demands attention to drug half-life. Decreasing lipophilicity will, on average, result in improvements in CL (and AUC), which in the majority of the cases will not translate in prolonged T1/2. When CL is decreased via modifications that increase lipophilicity, prolonged T1/2 is the most likely outcome. While MetID data characterizing each PK experiment analyzed in this study is not available, the outcome of the analysis suggests that decreasing lipophilicity when the structural modification does not address a metabolic soft-spot, will, on average, result in higher exposure (AUC) and shorter T1/2. Lowering lipophilicity while maintaining potency may in general be beneficial from a safety standpoint due to a decreased promiscuity; however, chemists who adopt this strategy should concomitantly focus on the effect of lowering lipophilicity on the projected T1/2 in human. Gains in a safety profile due to a compound’s low lipophilicity might be counterbalanced or trumped by the higher dose, Cmax, and peak to trough ratio needed to achieve the exposure necessary for drug efficacy. The use of slow release formulations to extend oral T1/2 was not explored in this study; this can be used as an additional strategy to compensate for show IV T1/2, ultimately lowering Cmax and dose.
This study was limited to neutral compounds in the LogD7.4 range 1–2.5 for practical reasons; consequently, these observations may not translate when lipophilicity is modulated via changes in the ionization state of the molecule. Similarly conclusions may be altered when dealing with highly lipophilic or hydrophilic molecules.
Tools to enable T1/2 efficient transformations may include simple qualitative considerations regarding the change in lipophilicity and in vitro CLint associated with a transformation. The process could be further simplified by adopting lipophilic metabolic efficiency as a strategy during optimization.23 Just like other efficiency tools, this metric may be valuable for education and intrascaffold trend analysis, but should not be over interpreted, particularly across the series, due to the lack of a proper mathematical basis. A more suitable alternative would be the use of human dose projection during early dose optimization.24
Acknowledgments
We are grateful to Ronitte Libedisnky for the editorial help.
Glossary
ABBREVIATIONS
- MMP
matched molecular pairs
- Vd,ss
total volume of distribution at steady state
- Vd,ss,u
unbound volume of distribution at steady state
- CL
clearance
- CLu
unbound clearance.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
References
- Smith D. A.; Beaumont K.; Maurer M. S.; Di L. Volume of Distribution in Drug Design. J. Med. Chem. 2015, 58 (15), 5691–5698. 10.1021/acs.jmedchem.5b00201. [DOI] [PubMed] [Google Scholar]
- Smith D. A.; Di L.; Kerns E. H. The effect of plasma protein binding on in vivo efficacy: misconceptions in drug discovery. Nat. Rev. Drug Discovery 2010, 9, 929–939. 10.1038/nrd3287. [DOI] [PubMed] [Google Scholar]
- Liu X.; Wright M.; Hop C. E. C. A. H. Rational Use of Plasma Protein and Tissue Binding Data in Drug Design. J. Med. Chem. 2014, 57 (20), 8238–8248. 10.1021/jm5007935. [DOI] [PubMed] [Google Scholar]
- Obach R. S.; Lombardo F.; Waters N. J. Trend Analysis of a Database of Intravenous Pharmacokinetic Parameters in Humans for 670 Drug Compounds. Drug Metab. Dispos. 2008, 36 (7), 1385–1405. 10.1124/dmd.108.020479. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Keserü G. M.; Leeson P. D.; Rees D. C.; Reynolds C. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discovery 2014, 13, 105–121. 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]
- Lin B.; Pease J. H. A Novel Method for High Throughput Lipophilicity Determination by Microscale Shake Flask and Liquid Chromatography Tandem Mass Spectrometry. Comb. Chem. High Throughput Screening 2013, 16, 817–825. 10.2174/1386207311301010007. [DOI] [PubMed] [Google Scholar]
- Ye Z.; Zetterberg C.; Gao H. Automation of plasma protein binding assay using rapid equilibrium dialysis device and Tecan workstation. J. Pharm. Biomed. Anal. 2017, 140, 210–214. 10.1016/j.jpba.2017.03.019. [DOI] [PubMed] [Google Scholar]
- Waters N. J.; Jones R.; Williams G.; Sohal B. Validation of a Rapid Equilibrium Dialysis Approach for the Measurement of Plasma Protein Binding. J. Pharm. Sci. 2008, 97 (10), 4586–4595. 10.1002/jps.21317. [DOI] [PubMed] [Google Scholar]
- Choo E. F.; Belvin M.; Boggs J.; Deng Y.; Hoeflich K. P.; Ly J.; Merchant M.; Orr C.; Plise E.; Robarge K.; Martini J. F.; Kassees R.; Aoyama R. G.; Ramaiya A.; Johnston S. H. Preclinical Disposition of GDC-0973 and Prospective and Retrospective Analysis of Human Dose and Efficacy Predictions. Drug Metab. Dispos. 2012, 40 (5), 919–927. 10.1124/dmd.111.043778. [DOI] [PubMed] [Google Scholar]
- Choo E. F.; Alicke B.; Boggs J.; Dinkel V.; Gould S.; Grina J.; West K.; Menghrajani K.; Ran Y.; Rudolph J.; Wenglowsky S. Preclinical assessment of novel BRAF inhibitors: integrating pharmacokinetic-pharmacodynamic modelling in the drug discovery process. Xenobiotica 2011, 41 (12), 1076–1087. 10.3109/00498254.2011.603384. [DOI] [PubMed] [Google Scholar]
- Halladay J. S.; Wong S.; Jaffer S. M.; Sinhababu A. K.; Khojasteh-Bakht S. C. Metabolic stability screen for drug discovery using cassette analysis and column switching. Drug Metab. Lett. 2006, 38, 149–149. 10.2174/187231207779814364. [DOI] [PubMed] [Google Scholar]
- Moka 2.6.5; Molecular Discovery: United Kingdom. http://www.moldiscovery.com.
- Rodgers T.; Rowland M. Mechanistic approaches to volume of distribution predictions: understanding the processes. Pharm. Res. 2007, 24 (5), 918–33. 10.1007/s11095-006-9210-3. [DOI] [PubMed] [Google Scholar]
- Griffen E.; Leach G. A.; Robb R. G.; Warner J. D. Matched Molecular Pairs as a Medicinal Chemistry Tool. J. Med. Chem. 2011, 54 (22), 7739–7750. 10.1021/jm200452d. [DOI] [PubMed] [Google Scholar]
- Kramer C.; Ting A.; Zheng H.; Hert J.; Schindler T.; Stahl M.; Robb G.; Crawford J. J.; Blaney J.; Montague S.; Leach G. A.; Dossetter G. A.; Griffen J. E. Learning Medicinal Chemistry Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) Rules from Cross-Company Matched Molecular Pairs Analysis (MMPA). J. Med. Chem. 2017, 10.1021/acs.jmedchem.7b00935. [DOI] [PubMed] [Google Scholar]
- Poulin P.; Haddad S. Microsome composition-based model as a mechanistic tool to predict nonspecific binding of drugs in liver microsomes. J. Pharm. Sci. 2011, 100 (10), 4501–4517. 10.1002/jps.22619. [DOI] [PubMed] [Google Scholar]
- Benet L.; Broccatelli F.; Oprea T. BDDCS applied to over 900 drugs. AAPS J. 2011, 13 (4), 519–547. 10.1208/s12248-011-9290-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KNIME Analytic Platform, version 3.2.2; Zurich, Switzerland. https://www.knime.org/.
- Vernalis Nodes for KNIME (trusted extension). https://tech.knime.org/book/vernalis-nodes-for-knime-trusted-extension.
- Penning T. D.; Talley J. J.; Bertenshaw S. R.; Carter J. S.; Collins P. W.; Docter S.; Graneto M. J.; Lee L. F.; Malecha J. W.; Miyashiro J. M.; Rogers R. S.; Rogier D. J.; Yu S. S.; Anderson G. D.; Burton E. G.; Cogburn J. N.; Gregory S. A.; Koboldt K. M.; Perkins W. E.; Seibert K.; Veenhuizen A. W.; Zhang Y. Y.; Isakson P. C. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: identification of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide (SC-58635, Celecoxib). J. Med. Chem. 1997, 40, 1347–1365. 10.1021/jm960803q. [DOI] [PubMed] [Google Scholar]
- Stamler D.; Bradbury M.; Brown F. The Pharmacokinetics and Safety of Deuterated-Tetrabenazine. Neurology 2013, 80, 210.23296131 [Google Scholar]
- Stepan A. F.; Kauffman G. W.; Keefer C. E.; Verhoest P. R.; Edwards M. Evaluating the differences in cycloalkyl ether metabolism using the deisgn parameter “lipophilic metabolic efficiency” (LipMetE) and a matched molecular pair analysis. J. Med. Chem. 2013, 56, 6985–6990. 10.1021/jm4008642. [DOI] [PubMed] [Google Scholar]
- McGinnity D. F.; Collington J.; Austin R. P.; Riley R. J. Evaluation of Human Pharmacokinetics, Therapeutic Dose and Exposure Predictions Using Marketed Oral Drugs. Curr. Curr. Drug Metab. 2007, 8 (5), 463–479. 10.2174/138920007780866799. [DOI] [PubMed] [Google Scholar]