

Abstract

Cortistatin A is a natural product isolated from the marine sponge Corticium simplex and was found to be a potent and selective inhibitor of CDK8. Many synthetic groups have reported total syntheses of Cortistatin A; however, these syntheses require between 16 and 30 steps and report between 0.012–2% overall yields, which is not amenable to large-scale production. Owing to similarities between the complex core of Cortistatin A and the simple steroid core, we initiated a campaign to design simple, more easily prepared CDK8 inhibitors based on a steroid scaffold that would be more convenient for large-scale synthesis. Herein, we report the discovery and optimization of JH-VIII-49, a potent and selective inhibitor of CDK8 with a simple steroid core that has an eight-step synthesis with a 33% overall yield, making it suitable for large-scale preparation. Using this scaffold, we then developed a bivalent small molecule degrader, JH-XI-10-02, that can recruit the E3 ligase CRL4Cereblon to promote the ubiquitination and proteosomal degradation of CDK8.
Keywords: CDK8, CDK19, Cortistatin A, kinase inhibitor, mediator complex, PROTAC, Cereblon, E3 ligase, degradation
CDK8 is a cyclin-dependent kinase that forms part of the mediator complex, a multiprotein assembly comprising up to 30 subunits that regulates gene transcription in multiple contexts including stem cell function, immune response, inflammation, cell adhesion, epithelial to mesenchymal transition, and development.1−3 Multiple substrates of CDK8 have been identified,4 including histone H3,5 RNA polymerase II (RNAPII) C-terminal domain (CTD),6,7 subunits of general transcription factors (GTFs),8,9 and certain transactivators.10−14 CDK8 plays important roles in oncogenic signaling pathways, including the Wnt-β-catenin pathway,15 the TGF-β signaling pathway,16 the p53 pathway,17,18 the serum and hypoxia response network,19,20 the Notch and STAT1 signaling pathway,11,21 thyroid hormone receptor,22 and those governed by SMADs.16 CDK8 gene expression is also associated with increased mortality in colorectal, breast, and ovarian cancers.23 In addition, CDK8 was found to be overexpressed and essential for cell proliferation in melanoma.24 Knock down of CDK8 by short hairpin RNA (shRNA) modestly suppressed proliferation in cancer cells and induced cell cycle arrest and apoptosis in both in vitro and in vivo models.15
Therefore, inhibition of CDK8 by small molecules may be a promising approach for cancer therapy. There have been a number of small molecule inhibitors of CDK8 reported in the literature, including the natural product Cortistatin A as well as synthetic heterocyclic ATP-competitive inhibitors.25,26 Cortistatin A is a sponge-derived natural product comprising an elaborated steroidal core substituted with a quinoline ring and is unlike any previously reported kinase inhibitors. Structural and mechanistic studies have revealed that it is a highly potent ATP-competitive CDK8 inhibitor (IC50 = 15 nM) that utilizes the quinoline to contact the kinase hinge segment and that the steroidal core makes extensive contacts with the ATP-binding cavity. Cortistatin A exhibits exceptional selectivity for CDK8 and the closely related kinase CDK19, with only 4.5% of 359 kinases inhibited at 35% of control.27 Crystal structure and mutagenesis studies demonstrate the unique steroidal architecture combined with a π–cation interaction with W105 as major contributors to the exceptional kinase selectivity.26 There have been a number of total syntheses reported for Cortistatin A requiring between 16 and 30 steps with reported overall yields of between 0.012 and 2%.28−30 We therefore set out to design analogues of Cortistatin A that would maintain the same combination of potency and selectivity but that would be easier to synthesize. Given the similarity between the core structure of Cortistatin A and naturally occurring steroids, we wondered if we could replace the complex core of Cortistatin A with a simple steroid structure, such as dehydroepiandrosterone (DHEA). We began by constructing a molecular overlay of Cortistatin A with DHEA containing the same 7-isoquinoline hinge binder and a β-dimethyl amino group at C-3 (19) (Figure 1). See SI for details regarding construction of the overlay. This suggested that the overall size of the molecules was nearly identical and that the dimethyl amino groups were in very similar positions. In addition, the co-crystal structure of Cortistatin A with CDK8 reveals that the C5–C8 ethane bridge and the C13 methyl group of Cortistatin A occupy a hydrophobic cleft, which overlays well with the C18 and C19 methyl groups of compound 19.
Figure 1.
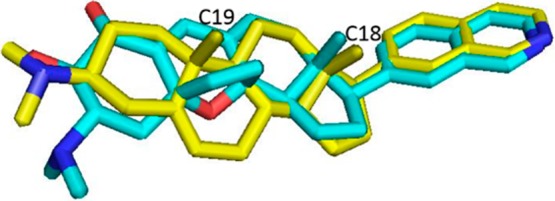
Overlay of 19 (yellow) with Cortistatin A (cyan).
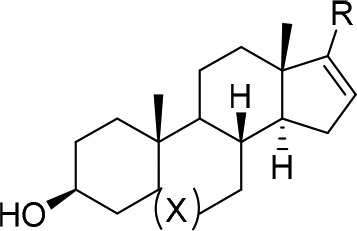
Based on the 3-D overlay, we felt reasonably confident that we could replace the core of Cortstatin A with that of a simple steroid. To test this hypothesis, we synthesized compound 1 from the commercially available 17-iodo-DHEA (17) and 7-isoquinoline boronic acid via Suzuki coupling according to Scheme 1. Compound 1 showed a modest biochemical IC50 of 164 nM against CDK8. Based on these promising results, we prepared the fully saturated 3β-androsterone analogue 2 to investigate the importance of the double bond at C5 and found that it was approximately 6-fold less potent. We also explored additional hinge-binding motifs containing additional hydrogen bond donors or acceptors as shown in Table 1 with the hope of increasing binding affinity. Compound 3 with a 6-thalazine hinge binder resulted in a drastic loss of potency with a biochemical IC50 > 10,000 nM. Compound 4 bearing a 6-quinazolin-2-amine also lost potency with a biochemical IC50 of 2300 nM. Compounds with 6–5-fused ring systems such as indazoles (5) and (6) also showed a dramatic loss in potency with IC50s > 10,000 nM. Introduction of 1H-indazol-3-amine (7) showed >10-fold increase in potency with respect to the previous indazoles 5 and 6; however, the potency was still much less than that of 1. Compound 8 with a 6-isoquinoline hinge binder showed a 50-fold decrease in potency compared to 1, with an IC50 of 8240 nM. This preliminary structure–activity relationship (SAR) suggested that the 7-isoquinoline group was optimal for potency. Compounds 3–8 were prepared in a similar manner as 1 and 2 using the appropriate boronic acid or boronic ester.
Scheme 1. Preparation of Compounds 1–16.

Reagents and conditions: (a) Pd(PPh3)2Cl2, isoquinolin-7-ylboronic acid, Na2CO3, dioxane/H2O, 80%; (b) KOOCN=NCOOK, AcOH, DMSO, THF, 60%.
Table 1. SAR of the 17 Position of Androsterone and DHEA Derivatives.

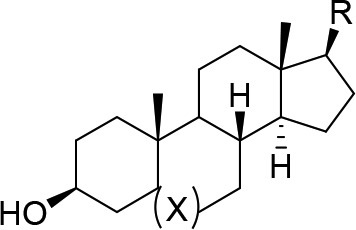
In order to investigate the importance of the benzylic olefin, we chemoselectively reduced it using KOOCN=NCOOK and acetic acid (Scheme 1) to give compounds 9–16 shown in Table 2. In general, reduction of the benzylic olefin resulted in a 2–4-fold increase in potency with the exception of compounds 11, 13, and 14, which remained completely inactive, and compound 12, which showed a 20-fold increase in potency. However, despite this increase in potency, compound 12 was still less potent than 9, so we decided to keep the 7-isoquinoline hinge binder in place.
Table 2. Biochemical IC50s of Reduced Androsterone and DHEA Derivatives.

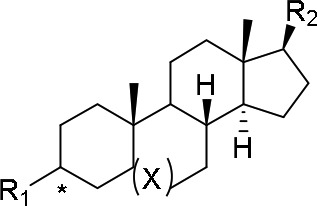
Having established 7-isoquinoline as the most potent hinge binder and the preference for the 16-dehydro steroid core, we chose to explore different groups at the 3-position. As previously mentioned, the dimethyl amino group of Cortistatin A was found to increase potency due to the formation of a π–cation interaction with W105 of CDK8. Therefore, we chose to install a variety of groups capable of forming this π–cation interaction, including the 3β- or 3α-dimethyl amino group (18–20), 3β-1,2,3-triazole (22), and 3β-morpholine (23). In addition, we installed a 3β-methylaminoacetamide group (24), which could form a π–π interaction with W105 through the carbonyl. As expected, compound 19 with a 3β-dimethylamino group on the DHEA backbone was the most potent with an IC50 of 16 nM. Surprisingly, compounds 18 showed similar potency to 19 despite having the saturated androsterone backbone, which was found to be 5–10-fold less potent with a 3β-hydroxy group (compare 1 with 2 and 9 with 10). We also prepared the 3α-dimethylamino analogue (20) to verify the stereochemical requirement at the 3-position and indeed found it to be 4-fold less potent than the 3β analogue (19) as shown in Table 3. We also prepared the 3β-methylamino analogue (21), which forms a weaker π–cation interaction, and found it to be 55-fold less potent than (18).
Table 3. SAR of the 3-Position of DHA and DHEA Cores.

Compounds 18–20 were prepared from commercially available DHEA or its 3α-epimer according to Scheme 2. 3α-DHEA was converted to acetal 26, which underwent a Mitsunobu reaction with DPPA to give the corresponding azide. The azide was reduced using trimethylphosphine to give the amine, which was dimethylated using formaldehyde to give 26. Compound 26 was deprotected with TsOH to furnish the ketone, which was converted to the triflate 27 with PhN(SO2CF3)2. The 7-isoquinoline group was installed via Suzuki coupling followed by diazocarboxylate-mediated reduction of the benzylic olefin to give 19 with a 33% overall yield. More importantly, three of the eight steps required no purification, further demonstrating the convenience of this synthesis. Synthetic schemes and detailed experimental procedures for compounds 18–24 can be found in the Supporting Information (SI).
Scheme 2. Preparation of Compounds 18–20.

Reagents and conditions: (a) ethylene glycol, toluene, TsOH, 100 °C, 98%; (b) DPPA, DEAD, PPh3, THF, 87%; (c) trimethylphosphine, THF/H2O, 85%; (d) formaldehyde, EtOAc, MeOH, 92%; (e) TsOH, acetone, 98%; (f) KHMDS, PhN(SO2CF3)2, THF, 95%; (g) Pd(PPh3)2Cl2, 7-isoquinoline boronic acid, Na2CO3, dioxane/H2O, 76%; (h) KOOCN=NCOOK, AcOH, DMSO, THF, 70%.
Based on these results, we chose compound 19 as our lead and initiated a molecular modeling study using the published co-crystal structure of Cortistatin A with CDK8 (Figure 2). See SI for details regarding construction of the molecular modeling study. Our model predicted that compound 19 makes the same single hinge contact through the nitrogen of the isoquinoline and the N–H of A100. Additionally, the 3β-dimethyl amino group of compound 19 was predicted to be in close proximity of W105 and capable of forming the same π–cation interaction.
Figure 2.
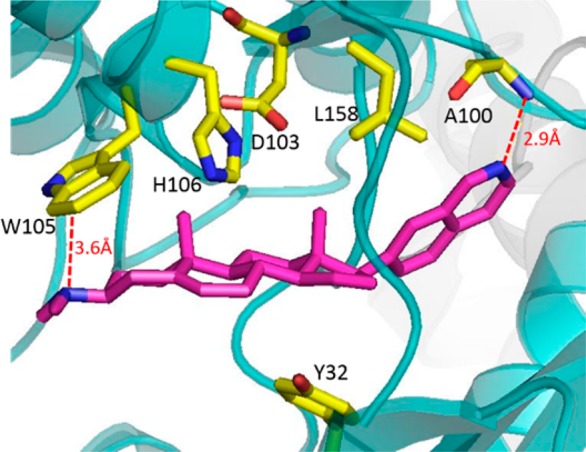
Molecular model of JH-VIII-49 with CDK8.
To evaluate the kinase selectivity, we performed KinomeScan binding analysis against a near comprehensive panel of 468 kinases at a concentration of 10 μM (Figure 3). This profiling revealed an extremely high level of selectivity for compound 19 with only four interactions greater than 90% inhibition including CDK19, CDK8, NEK1, and PIKFYVE (not shown). Dose–response analysis revealed an IC50 of 8 nM against CDK19 and no inhibition of NEK1 with an IC50 > 10,000 nM.
Figure 3.
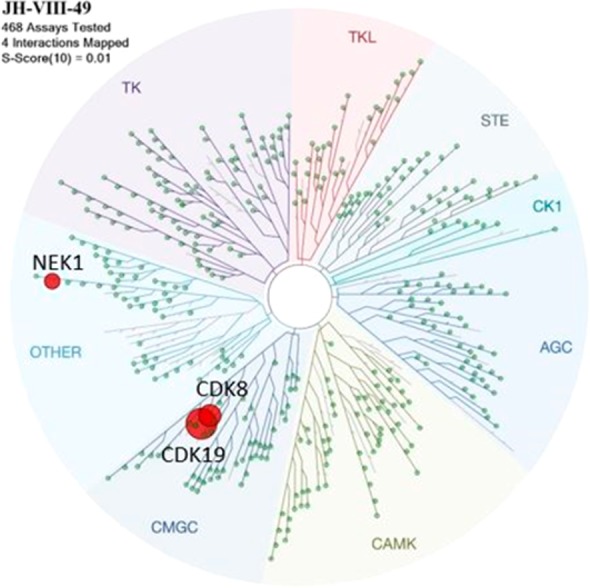
Kinomescan results for JH-VIII-49 at a concentration of 10 μM with a cutoff of 90% inhibition.
There are currently no commercial enzyme assays for PIKFYVE. Complete profiling results are provided in the SI.
We next examined the ability of 19 to inhibit CDK8 in a cellular context in comparison with Dehydrocortistatin A (DCA), which has an enzyme IC50 of 17 nM against CDK8, by monitoring phosphorylation of S727-STAT1, a known substrate of CDK8.21 Compound 19 and DCA induced a dose-dependent inhibition of S727 phosphorylation in HepG2 cells. Substantial dephosphorylation of S727 was observed at approximately 1 μM DCA, while similar effects were seen at 5 μM compound 19 (SI Figure S1).
Recently, several groups have pioneered a chemical-induced protein degradation strategy involving bivalent compounds that bind to a protein of interest (POI) and possess a linker to thalidomide or a close analog, which results in the recruitment of Cereblon (CRBN), a ubiquitously expressed E3 ligase receptor, to induce ubiquitination and proteasomal degradation of the POI.31 Potential theoretical advantages of induced target degradation versus inhibition include prolonged pharmacodynamic effects and an ability to abrogate nonenzyme-dependent or so-called “scaffolding” functions of the protein. In addition, because these compounds can potentially act catalytically to induce protein degradation, they may not require the intracellular concentrations necessary to achieve the level of target occupancy required by traditional reversible inhibitors. Using the molecular model of the inhibitor bound to CDK8, we designed a bivalent molecule by derivatizing the amino group of compound 18 with a polyethylene glycol (PEG) linker conjugated to pomalidomide to generate a series of compounds (Figure 4). We anticipated that such a degrader could be used both to confirm cellular target engagement and to explore the pharmacological consequences of degrading CDK8 in a cellular context. We chose to install the linker with an E3 ligase-targeting warhead on the 3β-amino group since this group points toward the solvent exposed region in our molecular model, which would allow the parent CDK8 inhibitor to bind to CDK8 while simultaneously allowing the E3-targeting warhead to bind to the E3 ligase. We began by installing an alkyl linker on the 3β amino group (Figure 4) since the 3β-dimethyl amino group provided the highest affinity for CDK8. In addition, we installed pomalidomide at the other end of the linker, which is known to target the E3 ligase Cereblon (CRBN). Our initial compound 28 showed no degradation of CDK8 in Jurkat cells after treatment for 6 or 24 h at a concentration of 0.1 or 1 μM as seen in Figure 5A. We then decided to switch to an N-methylacetamide-based linker since the parent inhibitor (24) was also potent with an IC50 of 34 nM. We prepared compounds with a Peg4 linker (29) and a Peg2 (30) linker (Figure 4) and were excited to find that compound 29 induced partial degradation of CDK8 in Jurkat cells upon treatment for 6 h at a concentration of 1 μM. More importantly, we observed significant degradation of CDK8 after treatment for 24 h at a concentration of 1 μM (Figure 5A).
Figure 4.

Structure of PROTAC compounds and biochemical IC50s.
Figure 5.

Cellular evaluation of the bivalent degrader molecules. (A) Jurkat cells were treated with 0.1 and 1 μM compounds 28 and 29; lysates were analyzed at 6 and 24 h for CDK8 levels by immunoblot. (B) WT Molt4 and CRBN null Molt4 cells were treated with increasing concentrations of 29; lysates were analyzed at 24 h for CDK8 levels by immunoblot. (C) Jurkat cells were treated with increasing concentrations of 30, and CDK8 levels were analyzed after 24 h by immunoblot. (D) Jurkat cells were treated with increasing concentrations of negative control 31, and CDK8 levels were analyzed after 24 h by immunoblot.
However, we observed no degradation of CDK8 upon treatment with compound 30, indicating a requirement for a longer linker to bind both CDK8 and CRBN (Figure 5C). To verify that degradation was in fact due to recruitment of CRBN, WT Molt4 cells and Molt4 cells where CRBN had been subject to CRISPER/CAS9-mediated deletion were treated with compound 29 at multiple concentrations for 24 h. While we observed degradation of CDK8 at 5 μM in WT Molt4 cells, we did not observe any degradation in CRBN null Molt4 cells at any concentration of 29 (Figure 5B). For further confirmation, we prepared compound 31, which contains an N-methyl glutarimide ring. This modification has been shown to disrupt a key hydrogen bond interaction of the glutarimide ring with Trp382 of CRBN, which greatly reduces the affinity for CRBN. As seen in Figure 5D, we observed no degradation of CDK8 in Jurkat cells treated with 31 at concentrations up to 10 μM for 24 h.
To further investigate the mechanism for loss of CDK8 protein induced by treatment with 29, we pretreated Jurkat cells with 10 μM proteasome inhibitor (bortezomib), a neddylation inhibitor (MLN4924), lenalidomide, or 31 followed by treatment with 29 (SI Figure S2). As expected, blockade of proteasome activity with bortezomib or CRL4 E3 ligase activity with MLN4924 abolished degradation of CDK8. Moreover, competition for binding with either CRBN (with excess lenalidomide) or CDK8 (with excess 31) also ablated CDK8 degradation, confirming that CDK8 degradation is induced by CRBN-mediated ubiquitination and subsequent proteasomal degradation of CDK8. Importantly, we observed no change in Cdk8 mRNA levels using two different primer sets for Cdk8 (relative to Actin) from Jurkat cells treated with 29 (1 μM) or vehicle control for 24 h (SI Figure S3), indicating that the loss of CDK8 was due to proteasomal degradation rather than changes in its transcription. To determine the limits of this approach, Jurkat cells were treated with increasing concentrations of 29 up to 20 μM for 24 h, which again resulted in significant degradation of CDK8 at concentrations of 1 and 10 μM; however, at 20 μM we observed a reduction in degradation, likely due to the hook effect32 (SI Figure S4). Moreover, we examined the effects of 29 on CDK19 protein levels in Jurkat cells treated with 29 for 24 h. We did not observe any change in CDK19 protein levels (SI Figure S5), indicating a high degree of selectivity for CDK8 degradation. Previous studies have reported that converting inhibitors into degraders can confer additional selectivity,33 which may explain why CDK19 remains unaffected. Additional experiments are currently underway to understand the effects of degrading CDK8 in a cellular context and will be reported in due time.
Synthetic schemes and detailed synthetic procedures describing the synthesis of compounds 29–31 are available in the Supporting Information.
In summary, we have discovered JH-VIII-49 (19), a much simplified analog of Cortistatin A that exhibits only slightly reduced potency against CDK8 in biochemical and cellular assays. Compound 19 can be prepared in eight steps with an overall yield of 33% making it suitable for large-scale preparation. In addition, 19 displays an extremely high level of selectivity binding to only four kinases >10% of control in a panel of 468 kinases at a concentration of 10 μM. Using this scaffold, we developed JH-XI-10-02 (29), a potent degrader of CDK8, by converting 19 into degrader molecules that simultaneously bind to CDK8 and recruit CRBN to induce degradation of CDK8. We observed significant degradation of CDK8 in Jurkat cells treated with 1 μM 29 for 24 h. We confirmed that degradation is mediated by CRBN by testing 29 in CRBN knockout Molt14 cells as well as using the negative control 31, which cannot bind CRBN. In both cases, we observed no degradation of CDK8. These CDK8 degraders offer a chemical means to modulate the protein levels of CDK8 and explore whether loss of CDK8 protein is a viable therapeutic strategy in cancer. Further elaboration of this scaffold and characterization of adsorption, distribution, metabolism, and excretion (ADME) will be reported in due course.
Acknowledgments
The authors wish to thank Philip Baran for providing DCA.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00011.
Full Ambit profiling data for JH-VIII-49, experimental details, effect on cellular pS727-STAT1 levels, dose–response degradation by 29 in Jurkat cells, rescue of degradation experiments, and CDK8 mRNA levels following treatment with 29 (PDF)
Author Contributions
Conceptualization: J.M.H. and N.S.G. Compound synthesis: J.M.H. Cellular experiments: E.W. Molecular modeling: T.S. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Allen B. L.; Taatjes D. J. The Mediator complex: a central integrator of transcription. Nat. Rev. Mol. Cell Biol. 2015, 16 (3), 155–166. 10.1038/nrm3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsten J. O. P.; Zhu X. F.; Gustafsson C. M. The multitalented Mediator complex. Trends Biochem. Sci. 2013, 38 (11), 531–537. 10.1016/j.tibs.2013.08.007. [DOI] [PubMed] [Google Scholar]
- Schiano C.; Casamassimi A.; Rienzo M.; de Nigris F.; Sommese L.; Napoli C. Involvement of Mediator complex in malignancy. Biochim. Biophys. Acta, Rev. Cancer 2014, 1845 (1), 66–83. 10.1016/j.bbcan.2013.12.001. [DOI] [PubMed] [Google Scholar]
- Poss Z. C.; Ebmeier C. C.; Odell A. T.; Tangpeerachaikul A.; Lee T.; Pelish H. E.; Shair M. D.; Dowell R. D.; Old W. M.; Taatjes D. J. Identification of Mediator Kinase Substrates in Human Cells using Cortistatin A and Quantitative Phosphoproteomics. Cell Rep. 2016, 15 (2), 436–450. 10.1016/j.celrep.2016.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer K. D.; Donner A. J.; Knuesel M. T.; York A. G.; Espinosa J. M.; Taatjes D. J. Cooperative activity of cdk8 and GCN5L within Mediator directs tandem phosphoacetylation of histone H3. EMBO J. 2008, 27 (10), 1447–1457. 10.1038/emboj.2008.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuesel M. T.; Meyer K. D.; Donner A. J.; Espinosa J. M.; Taatjes D. J. The Human CDK8 Subcomplex Is a Histone Kinase That Requires Med12 for Activity and Can Function Independently of Mediator. Mol. Cell. Biol. 2009, 29 (3), 650–661. 10.1128/MCB.00993-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickert P.; Corden J. L.; Lees E. Cyclin C CDK8 and cyclin H CDK7 p36 are biochemically distinct CTD kinases. Oncogene 1999, 18 (4), 1093–1102. 10.1038/sj.onc.1202399. [DOI] [PubMed] [Google Scholar]
- Akoulitchev S.; Chuikov S.; Reinberg D. TFIIH is negatively regulated by cdk8-containing mediator complexes. Nature 2000, 407 (6800), 102–106. 10.1038/35024111. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Kung C.; Fishburn J.; Ansari A. Z.; Shokat K. M.; Hahn S. Two cyclin-dependent kinases promote RNA polymerase II transcription and formation of the scaffold complex. Mol. Cell. Biol. 2004, 24 (4), 1721–1735. 10.1128/MCB.24.4.1721-1735.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi Y.; Huddleston M. J.; Zhang X. L.; Young R. A.; Annan R. S.; Carr S. A.; Deshaies R. J. Negative regulation of Gcn4 and Msn2 transcription factors by Srb10 cyclin-dependent kinase. Genes Dev. 2001, 15 (9), 1078–1092. 10.1101/gad.867501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer C. J.; White J. B.; Jones K. A. Mastermind recruits CycC: CDK8 to phosphorylate the notch ICD and coordinate activation with turnover. Mol. Cell 2004, 16 (4), 509–520. 10.1016/j.molcel.2004.10.014. [DOI] [PubMed] [Google Scholar]
- Hirst M.; Kobor M. S.; Kuriakose N.; Greenblatt J.; Sadowski I. GAL4 is regulated by the RNA polymerase II holoenzyme-associated cyclin-dependent protein kinase SRB10/CDK8. Mol. Cell 1999, 3 (5), 673–678. 10.1016/S1097-2765(00)80360-3. [DOI] [PubMed] [Google Scholar]
- Nelson C.; Goto S.; Lund K.; Hung W.; Sadowski I. Srb10/Cdk8 regulates yeast filamentous growth by phosphorylating the transcription factor Ste12. Nature 2003, 421 (6919), 187–190. 10.1038/nature01243. [DOI] [PubMed] [Google Scholar]
- Vincent O.; Kuchin S.; Hong S. P.; Townley R.; Vyas V. K.; Carlson M. Interaction of the Srb10 kinase with Sip4, a transcriptional activator of gluconeogenic genes in Saccharomyces cerevisiae. Mol. Cell. Biol. 2001, 21 (17), 5790–5796. 10.1128/MCB.21.17.5790-5796.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestein R.; Bass A. J.; Kim S. Y.; Dunn I. F.; Silver S. J.; Guney I.; Freed E.; Ligon A. H.; Vena N.; Ogino S.; Chheda M. G.; Tamayo P.; Finn S.; Shrestha Y.; Boehm J. S.; Jain S.; Bojarski E.; Mermel C.; Barretina J.; Chan J. A.; Baselga J.; Tabernero J.; Root D. E.; Fuchs C. S.; Loda M.; Shivdasani R. A.; Meyerson M.; Hahn W. C. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature 2008, 455 (7212), 547–U60. 10.1038/nature07179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcon C.; Zaromytidou A. I.; Xi Q. R.; Gao S.; Yu J. Z.; Fujisawa S.; Barlas A.; Miller A. N.; Manova-Todorova K.; Macias M. J.; Sapkota G.; Pan D. J.; Massague J. Nuclear CDKs Drive Smad Transcriptional Activation and Turnover in BMP and TGF-beta Pathways. Cell 2009, 139 (4), 757–769. 10.1016/j.cell.2009.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donner A. J.; Szostek S.; Hoover J. M.; Espinosa J. M. CDK8 is a stimulus-specific positive coregulator of p53 target genes. Mol. Cell 2007, 27 (1), 121–133. 10.1016/j.molcel.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donner A. J.; Hoover J. M.; Szostek S. A.; Espinosa J. M. Stimulus-specific transcriptional regulation within the p53 network. Cell Cycle 2007, 6 (21), 2594–2598. 10.4161/cc.6.21.4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donner A. J.; Ebmeier C. C.; Taatjes D. J.; Espinosa J. M. CDK8 is a positive regulator of transcriptional elongation within the serum response network. Nat. Struct. Mol. Biol. 2010, 17 (2), 194–U9. 10.1038/nsmb.1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbraith M. D.; Allen M. A.; Bensard C. L.; Wang X. X.; Schwinn M. K.; Qin B.; Long H. W.; Daniels D. L.; Hahn W. C.; Dowell R. D.; Espinosa J. M. HIF1A Employs CDK8-Mediator to Stimulate RNAPII Elongation in Response to Hypoxia. Cell 2013, 153 (6), 1327–1339. 10.1016/j.cell.2013.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bancerek J.; Poss Z. C.; Steinparzer I.; Sedlyarov V.; Pfaffenwimmer T.; Mikulic I.; Dolken L.; Strobl B.; Muller M.; Taatjes D. J.; Kovarik P. CDK8 Kinase Phosphorylates Transcription Factor STAT1 to Selectively Regulate the Interferon Response. Immunity 2013, 38 (2), 250–262. 10.1016/j.immuni.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbraith M. D.; Donner A. J.; Espinosa J. M. CDK8. Transcription 2010, 1 (1), 4–12. 10.4161/trns.1.1.12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter D. C.; Liang J. X.; Kaza V.; Chumanevich A. A.; Altilia S.; Farmaki E.; Chen M. Q.; Schools G. P.; Chatzistamou I.; Pena M. M.; Friedhoff L. T.; Wentland M. P.; Broude E.; Kiaris H.; Roninson I. B. Targeting the seed and the soil of cancers with selective small-molecule inhibitors of CDK8/19: Chemopotentiating, chemopreventive, anti-invasive and anti-metastatic activities. Cancer Res. 2014, 74 (19), 4879. 10.1158/1538-7445.AM2014-4879. [DOI] [Google Scholar]
- Kapoor A.; Goldberg M. S.; Cumberland L. K.; Ratnakumar K.; Segura M. F.; Emanuel P. O.; Menendez S.; Vardabasso C.; LeRoy G.; Vidal C. I.; Polsky D.; Osman I.; Garcia B. A.; Hernando E.; Bernstein E. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 2010, 468 (7327), 1105–U509. 10.1038/nature09590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzymski T.; Mikula M.; Wiklik K.; Brzozka K. CDK8 kinase-An emerging target in targeted cancer therapy. Biochim. Biophys. Acta, Proteins Proteomics 2015, 1854 (10), 1617–1629. 10.1016/j.bbapap.2015.05.011. [DOI] [PubMed] [Google Scholar]
- Pelish H. E.; Liau B. B.; Nitulescu II; Tangpeerachaikul A.; Poss Z. C.; Da Silva D. H.; Caruso B. T.; Arefolov A.; Fadeyi O.; Christie A. L.; Du K.; Banka D.; Schneider E. V.; Jestel A.; Zou G.; Si C.; Ebmeier C. C.; Bronson R. T.; Krivtsov A. V.; Myers A. G.; Kohl N. E.; Kung A. L.; Armstrong S. A.; Lemieux M. E.; Taatjes D. J.; Shair M. D. Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature 2015, 526 (7572), 273. 10.1038/nature14904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cee V. J.; Chen D. Y. K.; Lee M. R.; Nicolaou K. C. Cortistatin A is a High-Affinity Ligand of Protein Kinases ROCK, CDK8, and CDK11. Angew. Chem., Int. Ed. 2009, 48 (47), 8952–8957. 10.1002/anie.200904778. [DOI] [PubMed] [Google Scholar]
- Lee H. M.; Nieto-Oberhuber C.; Shair M. D. Enantioselective Synthesis of (+)-Cortistatin A, a Potent and Selective Inhibitor of Endothelial Cell Proliferation. J. Am. Chem. Soc. 2008, 130 (50), 16864. 10.1021/ja8071918. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Peng X. S.; Sun Y. P.; Polet D.; Zou B.; Lim C. S.; Chen D. Y. K. Total Synthesis and Biological Evaluation of Cortistatins A and J and Analogues Thereof. J. Am. Chem. Soc. 2009, 131 (30), 10587–10597. 10.1021/ja902939t. [DOI] [PubMed] [Google Scholar]
- Shi J.; Manolikakes G.; Yeh C. H.; Guerrero C. A.; Shenvi R. A.; Shigehisa H.; Baran P. S. Scalable Synthesis of Cortistatin A and Related Structures. J. Am. Chem. Soc. 2011, 133 (20), 8014–8027. 10.1021/ja202103e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai A. C.; Crews C. M. Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discovery 2017, 16 (2), 101–114. 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X. D.; Dixit V. M. Drugging the undruggables: exploring the ubiquitin system for drug development. Cell Res. 2016, 26 (4), 484–498. 10.1038/cr.2016.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson C. M.; Jiang B. S.; Erb M. A.; Liang Y. K.; Doctor Z. M.; Zhang Z. N.; Zhang T. H.; Kwiatkowski N.; Boukhali M.; Green J. L.; Haas W.; Nomanbhoy T.; Fischer E. S.; Young R. A.; Bradner J. E.; Winter G. E.; Gray N. S. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat. Chem. Biol. 2018, 14 (2), 163. 10.1038/nchembio.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.