

Abstract
Toll-like receptor 4 interactor with leucine-rich repeats (Tril) functions as a coreceptor for Toll-like receptors (Tlrs) to mediate innate immune responses in adults. In embryos, Tril signals to promote degradation of the Bmp inhibitor, Smad7, to allow for blood formation. It is not known whether this function requires, or is independent of, Tlrs. In the current studies, we performed a structure–function analysis, which indicated that the fibronectin type III (FN) domain and the intracellular domain of Tril are required to trigger Smad7 degradation in Xenopus embryos. Furthermore, we found evidence suggesting that a Tril deletion mutant lacking the FN domain (Tril∆FN) can dominantly inhibit signaling by endogenous Tril when overexpressed. This finding raises the possibility that the FN domain functions to bind endogenous Tril ligands. We also show that Tril cycles between the cell surface and endosomes and that the Tril extracellular domain, as well as cadherin based cell–cell adhesion, are required for cell surface retention, while the intracellular domain is required for internalization in Xenopus ectodermal explants. Using a CHO cell aggregation assay, we show that, unlike other transmembrane proteins that contain leucine-rich repeats, Tril is not sufficient to mediate homophilic adhesion.
INTRODUCTION
Bone morphogenetic proteins (Bmps) play a critical role in specifying ventral and posterior fates during early development in all vertebrates (Tuazon and Mullins, 2015). Bmps activate transmembrane serine/threonine receptors that phosphorylate the cytoplasmic proteins Smad1, 5, and 8 (Weiss and Attisano, 2013). Phosphorylated Smads (pSmad1/5/8) then recruit the co-Smad, Smad4, and translocate to the nucleus to induce target gene expression. During gastrulation, Bmps induce expression of hematopoietic transcription factors that are necessary and sufficient to specify primitive erythroid fate (Mead et al., 1998; Lengerke et al., 2008). Smad7 is another target gene that is induced by pSmad1/5/8 and is a central hub for negative regulation of activated Bmp receptors (Yan and Chen, 2011). Smad7 recruits E3 ubiquitin ligases to activated Bmp receptors, targeting them for proteosomal degradation and thereby dampening Bmp signal transduction.
We have recently shown that the transmembrane protein Toll-like receptor 4 (Tlr4) interactor with leucine-rich repeats (Tril) is required to augment Bmp signaling during gastrulation in Xenopus embryos (Green et al., 2016). Tril activates a yet-to-be-defined signaling pathway that triggers degradation of the Bmp inhibitor, Smad7, thereby relieving repression of endogenous Bmp signaling. This is essential to enable the mesoderm to commit to a hematopoietic fate during gastrulation. When expression of endogenous Tril is reduced in Xenopus embryos using antisense morpholinos, high levels of Smad7 protein accumulate with no change in levels of Smad7 RNA (Green et al., 2016). As a consequence, pSmad1/5/8 levels are reduced, and blood development is impaired.
The signaling pathway that Tril activates to degrade Smad7 is unknown. Tril was initially characterized as a coreceptor for Tlr3 and Tlr4 and is required to mediate innate immune responses in the brain of adult mammals (Carpenter et al., 2009; Wochal et al., 2014). Tlrs bind microbial products or nucleic acids, initiating a signaling cascade involving cytoplasmic adaptors, kinases, and ubiquitin ligases that generally culminates in activation of transcriptional regulators such as NF-κB, AP-1 and interferon regulatory factors (Brubaker et al., 2015). It is unknown, at present, whether Tril functions independent of Tlrs to promote degradation of Smad7 or whether it serves as a coreceptor for Tlrs in the context of early development to activate a noncanonical signaling pathway. While Tlrs are known to play nonimmune roles in developmental patterning and morphogenesis in insects, these roles do not appear to be conserved during early vertebrate development (Leulier and Lemaitre, 2008). Regulatory crosstalk between Tgf-β/Bmp and Tlr signaling plays a critical role in restraining inflammation in adults (Choi et al., 2006; Seth and Chen, 2007), but there is no evidence that activation of Tlr signaling can promote degradation of Smad7.
In the current studies, we performed a structure–function analysis to identify domains of Tril that are required to trigger degradation of Smad7 in the context of the early embryo. We also identified protein domains and cellular factors that drive the dynamic subcellular localization of Tril in embryos.
RESULTS AND DISCUSSION
In vivo activity of wild-type and deletion mutant forms of Xenopus Tril
We have previously shown that endogenous Tril induces degradation of Smad7 protein (Green et al., 2016). In the current studies, we repeated our analysis of steady-state levels and subcellular localization of Smad7 in Xenopus embryos in which Tril expression was reduced by injection of a well-characterized translation blocking antisense morpholino (MO) (Green et al., 2016). Two-cell embryos were injected with smad7myc RNA (100 pg) together with control or Tril MOs (35 ng). At the midgastrula stage (st. 11), 15 embryos in each group were harvested for immunoblot analysis, and ectoderm was explanted from an additional 10 embryos in each group for immunostaining. Steady-state levels of Smad7myc protein were 3- to 10-fold higher in Tril morphants than in controls in three independent experiments (Figure 1A, top panel). Furthermore, Smad7myc protein accumulated predominantly in nuclei of Tril morphant embryos, whereas it was diffusely localized throughout the cell and at the membrane in control embryos (Figure 1A, bottom panel). These results replicate our published studies showing that endogenous Tril is required to promote degradation of Smad7 protein.
FIGURE 1:

Structure–function analysis of Tril. (A) Embryos were injected with RNA (100 pg) encoding Smad7myc together with control or Tril MOs (35 ng). Immunoblots of lysates from stage 11 embryos (10 per group) were probed with anti-Myc antibodies and then reprobed for β-actin. Relative level of Smad7myc, normalized to actin and reported relative to that in control embryos is indicated below each lane. Ectoderm was explanted from 5–10 embryos in each group at stage 11 and immunostained for Myc (all images taken under identical conditions). (B, E, I, J) RNA (100 pg) encoding Smad7Myc was injected into two-cell embryos alone or together with RNA encoding wild-type or deletion mutant forms of Tril. Immunoblots of lysates from stage 11 embryos (15 per group) were probed with anti-Myc antibodies and then reprobed for β-actin. Representative blots are shown. The relative level of Smad7myc, normalized to actin and reported relative to that in embryos injected with Smad7myc alone is indicated below each lane and quantitated and graphed below each blot (mean ± SD, n is the number of replicates as indicated on each column). In E, all lanes are from the same immunoblot, aligned following removal of an intervening lane (following the third lane, marked by a black bar). (C, D, F, G) smad7myc RNA was injected alone or together with increasing doses of tril (C, D) or 500 pg of tril∆FN RNA (F, G) into one cell of two-cell embryos. Ectoderm was explanted from 7–15 embryos in each group at stage 11 and immunostained for Myc. Representative immunostaining is shown in C and F (all images taken under identical conditions), and results are quantified in D and G. Results are pooled from three independent experiments. The total number of explants analyzed is indicated on each column. (H) Embryos were injected with RNA (100 pg) encoding Smad7myc together with control or Tril MOs (35 ng) and tril or tril∆fn RNA (100 pg) as indicated at the top of each lane. Immunoblots of lysates from stage 11 embryos (10 per group) were probed with anti-Myc antibodies and then reprobed for β-actin. Relative level of Smad7myc, normalized to actin and reported relative to that in control embryos is indicated below each lane. Results were replicated in three independent experiments.
We next used a gain of function assay to ask whether ectopic Tril has the opposite effect of reducing Smad7 protein levels. We injected RNA encoding Smad7myc (100 pg) alone, or together with increasing doses of RNA encoding wild-type Tril (25 pg–1 ng), into one cell of two cell embryos and analyzed Smad7myc protein levels and subcellular localization at st. 11. Steady-state levels of Smad7myc protein were reduced (Figure 1B and Supplemental Figure S1A), but levels of smad7myc RNA were unchanged (Supplemental Figure S1B) in embryos coinjected with low doses of tril RNA (25–100 pg). In embryos injected with smad7myc RNA alone, Smad7myc was evenly distributed throughout the cytoplasm, nucleus, and membrane or was slightly enriched in the nucleus (Figure 1, C and D). In explants isolated from embryos coinjected with 25 or 50 pg of tril RNA, Smad7myc staining was barely detectable when imaged under identical conditions (Figure 1C) and was evenly distributed throughout the nucleus and cytoplasm in most explants (Figure 1D). These results demonstrate that ectopic Tril, like endogenous Tril, can activate downstream signaling pathways that lead to degradation of Smad7.
Surprisingly, in embryos injected with the highest dose of tril RNA (1 ng), steady-state levels of Smad7myc were reproducibly enhanced (Figure 1B), and Smad7myc was detected primarily in nuclei (Figure 1, C and D). This is identical to what is observed when expression of Tril is reduced (Figure 1A). This result raises the possibility that supraphysiological levels of ectopic Tril can squelch signaling downstream of endogenous Tril, perhaps by disrupting the normal stoichiometry of upstream or downstream binding partners that are required to transduce signals necessary for degradation of Smad7. There are many examples in which overexpression of wild-type gene products involved in macromolecular complexes dominantly inhibit signaling downstream of the endogenous protein and generate a loss-of-function phenotype (Veitia, 2007).
We next used our gain of function assay to identify domains of Tril that are required to trigger degradation of Smad7 protein. Tril is a single-pass transmembrane protein with a large extracellular domain (ECD) that contains multiple leucine-rich repeats and a fibronectin type III (FN) domain as well as a small intracellular domain (ICD) that lacks any obvious signaling motifs (illustrated above Figure 1B). We analyzed steady-state levels of Smad7myc in embryos expressing increasing doses of deletion mutant forms of Tril lacking putative functional domains. Notably, injection of increasing doses of RNA encoding wild-type or deletion mutant forms of epitope (HA)-tagged Tril led to a dose-dependent increase in Tril-HA proteins (Supplemental Figure S2).
We first analyzed Smad7 levels in embryos expressing a Tril deletion mutant lacking the FN domain (Tril∆FN). Smad7myc protein levels were not reproducibly altered in embryos injected with 25 pg (Figure 1E) or 50 pg (Supplemental Figure S1C) of RNA encoding Tril∆FN, suggesting that the FN domain is essential for Tril function. By contrast, embryos injected with 100 pg or more of tril∆fn RNA accumulated high levels of Smad7myc protein (Figure 1E and Supplemental Figure S1C) that was found primarily in the nuclei of cells (Figure 1, F and G) but showed no change in levels of smad7myc RNA (Supplemental Figure S1D). This is identical to what is observed in Tril morphants (Green et al., 2016). Furthermore, whereas injection of tril RNA was sufficient to rescue, and even reduced the elevated levels of Smad7myc protein found in Tril morphants, no change in levels of Smad7myc were observed in Tril morphants injected with the same dose of tril∆fn RNA (Figure 1H). These findings demonstrate that the FN domain of Tril is required for Tril function and suggest that ectopic Tril∆FN can dominantly suppress signaling by endogenous Tril.
We next analyzed Smad7 levels in embryos ectopically expressing mutant forms of Tril lacking the ECD (Tril∆ECD) or the ICD (Tril∆ICD). Levels of Smad7myc were not reproducibly changed in embryos injected with 10, 20, or 200 pg of tril∆ECD RNA (the molar equivalent of 50, 100, or 1000 pg of wild-type Tril) (Figure 1I and Supplemental Figure S1E), nor were they reproducibly changed in embryos injected with 25, 50, or 1000 pg of RNA encoding Tril∆ICD (Figure 1J). Thus, the ECD and the ICD of Tril are essential for signal transduction.
Our previous studies have shown that endogenous Tril is required for blood formation (Green et al., 2016). As a further test of whether ectopically expressed wild-type or deletion mutant forms of Tril can function as dominant mutants, we asked whether any of these proteins can phenocopy the loss of blood that is observed in Tril morphant embryos. We injected 1 ng of RNA encoding Tril, Tril∆FN, or Tril∆ICD or the molar equivalent (200 pg) of RNA encoding Tril∆ECD into each ventral blastomere near the marginal zone of four-cell embryos. We then analyzed expression of the red blood cell (RBC) differentiation marker hba3.L (α-globin) at stage 34 using either whole mount in situ hybridization (WMISH) (Figure 2, A and B) or quantitative real-time PCR (qPCR) analysis (Figure 2C). Embryos injected with a total of 2 ng of tril or tril∆FN RNA showed a reproducible reduction in expression of hba3.L in the posterior ventral blood island (pVBI), which is the region derived from the ventral cells that were injected. By contrast, embryos injected with an equivalent dose of tril∆ECD (400 pg) or tril∆ICD (2 ng) RNA did not show a reproducible loss of blood (Figure 2, B and C). Notably, in two experiments, embryos made to overexpress Tril∆ICD showed a partial reduction in levels of hba3.L relative to embryos injected with red fluorescent protein (RFP), but this was not reproduced in five additional experiments. Collectively, our results showing that low doses of wild-type Tril can induce degradation of Smad7, whereas comparable doses of Tril lacking the FN domain, the ECD, or the ICD cannot induce degradation of Smad7 suggest that each of these domains are essential to transduce Tril signals. Furthermore, our finding that ectopic expression of high doses of wild-type Tril, or Tril∆FN phenocopies the accumulation of Smad7myc and loss of blood that is observed in Tril morphants, suggests that these constructs can interfere with the ability of endogenous Tril to transduce signals necessary for blood formation.
FIGURE 2:
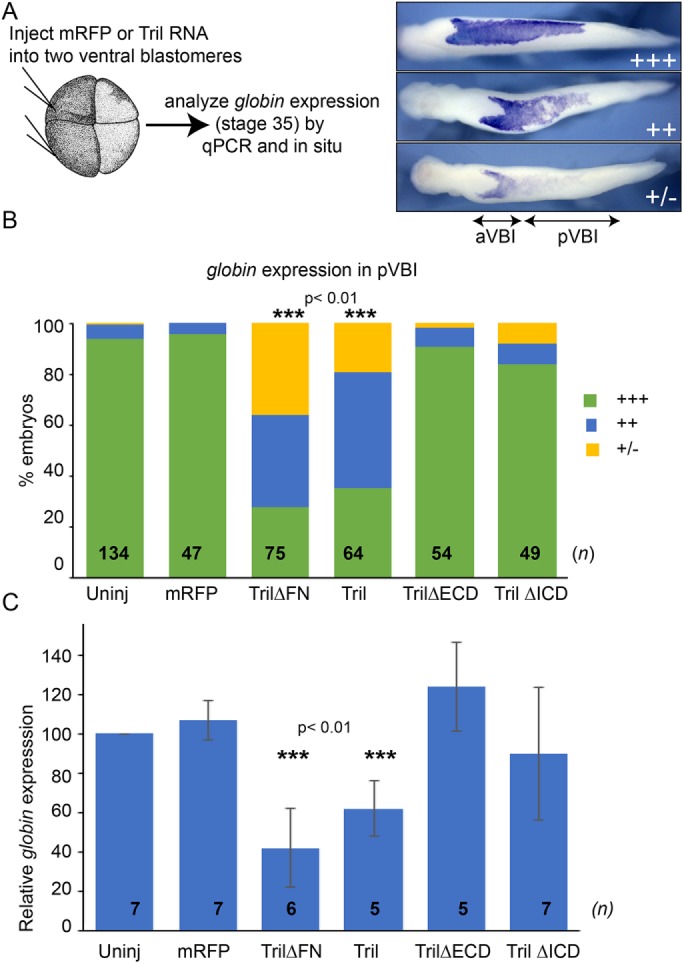
Overexpression of Tril or Tril∆FN inhibits blood formation. (A, B) RNA encoding mRFP or wild-type or deletion mutant forms of Tril was injected into both ventral cells of four-cell embryos, and expression of hba3.L was analyzed by WMISH at stage 34 in three experiments. hba3.L staining in the posterior VBI (pVBI) was scored as absent or very weak (+/–), weak (++), or strong (+++), as illustrated. aVBI, anterior VBI. n represents the number of embryos analyzed in each group, pooled from three independent biological replicates. (C) RNA encoding mRFP or wild-type or deletion mutant forms of Tril was injected into both ventral cells of four-cell embryos and expression of hba3.L was analyzed in 15 pooled embryos from each group by qPCR at stage 34. Mean ± SD are shown. n is the number of biological replicates, indicated on each column. ***p < 0.01 by two-tailed t test.
Our finding that Tril∆FN is unable to initiate signaling suggests a critical role for the fibronectin type III domain in receptor activation. Functions of fibronectin type III domains are not well understood but can include interactions with other extracellular matrix (ECM) or cell surface proteins (Krammer et al., 2002). Interestingly, there is growing evidence that Tlrs can respond to a variety of endogenous ligands including ECM degradation products such as fibronectin and hyaluronin (Yu et al., 2010). It is possible that the FN domain of Tril binds to ECM components that function as ligands to activate signaling downstream of Tril either independent of, or together with, Tlrs. This scenario provides a possible explanation for the mechanism by which Tril∆FN dominantly inhibits signaling by endogenous Tril. If the ectodomain of Tril∆FN can interact with Tlr4 but cannot interact productively with ligands required to initiate signaling, then this could potentially sequester endogenous Tlrs at the membrane in an inactive conformation. Alternatively, the intact ICD of Tril∆FN might sequester cytoplasmic effectors at the membrane and/or in inappropriate subcellular compartments so that they are not available to transduce signals downstream of endogenous Tril. Testing these possibilities will require further analysis of whether Tril functions as a Tlr coreceptor in embryos.
Tril localizes to the plasma membrane and intracellular vesicles in Xenopus embryos, and the extracellular domain is required for plasma membrane retention
We next examined the subcellular localization of HA-tagged wild-type and deletion mutant forms of Tril in Xenopus ectodermal explants. RNA encoding Tril variants (100 pg) was injected together with membrane-localized RFP (memRFP) RNA (50 pg) into a single animal pole blastomere of four-cell embryos. Ectoderm was explanted at stage 11 and immunostained with antibodies specific for the HA epitope and RFP. Wild-type Tril was strongly detected at the plasma membrane but was also present in cytoplasmic puncta (Figure 3, A–A‴). By contrast, Tril∆ICD was detected only at the plasma membrane (Figure 3, B–B‴), suggesting that the ICD is required for localization of wild-type Tril in vesicles. Surprisingly, Tril∆ECD was detected almost exclusively in intracellular puncta and not at the cell surface (Figure 3, C–C‴) despite the presence of an intact transmembrane domain. Tril∆FN was abundantly detected in cytoplasmic puncta and also at the plasma membrane (Figure 3, D–D‴).
FIGURE 3:

The extracellular domain of Tril is required to retain it at the plasma membrane while the intracellular domain is required for internalization. (A–D‴) RNAs encoding wild-type or deletion mutant forms of Tri-HA (illustrated to the left of each panel) and membrane-localized RFP (memRFP) were injected into a single animal pole blastomere of four-cell embryos. Ectoderm was explanted from 5–10 embryos in each group at stage 11 and immunostained for HA and RFP. (A–D″) Projections viewed from the xy-axis. (A‴–D‴) Projections viewed from the z-axis. Results were replicated in three independent experiments. (E) CHO cells transfected with GFP (negative control), cadherin6-GFP (Cad6-GFP) (positive control), or Tril-GFP were tested for adhesion. Only cells expressing cadherin6 formed aggregates. The aggregation index was calculated by dividing the total GFP fluorescence in cell aggregates by the total GFP fluorescence in the well. Mean ± SD are shown, n = 3, ***p < 0.01 by two-tailed t test.
The observation that the ECD is required to retain Tril at the cell surface raises the possibility that it does so through homophilic interactions with the ECD of other Tril molecules or through heterophilic interactions with other cell surface proteins on adjacent cells. Consistent with this possibility, the ECD of several other LRR-containing proteins have been shown to interact and to mediate homophilic or heterophilic adhesion (Krantz and Zipursky, 1990; Nose et al., 1992; Ko, 2012; Pare et al., 2014). To test whether Tril is sufficient to mediate cell adhesion, we transiently transfected nonadherent CHO cells with human Tril-GFP, cadherin6-green fluorescent protein (GFP) (as a positive control), or GFP (as a negative control) and compared the ability of cells to aggregate. As shown in Figure 3E, only cells expressing cadherin6 formed aggregates. Thus, the ECD of Tril promotes neither homophilic adhesion nor heterophilic adhesion with endogenous proteins expressed on the surface of CHO cells.
Tril is retrieved from the cell surface and resides mainly in endosomes in HeLa cells
Imaging of HeLa cells transiently transfected with human Tril-GFP revealed weak punctate expression of Tril at the cell surface and more abundant expression in intracellular vesicles (Figure 4A). Double-label immunostaining of HeLa cell transiently transfected with Tril-Flag revealed that the Tril-containing vesicles partially colocalize with the early endosomal marker Rab5 (Figure 4B).
FIGURE 4:

Tril is retrieved from the cell surface into endosomes in HeLa cells. (A) HeLa cells were transiently transfected with DNA encoding human Tril-GFP. The plasma membrane was stained with CellMask 24 h later, and GFP was imaged in unfixed cells. (B) HeLa cells were transfected with DNA encoding human Tril-Flag. Cells were fixed 24 h later and double-label immunostained with antibodies specific for Flag and Rab. All results were replicated in three experiments. (C–F) Hela cells were transiently transfected with DNA encoding HA- and Flag-epitope tagged Xenopus Tril (illustrated above panels). Cells were either permeabilized and double-label immunostained with antibodies specific for both epitopes (C–C″) or were incubated at 4°C with anti-HA antibodies for 1 h and then either permeabilized immediately (D–D″) or incubated at 4°C (E–E″) or at 37°C (F–F″) for 1 h before permeabilization and addition of antibodies specific for the Flag tag. HA and Flag epitopes were visualized with species specific fluorescent secondary antibodies. The white arrowheads in D–D″ denote Tril present at the cell surface while the white arrows in F–F″ indicate Tril present in intracellular compartments. Results were replicated in three independent experiments.
To test whether the difference in subcellular localization of Tril in Xenopus embryos (primarily at the cell surface) versus in HeLa cells (primarily in intracellular vesicles) is due to differences in the structure of Xenopus versus human Tril, HeLa cells were transiently transfected with DNA encoding Xenopus Tril containing an HA epitope tag in the ECD (yellow bar in illustration) and a Flag epitope tag at the end of the ICD (red bar). After 24 h, cells were permeabilized, and double-label immunofluorescence was used to detect the HA and Flag epitopes. Under steady-state conditions, both antibodies recognize ectopic Xenopus Tril primarily in vesicles throughout the cell (Figure 4, C–C″). Thus, the sparse presence of Tril at the surface of HeLa cells reflects differences intrinsic to the cell type rather than specifies differences in Tril.
We used an antibody uptake assay (Jean and Kiger, 2016) to test whether the Tril protein detected in intracellular vesicles is retrieved from the cell surface as opposed to being present in vesicles en route to the plasma membrane. Cells were incubated at 4°C for 1 h together with antibodies specific for the HA tag. This allows the HA antibody to bind to the extracellular domain of Tril molecules that are located on the cell surface. Cells were then washed to remove unbound antibody and were fixed immediately (0 min) or were incubated at either 4°C (which prohibits endocytosis) or at 37°C (which is permissive for endocytosis) for 1 h before fixation. Cells were then permeabilized and incubated with antibodies specific for the Flag tag, and the Flag and HA epitopes were visualized using Alexa-488 and Alex-568 coupled secondary antibodies (illustrated above Figure 4, C–F). Immediately after incubation at 4°C (Figure 4, D–D″) or following an additional 1-h incubation at 4°C (Figure 4, E–E″) the HA epitope tag was detected only at the cell surface, demonstrating that the block to endocytosis was complete, while the Flag epitope tag was detected primarily intracellularly. After 60 min at 37°C, the HA antibody bound to Tril was no longer detected at the cell surface but was instead present in vesicles that were also detected using the Flag antibody (Figure 4, F–F″). Thus, Tril is trafficked to the plasma membrane of cells and is retrieved into endosomal compartments by endocytosis.
Cadherin-mediated cell–cell adhesion is required for cell surface retention of Tril in Xenopus explants
The observation that Tril∆ECD is largely absent from the cell surface in Xenopus ectodermal explants suggests that interactions between the ECD of Tril and heterologous cell surface proteins and/or components of the ECM are required to prevent constitutive internalization in the absence of pathway stimulation. To begin to examine these possibilities, we first asked whether Tril colocalizes with the ECM molecule fibronectin and/or with the cell surface molecule E-cadherin in Xenopus. RNA encoding Tril-HA (100 pg) was injected into a single animal pole blastomere of four-cell-stage embryos. Ectoderm was explanted at stage 11 and immunostained with antibodies specific for HA and for endogenous fibronectin or E-cadherin. While there was no overlap in staining for Tril-HA and fibronectin (Figure 5, A–A‴), some overlap between Tril-HA and E-cadherin was observed (Figure 5, B–B‴). To test whether cadherin-dependent cell–cell adhesion is required for cell surface retention of Tril, RNA encoding a truncated form of Xenopus N-cadherin (N-cad∆E-myc) (500 pg) that lacks most of the extracellular domain was coinjected along with tril-HA RNA. This mutant dominantly interferes with the function of all cadherins and completely inhibits cell adhesion in Xenopus embryos (Kintner, 1992). In cells expressing N-cad∆Emyc, Tril was detected exclusively in intracellular puncta and not at the cell surface (Figure 5, C–C‴). Thus, cell surface retention of Tril requires close cell–cell contact. This finding raises the possibility that the Tril ECD interacts in trans with cell surface molecules on neighboring cells or interacts in cis with cadherins present in adherens junctions on Tril expressing cells and that this is required to prevent constitutive internalization. This finding provides a possible explanation for why Tril is present primarily at the plasma membrane in stably transfected HEK cells (Carpenter et al., 2011) and in intact Xenopus ectodermal explants, both of which have functional adherens junctions (Kintner, 1992; Inada et al., 2016), whereas it is present primarily in endosomes in HeLa cells. Although HeLa cells express Tlr3 and Tlr4 (Kurt-Jones et al., 2004), both of which can interact with Tril (Carpenter et al., 2009), they are poorly adhesive, express low levels of N- but not E-cadherin, and do not form strong adherens junctions (Doyle et al., 1995).
FIGURE 5:

Cadherin-mediated cell–cell adhesion is required for cell surface retention of Tril in Xenopus explants. (A–D‴) RNA encoding Tril-HA (100 pg) was injected into a single animal pole blastomere of four-cell embryos. Ectoderm was explanted from 5–10 embryos in each group at stage 11 and immunostained for HA and Fibronectin (A–A‴) or E-cadherin (B–B‴). (C–C‴) RNA encoding Tri-HA (100 pg) and N-cad∆Emyc was coinjected into a single animal pole blastomere of four-cell embryos. Ectoderm was explanted from 5–10 embryos in each group at stage 11 and immunostained for HA and myc. Results were replicated in three independent experiments. A–C″″ show projections viewed from the xy-axis and A‴–D‴ viewed from the z-axis.
In the context of innate immunity, the extracellular domain of Tril interacts directly with Tlr4, and with its ligand LPS, suggesting that it functions primarily to enhance ligand–receptor affinity (Carpenter et al., 2009). By contrast, our structure–function analysis shows that the ICD of Tril is indispensable for downstream signaling. In addition, we show that the ICD is required for internalization of Tril. The ICD of Tril is highly conserved across species; Xenopus and human Tril share 77% amino acid identity in this domain (Supplemental Figure S3). Although the ICD of Tril lacks any recognizable signaling motifs, it does include di-leucine–like motifs with upstream and downstream acidic residues that may function to direct endocytosis (Kozik et al., 2010). It is possible that the ICD mediates endocytic retrieval to enable Tlrs to signal from internal compartments within the cell. Tlrs recruit a variety of obligate accessory molecules, some of which act as chaperones to traffic receptors to signaling competent subcellular locations (Tan and Kagan, 2017). For example, the adaptor protein CD14 controls the LPS-induced endocytosis of Tlr4 (Zanoni et al., 2011), which is critical to activate some signaling networks (Kagan et al., 2008). If Tril does function as a Tlr coreceptor in embryos, then in this context Tlrs must activate a noncanonical signaling cascade that culminates in degradation of Smad7 rather than in activation of NF-κB. An alternate possibility is that the ICD directly transmits signals required for degradation of Smad7, independent of Tlrs. If so, then this must require additional adaptor proteins, since we are unable to detect interactions between Tril and Smad7. Ongoing studies are aimed at identifying the proteins that interact with, and transduce signals downstream of, Tril during early development.
MATERIALS AND METHODS
Xenopus embryo culture and manipulation
Animal procedures followed protocols approved by the University of Utah Institutional Animal Care and Use Committee. Embryos were obtained, microinjected, and cultured as described (Mimoto and Christian, 2011). Embryos were processed for in situ hybridization as described (Harland, 1991), except that the vitelline coat was not removed prior to fixation and BM purple (Roche) was used as a substrate.
cDNA constructs and MOs
Sequence encoding an HA epitope tag was inserted into the ECD of Xenopus tril.S between amino acids 499 and 500 to generate Tril-HA and/or sequence encoding a Flag epitope tag was appended following the last amino acid to generate Tril-HAFlag/Tril-Flag using a PCR-based approach. Tril-HAFlag can rescue blood formation in embryos in which translation of endogenous Tril is blocked, demonstrating that the epitope tags do not interfere with Tril function (Green et al., 2016). cDNAs encoding deletion mutant forms of tril were made using the splicing by overlap extension method (Horton et al., 1990) or by PCR-mediated introduction of appropriate restriction sites. Tril∆FN lacks amino acids 650–730, Tril∆ECD lacks amino acids 45–744, and Tril∆CT lacks amino acids 792–863 of Tril. All constructs were fully sequenced before use.
Transient transfection
HeLa cells were purchased from the American Type Culture Collection (ATCC) and grown in DMEM in the presence of 10% fetal bovine serum (FBS) and penicillin–streptomycin. Cells were transiently transfected with DNA encoding Tril-HAFlag in pCS2+ using Lipofectamine 2000 (Invitrogen), and cells were cultured in OptiMEM-I (Invitrogen) for 24 h before harvesting for immunoblots or immunostaining.
Immunoblots
Embryos were lysed in Triton X-100 lysis buffer (Choi and Han, 2005) containing HALT protease inhibitor and phosphatase cocktail (Thermo Scientific). Immunoblots were performed as described (Kwon and Christian, 2011) with the following antibodies: rat anti- HA (Roche rat monoclonal 3F10, cat. no. 11 867 423 001, lot # 12213, 1:1000), mouse anti-Myc (Developmental Hybridoma bank, 9E10, 1:1,000), and rabbit anti-β-actin (Abcam ab82272, lot # GR265013-1, 1:10,000). Uninjected embryos were included on all anti-myc blots as a negative control to ensure antibody specificity.
Immunostaining
Ectodermal explants were immunostained with antibodies specific for HA (Roche rat monoclonal 3F10, cat. no. 11 867 423 001, lot # 12213, 1:500) or myc (Cell Signaling, 7D10 rabbit mAb #2278, lot # 5, 1:250) as described (Green et al., 2016). HeLa cells were transiently transfected with 10 ng of DNA encoding Tril-HAFlag in pCS2+. After 24 h, cells were fixed in 4% paraformaldehyde/phosphate-buffered saline for 5 min and then immunostained with antibodies specific for Flag (Sigma mouse mAb clone M2, F1804, lot # SLBK1346V, 1:500) and/or HA (Roche, rat monoclonal 3F10, cat. no. 11 867 423 001, lot # 12213, 1:500). Primary antibodies were detected with Alexa 488-conjugated anti-rabbit (Invitrogen, A11008, lot # 1408830) or anti-rat (Invitrogen, A21208, lot #1701951), or Alexa 568-conjugated anti-mouse (Invitrogen, A11031, lot # 6592-1) secondary antibodies used at a dilution of 1:1000.
For antibody uptake assays, transfected cells were incubated with 1:200 rat anti-HA antibody for 1 h at 4°C and then were either fixed prior to addition of anti-Flag antibodies or were incubated for an additional 60 min at 4° or 37°C prior to fixation and immunostaining with Flag as described above. To detect Smad7myc, explants were imaged using a Leica DM2500 compound microscope and a Leica DFC425 C digital camera. For all other immunostaining, explants and cells were imaged using an Olympus FV1000 confocal microscope.
Analysis of RNA
Total RNA was isolated, and semiquantitative reverse transcription (RT)-PCR (Nakayama et al., 1998) and qPCR (Mimoto et al., 2015) were performed as described using an annealing temperature of 58°C. Primer sequences have been reported previously (Green et al., 2016).
CHO cell aggregation assay
CHO cells were purchased from the ATCC and transfected with hTril-GFP/pCDNA3.1 (5 μg), cadherin6-GFP/pCAG (5 μg), or GFP/pCS2+ (5 μg) using polyethylenimine (PEI). Forty-eight hours later, cells were washed with HEPES Mg2+ free (HMF) buffer (137 mM NaCl, 5.4 mM KCl, 1 mM CaCl2, 0.34 mM Na2HPO4, 5.5 mM glucose, 10 mM HEPES, pH 7.4, adjusted with NaOH) and detached from the dishes using 0.01% trypsin in HMF. Detached cells were spun down, resuspended in HMF, and counted, and 100,000 cells were pipetted into single wells of 24-well plates precoated with 1% bovine serum albumin in HMF. Subsequently, the plates were placed on a nutator for 90 min at 37°C. The cells were then fixed with paraformaldehyde (PFA) (4% final concentration), transferred to a 96-well glass bottom plate, and imaged using an Olympus FV1000 confocal microscope. The aggregation index was calculated by dividing the total GFP fluorescence in cell aggregates by the total GFP fluorescence in the well. Analysis was done using ImageJ.
Statistical analysis
NIH ImageJ software was used to quantify band intensities. A two-tailed Student’s t test was used to compare differences between two groups. Differences in subcellular localization of Smad7 and in relative globin expression (WMISH) were scored blind and analyzed using GraphPad software to conduct two-way analysis of variance followed by a Bonferroni multiple comparisons test. Differences with p < 0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
We thank Anne Martin for help with troubleshooting the adhesion assay in CHO cells, Luke O’Neill for the human Tril-GFP construct, and Christine Holt for the N-cad∆E-myc construct. This work was supported by the National Institute of Child Health and Human Development (RO1HD067473 to J.L.C.), the Huntsman Cancer Foundation (150203 to J.L.C.), and the National Cancer Institute (P30CA042014). This work utilized DNA, peptide, and imaging shared resources supported by the Huntsman Cancer Foundation and the National Cancer Institute (P30CA042014). The content is solely the responsibility of the authors and does not represent the official views of the National Institutes of Health.
Abbreviations used:
- Bmps
bone morphogenetic proteins
- ECD
extracellular domain
- ECM
extracellular matrix
- FN
fibronectin type III
- ICD
intracellular domain
- Tril
Toll-like receptor 4 interactor with leucine-rich repeats
- Tlr
Toll-like receptors
- WMISH
whole mount in situ hybridization.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-07-0446) on January 3, 2018.
REFERENCES
- Brubaker SW, Bonham KS, Zanoni I, Kagan JC. (2015). Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol , 257-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter S, Carlson T, Dellacasagrande J, Garcia A, Gibbons S, Hertzog P, Lyons A, Lin LL, Lynch M, Monie T, et al. (2009). TRIL, a functional component of the TLR4 signaling complex, highly expressed in brain. J Immunol , 3989-3995. [DOI] [PubMed] [Google Scholar]
- Carpenter S, Wochal P, Dunne A, O’Neill LA. (2011). Toll-like receptor 3 (TLR3) signaling requires TLR4 Interactor with leucine-rich REPeats (TRIL). J Biol Chem , 38795-38804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KC, Lee YS, Lim S, Choi HK, Lee CH, Lee EK, Hong S, Kim IH, Kim SJ, Park SH. (2006). Smad6 negatively regulates interleukin 1-receptor-Toll-like receptor signaling through direct interaction with the adaptor Pellino-1. Nat Immunol , 1057-1065. [DOI] [PubMed] [Google Scholar]
- Choi SC, Han JK. (2005). Rap2 is required for Wnt/beta-catenin signaling pathway in Xenopus early development. EMBO J , 985-996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JP, Stempak JG, Cowin P, Colman DR, D’Urso D. (1995). Protein zero, a nervous system adhesion molecule, triggers epithelial reversion in host carcinoma cells. J Cell Biol , 465-482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green YS, Kwon S, Mimoto MS, Xie Y, Christian JL. (2016). Tril targets Smad7 for degradation to allow hematopoietic specification in Xenopus embryos. Development , 4016-4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harland RM. (1991). In situ hybridization: an improved whole-mount method for Xenopus embryos. Methods Cell Biol , 685-695. [DOI] [PubMed] [Google Scholar]
- Horton RM, Cai ZL, Ho SN, Pease LR. (1990). Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques , 528-535. [PubMed] [Google Scholar]
- Inada M, Izawa G, Kobayashi W, Ozawa M. (2016). 293 cells express both epithelial as well as mesenchymal cell adhesion molecules. Int J Mol Med , 1521-1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean S, Kiger AA. (2016). VAMP8–3xHA uptake assay in HeLa cells. Bio Protoc . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. (2008). TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat Immunol , 361-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kintner C. (1992). Regulation of embryonic cell adhesion by the cadherin cytoplasmic domain. Cell , 225-236. [DOI] [PubMed] [Google Scholar]
- Ko J. (2012). The leucine-rich repeat superfamily of synaptic adhesion molecules: LRRTMs and Slitrks. Mol Cells , 335-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozik P, Francis RW, Seaman MN, Robinson MS. (2010). A screen for endocytic motifs. Traffic , 843-855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krammer A, Craig D, Thomas WE, Schulten K, Vogel V. (2002). A structural model for force regulated integrin binding to fibronectin’s RGD-synergy site. Matrix Biol , 139-147. [DOI] [PubMed] [Google Scholar]
- Krantz DE, Zipursky SL. (1990). Drosophila chaoptin, a member of the leucine-rich repeat family, is a photoreceptor cell-specific adhesion molecule. EMBO J , 1969-1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurt-Jones EA, Sandor F, Ortiz Y, Bowen GN, Counter SL, Wang TC, Finberg RW. (2004). Use of murine embryonic fibroblasts to define Toll-like receptor activation and specificity. J Endotoxin Res , 419-424. [DOI] [PubMed] [Google Scholar]
- Kwon S, Christian JL. (2011). Sortilin associates with transforming growth factor-beta family proteins to enhance lysosome-mediated degradation. J Biol Chem , 21876-21885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengerke C, Schmitt S, Bowman TV, Jang IH, Maouche-Chretien L, McKinney-Freeman S, Davidson AJ, Hammerschmidt M, Rentzsch F, Green JB, et al. (2008). BMP and Wnt specify hematopoietic fate by activation of the Cdx-Hox pathway. Cell Stem Cell , 72-82. [DOI] [PubMed] [Google Scholar]
- Leulier F, Lemaitre B. (2008). Toll-like receptors–taking an evolutionary approach. Nat Rev Genet , 165-178. [DOI] [PubMed] [Google Scholar]
- Mead PE, Kelley CM, Hahn PS, Piedad O, Zon LI. (1998). SCL specifies hematopoietic mesoderm in Xenopus embryos. Development , 2611-2620. [DOI] [PubMed] [Google Scholar]
- Mimoto MS, Christian JL. (2011). Manipulation of gene function in Xenopus laevis. Methods Mol Biol , 55-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimoto MS, Kwon S, Green YS, Goldman D, Christian JL. (2015). GATA2 regulates Wnt signaling to promote primitive red blood cell fate. Dev Biol , 1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama T, Gardner H, Berg LK, Christian JL. (1998). Smad6 functions as an intracellular antagonist of some TGF-beta family members during Xenopus embryogenesis. Genes Cells , 387-394. [DOI] [PubMed] [Google Scholar]
- Nose A, Mahajan VB, Goodman CS. (1992). Connectin: a homophilic cell adhesion molecule expressed on a subset of muscles and the motoneurons that innervate them in Drosophila. Cell , 553-567. [DOI] [PubMed] [Google Scholar]
- Pare AC, Vichas A, Fincher CT, Mirman Z, Farrell DL, Mainieri A, Zallen JA. (2014). A positional Toll receptor code directs convergent extension in Drosophila. Nature , 523-527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth RB, Chen ZJ. (2007). Smads keep TABs on inflammation. Nat Immunol , 477-478. [DOI] [PubMed] [Google Scholar]
- Tan Y, Kagan JC. (2017). Microbe-inducible trafficking pathways that control Toll-like receptor signaling. Traffic , 6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuazon FB, Mullins MC. (2015). Temporally coordinated signals progressively pattern the anteroposterior and dorsoventral body axes. Semin Cell Dev Biol , 118-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veitia RA. (2007). Exploring the molecular etiology of dominant-negative mutations. Plant Cell , 3843-3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss A, Attisano L. (2013). The TGFbeta superfamily signaling pathway. Wiley Interdiscip Rev Dev Biol , 47-63. [DOI] [PubMed] [Google Scholar]
- Wochal P, Rathinam VA, Dunne A, Carlson T, Kuang W, Seidl KJ, Hall JP, Lin LL, Collins M, Schattgen SA, et al. (2014). TRIL is involved in cytokine production in the brain following Escherichia coli infection. J Immunol , 1911-1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Chen YG. (2011). Smad7: not only a regulator, but also a cross-talk mediator of TGF-beta signalling. Biochem J , 1-10. [DOI] [PubMed] [Google Scholar]
- Yu L, Wang L, Chen S. (2010). Endogenous toll-like receptor ligands and their biological significance. J Cell Mol Med , 2592-2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanoni I, Ostuni R, Marek LR, Barresi S, Barbalat R, Barton GM, Granucci F, Kagan JC. (2011). CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell , 868-880. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.