

Abstract
Mutant Tau (MAPT) can lead to frontotemporal lobar degeneration (FTLD). Previous studies associated MAPT mutations and altered function with aneuploidy and chromosome instability in human lymphocytes and in Drosophila development. Here we examine whether FTLD-causing mutations in human MAPT induce aneuploidy and apoptosis in the mammalian brain. First, aneuploidy was found in brain cells from MAPT mutant transgenic mice expressing FTLD mutant human MAPT. Then brain neurons from mice homozygous or heterozygous for the Tau (Mapt) null allele were found to exhibit increasing levels of aneuploidy with decreasing Tau gene dosage. To determine whether aneuploidy leads to neurodegeneration in FTLD, we measured aneuploidy and apoptosis in brain cells from patients with MAPT mutations and identified both increased aneuploidy and apoptosis in the same brain neurons and glia. To determine whether there is a direct relationship between MAPT-induced aneuploidy and apoptosis, we expressed FTLD-causing mutant forms of MAPT in karyotypically normal human cells and found that they cause aneuploidy and mitotic spindle defects that then result in apoptosis. Collectively, our findings reveal a neurodegenerative pathway in FTLD-MAPT in which neurons and glia exhibit mitotic spindle abnormalities, chromosome mis-segregation, and aneuploidy, which then lead to apoptosis.
INTRODUCTION
Frontotemporal lobar degeneration (FTLD), also termed frontotemporal dementia (FTD), is most often an early-onset neurodegenerative disease in which a subset of cases has tau-positive neuronal and glial inclusions in the absence of Alzheimer’s disease (AD)-like amyloid deposits, whereas other cases have TDP-43 neuronal and glial inclusions (Rademakers et al., 2012). Familial forms of FTLD are associated with mutations in several genes, and 50–80% of FTLD cases are sporadic. Some autosomal-dominant familial forms of FTLD and Parkinsonism are linked to chromosome 17 (FTDP-17) and are caused by mutations in one of two nearby genes, the tau gene (MAPT) or the progranulin gene (PGRN). A large proportion of familial FTLD and of amyotrophic lateral sclerosis (ALS) is also caused by abnormal expansions of a hexanucleotide repeat in the C9orf72 gene (Rademakers et al., 2012).
A potential mechanism by which FTLD arises in patients with MAPT mutations is suggested by work on AD. We previously proposed that trisomy 21 mosaicism may underlie the development of AD (Potter, 1991), and subsequent studies demonstrated that people with both sporadic and familial forms of AD exhibit trisomy 21 mosaicism and other aneuploidy in all tissues examined, including in brain neurons and glia (Potter et al., 1995; Migliore et al., 1997; Geller and Potter, 1999; Kingsbury et al., 2006; Mosch et al., 2007; Thomas and Fenech, 2008; Iourov et al., 2009; Arendt et al., 2010, 2015; Arendt, 2012). In addition, AD-causing mutations in the amyloid precursor protein (APP) and presenilin (PS) genes as well as exposure to amyloid-beta (Aβ) lead to mitotic spindle abnormalities and chromosome mis-segregation that depend on MAPT/Tau (Boeras et al., 2008; Granic et al., 2010). Furthermore, we discovered that a likely mechanism by which Aβ causes spindle abnormalities and aneuploidy is through its competitive inhibition of the microtubule-dependent motor Kinesin-5/Eg5, which can be modulated by addition of MAPT/Tau (Borysov et al., 2011). These findings suggested that an FTLD-associated alteration in MAPT function may include chromosome mis-segregation as a contributing or even a necessary step in the neurodegenerative pathway. Indeed, lymphocytes from FTLD patients or from transgenic mice expressing human MAPT harboring FTLD-causing mutations exhibit chromosome instability, including aneuploidy (usually chromosome loss) (Rossi et al., 2008). However, whether and how FTLD-causing mutations in MAPT affect mitosis, whether they affect chromosome segregation in the brain, and whether such cell-cycle defects contribute to neurodegeneration in FTLD are unknown.
Herein we examined the effects of FTLD-causing MAPT mutations and loss of MAPT function in brain cell populations and/or in transfected cells and determined that defects in MAPT lead to aberrant mitotic spindle function, abnormal chromosome segregation, and apoptosis. Together, the data indicate that, as in AD, aneuploid neurons arise in the FTLD-MAPT brain, are prone to apoptosis, and thus can contribute to the development of neurodegeneration and dementia.
RESULTS
Aneuploidy induced by expression of human MAPT harboring FTLD-causing mutations in mice
Although overexpression of the human MAPT gene harboring familial FTLD-causing mutations (P301L or P301S) in mice has been shown to result in increased aneuploidy in splenic lymphocytes (Rossi et al., 2014), a key unanswered question with high disease relevance is whether FTLD-causing MAPT mutations also lead to aneuploidy in brain cells. We generated brain cell suspensions from young and older transgenic mice expressing human MAPT P301S or P301L and from age-matched control mice and determined aneuploidy levels by fluorescence in situ hybridization (FISH) using a bacterial artificial chromosome (BAC) probe for mouse chromosome 16 (Kulnane et al., 2002), which is partially syntenic with human chromosome 21. The data showed that expression of human mutant MAPT P301L induces a small but significant increase in chromosome 16 trisomy and total aneuploidy in the brain as early as 8 weeks of age (Figure 1) with nearly undetectable levels of TUNEL-positive (apoptotic) cells (data not shown). A similar small but significant increase in aneuploidy was also detected in both brain cells (Figure 2A) and splenocytes (Figure 2B) from 8-mo-old transgenic mice expressing mutant human MAPT P301S relative to nontransgenic control mice.
FIGURE 1:
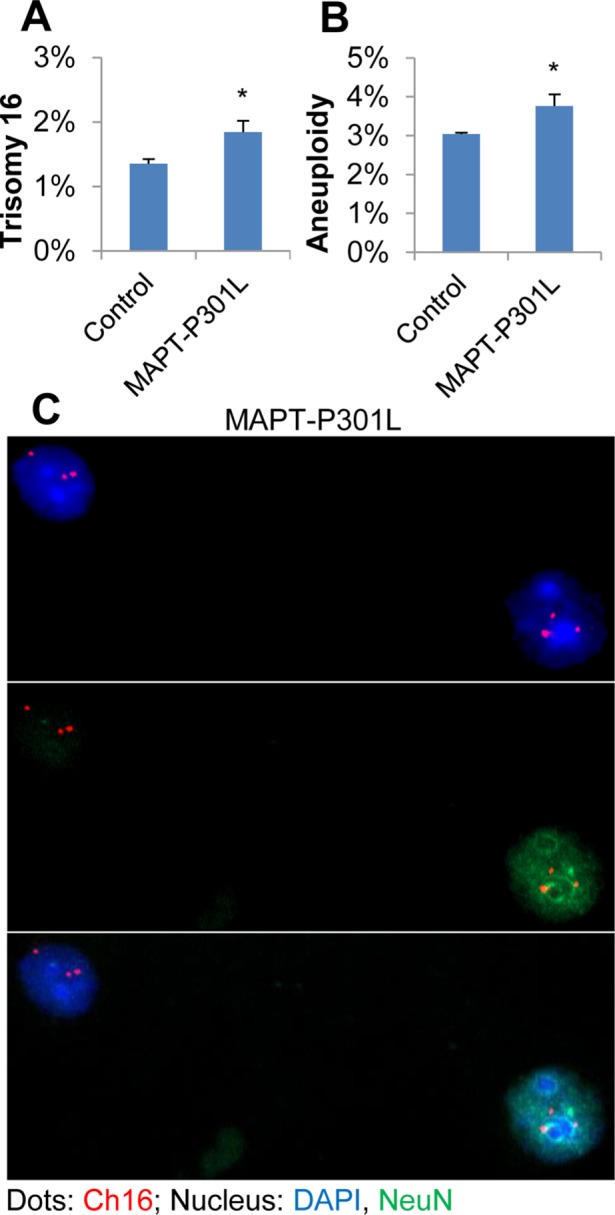
Increased percentage of cells with abnormal chromosome numbers in brain tissues from young transgenic mice expressing a human mutant FTLD-MAPT gene. FISH analysis using a mouse chromosome 16 probe was carried out using single-nuclei suspensions prepared with brain tissues from 8-wk-old transgenic mice expressing a human mutant MAPT transgene (MAPT-P301L, n = 4). Brain tissues from the transgenic mice exhibited elevated levels of trisomy 16 (A) and total chromosome 16 aneuploidy (including monosomy plus trisomy) (B) in comparison to control nontransgenic mice (control, n = 4). Both neurons [NeuN(+), green] and nonneuronal cells [NeuN(–)] showed abnormal chromosome copy numbers in the FTLD-MAPT mice (C). For statistical analyses, more than 300 cells per slide and three slides per brain sample were counted. Statistical analyses were conducted using a Student’s t test. Error bars indicate SEM, and * indicates p < 0.05.
FIGURE 2:
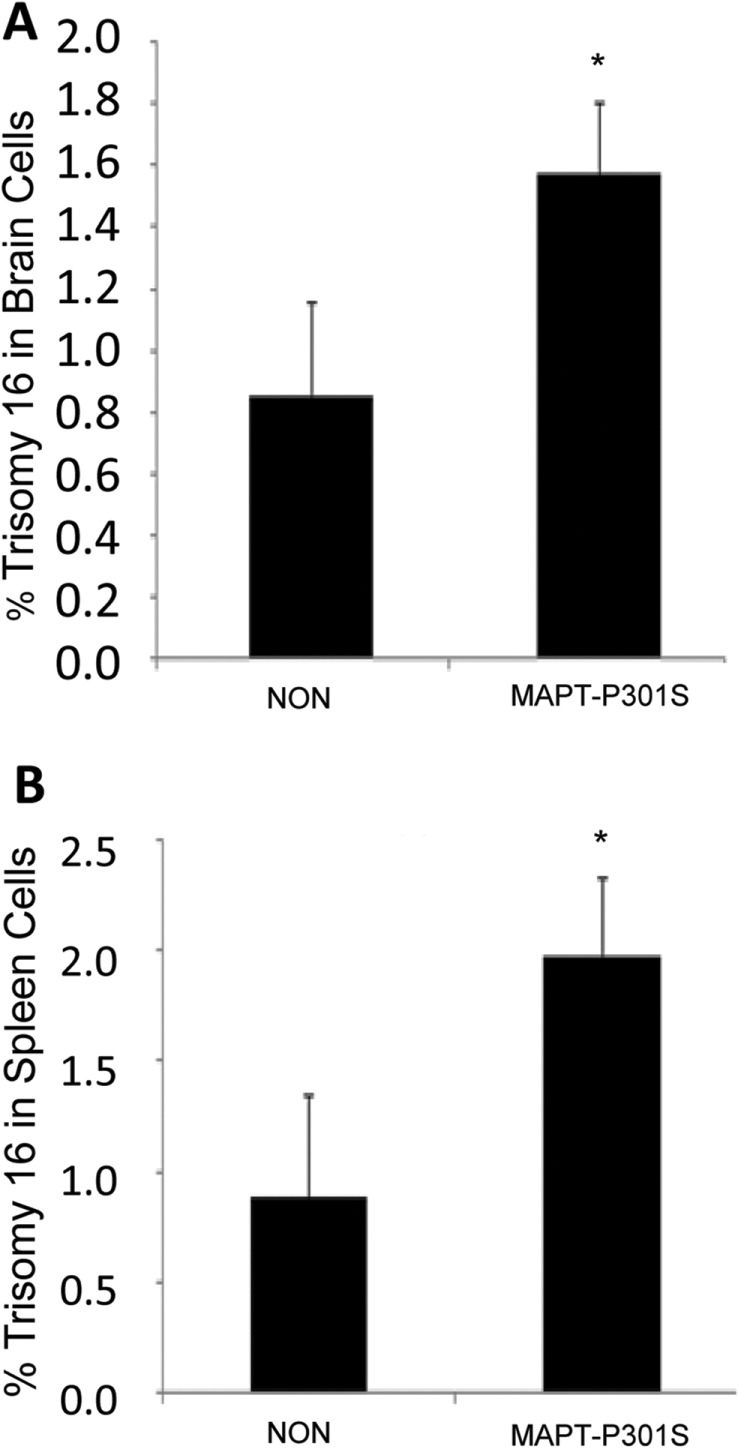
Trisomy 16 induced by the expression of human MAPT harboring the P301S FTLD-causing mutation in brain and spleen cells from older mice. Brains and spleens were harvested from 8-mo-old transgenic mice expressing the human MAPT gene harboring the FTLD-causing P301S mutation (MAPT-P301S) and from age-matched control mice (NON). Single-brain-cell suspensions and splenocyte cultures were prepared and analyzed for aneuploidy by FISH using a mouse chromosome 16 BAC probe. The data show that expression of MAPT-P301S induces chromosome mis-segregation in brain cells (A) and in splenocytes (B) (independent t test, one-tailed; effect size Cohen’s d of 1.54 and 1.69, respectively). Error bars indicate SEM, *p < 0.05.
Total or partial loss of tau function induces aneuploidy in mouse brain neurons
Our finding that expression in mice of human MAPT harboring FTLD-causing mutations resulted in increased levels of chromosome 16 aneuploidy in brain cells provided the first evidence that this mutation is not only associated with aneuploidy in peripheral cells (Rossi et al., 2008) but also with aneuploidy in the brain. Previously, we showed that splenocytes from tau (mapt) heterozygous (tau+/–) or homozygous (tau–/–) knockout mice exhibited 2.5% or 4.9% trisomy 16, respectively, compared with 1.1% stochastic trisomy in splenocytes from age-matched tau+/+ control mice (Granic et al., 2010). To further investigate the role of wild-type mouse Tau in chromosome segregation in the brain during neurogenesis and during postnatal neuronal development, we prepared single-cell suspensions from brains of tau+/+, tau+/–, and tau–/– mice (aged 6–7 mo) and analyzed both neurons and nonneuronal cells for chromosome 16 aneuploidy using chromosome-specific FISH and NeuN immunocytochemistry. Knocking out one copy, or, even more effectively, both copies of Tau led to a significant increase in aneuploid neurons (Figure 3, A and B). This result shows that a total or even partial loss of Tau function, suggesting possible haploinsufficiency, in mice leads to a cell-cycle defect in neuronal precursor cells that results in chromosome mis-segregation and mosaic aneuploidy.
FIGURE 3:
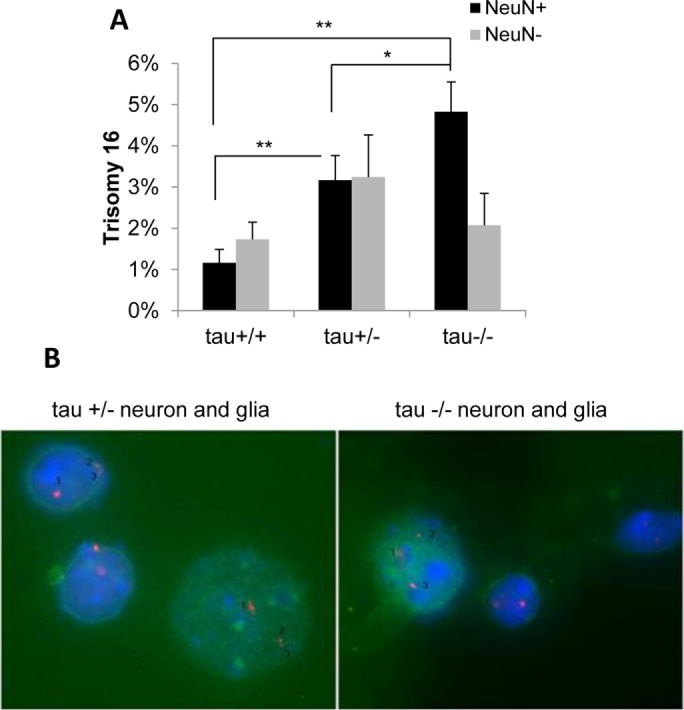
Complete or partial loss of tau function induces trisomy 16 in mouse neurons. Single-cell suspensions of brain cells from tau+/+, tau+/–, and tau–/– mice were analyzed by FISH using a mouse chromosome 16 BAC DNA probe and costained with the anti-NeuN antibody to identify neurons. Quantitative analysis of BAC signals (Spectrum Orange d-UTPs) revealed a significant increase in trisomy 16 in neurons from tau+/– and tau–/– mice compared with tau+/+ mice (A). The graph shows mean difference between groups using ANOVA with Tukey HSD post hoc (effect size η2 = 0.75 for neurons). Representative examples of tau+/– and tau–/– neurons [NeuN(+), green] and glia [NeuN(–)] are shown (B). *p < 0.05, **p < 0.005.
Elevated levels of mosaic aneuploidy in brain neurons and glia from FTLD patients with MAPT mutations
Although we observed aneuploidy in mouse brain cells that was induced by expression of the P301L or P301S FTLD-causing form of human MAPT and in mouse brain neurons with complete or partial loss of Tau (Mapt) function, whether FLTD-associated mutations in MAPT lead to aneuploidy and subsequent apoptosis in the human brain, where they could affect neurodegeneration, is unknown. To address this question, we prepared single-brain-cell suspensions using frozen samples of cortex from familial FTLD patients with one of three different MAPT mutations, specifically MAPT P301L, MAPT N279K, or MAPT E372G (Table 1), or from normal controls and determined aneuploidy levels using a dual-color DNA probe set to visualize chromosomes 12 and 21 via FISH. We found that cortical brain cells from FTLD patients with MAPT mutations exhibit significantly higher levels of trisomy 12 and trisomy 21 relative to cortical brain cells from normal controls (Figure 4A). Furthermore, we observed significantly higher levels of mosaic aneuploidy (i.e., either monosomy or trisomy) for chromosomes 12 and 21 in both neuronal [NeuN(+)] and nonneuronal [NeuN(–)] cells from the cortex relative to normal controls (Figure 4B). These results show that familial FTLD caused by MAPT mutations is characterized by mosaic aneuploidy, including trisomy 21, in both neurons and glia in the brain.
TABLE 1:
Demographics of familial MAPT FTLD cases and age-matched control individuals without neurologic disease.
| Diagnosis | MAPT mutation | Age (yr) | Sex |
|---|---|---|---|
| FTLD | P301L | 52 | M |
| P301L | 62 | M | |
| N279K | 51 | F | |
| E372G | 58 | M | |
| Controls | WT | 59 | F |
| WT | 61 | M | |
| WT | 63 | M | |
| WT | 63 | M | |
| WT | 53 | M | |
| WT | 79 | M | |
| WT | 74 | M |
FIGURE 4:
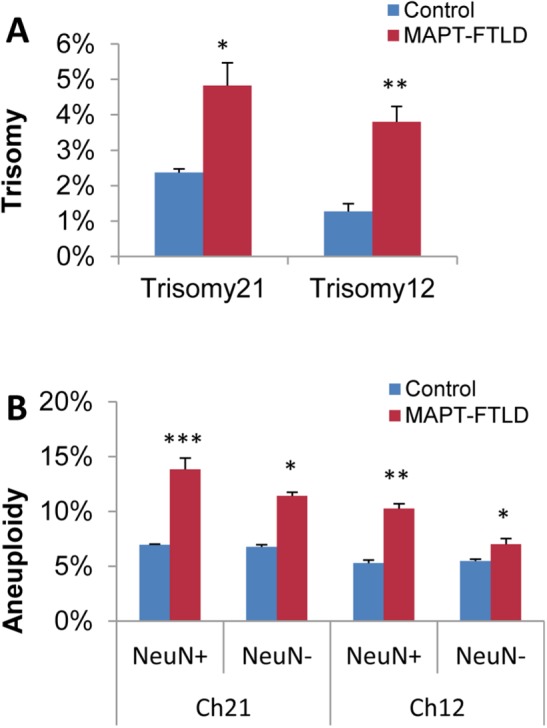
Increased levels of mosaic aneuploidy in brain neurons and glia from FTLD patients with MAPT mutations. Quantitative FISH analysis of single-cell suspensions prepared from frozen cortical brain samples of individuals genetically identified as having familial FTLD with a mutation in MAPT (MAPT-FTLD) (n = 4) compared with normal controls (n = 7) revealed significantly higher percentages of brain cells with trisomy 12 or 21 in FTLD-MAPT patients (A). Separated analysis of neurons [NeuN(+)] from other brain cells [NeuN(–)] showed increased total aneuploidy (i.e., monosomy or trisomy for chromosome 12 or 21) in both cortical neurons and glia from familial FTLD patients relative to normal controls (B). The graphs show mean difference and significance (*) using one-way ANOVA with Tukey post hoc test. Error bars indicate SEM. *p < 0.05, **p < 0.005, and ***p < 0.0005. A minimum of ∼1000 nuclei/slide and three slides/sample were counted and recorded for statistical analyses.
Increased apoptosis in brain neurons and glia from FTLD patients with MAPT mutations correlates with induced aneuploidy
To ask directly whether aneuploidy and neurodegeneration are linked in FTLD, we measured apoptosis in cortical brain cells from the patients with MAPT mutations. Simultaneous terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) to detect apoptotic cells, NeuN staining to identify neurons, and chromosome 12 and 21 FISH staining of brain samples from normal control subjects and familial FTLD patients with a MAPT mutation, including MAPT P301L, MAPT N279K, or MAPT E372G (Table 1), showed that apoptosis was significantly increased in cortical brain cells from FTLD-MAPT patients relative to normal controls (Figure 5, A and E). Furthermore, the proportion of TUNEL(+)/apoptotic cells that were aneuploid (i.e., either monosomic or trisomic) for chromosome 12 or 21 reached around 80% in normal control brain cells and in brain cells from FTLD-MAPT patients (Figure 5B), despite the significant difference in the percentages of both aneuploid and apoptotic cells between control and FTLD brains, and this was true for both NeuN(+) neurons (Figure 5C) and NeuN(–) nonneuronal cells (Figure 5D). The large percentage of apoptotic cells that were aneuploid (i.e., 80%) contrasts with the much smaller percentage of aneuploid brain cells that were apoptotic (i.e., 20%) in both FTLD-MAPT patients and in normal control subjects (Figure 5F). Thus, while the normal control subjects had significantly lower overall levels of both apoptosis and aneuploidy (Figure 5A), our finding that the majority of apoptotic cells were aneuploid suggests that aneuploidy is a cause, rather than a consequence, of apoptosis. If aneuploidy was a consequence, rather than a cause, of apoptosis, then we would have expected to find that the majority of aneuploid brain cells were apoptotic/TUNEL(+). Thus, our findings provide evidence that familial FTLD associated with MAPT mutations causes neurons and glia to become aneuploid and that the aneuploid neurons and glia are then prone to undergoing apoptosis. These results mirror previous findings in AD showing that aneuploid neurons are particularly susceptible to degeneration (Arendt et al., 2010), but that some aneuploid neurons retain sufficient function to be incorporated into brain circuits (McConnell et al., 2013). In our experiments, chromosome segregation clearly affects both neuronal and nonneuronal cells in familial FTLD patients with MAPT mutations.
FIGURE 5:

Apoptosis in aneuploid brain neurons and glia from FTLD patients with MAPT mutations. Costaining for TUNEL, NeuN, and FISH for chromosomes 12 and 21 revealed higher levels of apoptosis, indicated by TUNEL(+) staining, in cortical brain cells from FTLD patients with MAPT mutations (MAPT-FTLD) relative to normal control subjects (Control) (A). Quantitative analysis showed that the majority of cells that were TUNEL(+)/apoptotic were aneuploid (i.e., monosomic or trisomic for chromosome 12 or 21), regardless of whether they were from normal control subjects or from FTLD-MAPT patients (B). Further, a similarly large percentage of the TUNEL(+), NeuN(+) neuronal cells (C) and TUNEL(+), NeuN(–) glial cells (D) were aneuploid in both normal control subjects and FTLD-MAPT patients. (E) Images of an aneuploid FTLD-MAPT (P301L) cell with three copies of chromosome 21 and one copy of chromosome 12 (I) showing NeuN (II); and TUNEL staining (III); FISH for chromosomes 12 and 21 with DAPI (IV); TUNEL, NeuN, and FISH for chromosomes 12 and 21 (V); and a merged image of TUNEL (red), NeuN (green), FISH for chromosomes 12 (green) and 21 (orange/red), and DAPI (blue) (VI). Notably, although the probe signals are very distinct and easily distinguishable when viewed through the eyepieces of the microscope, it is difficult to show this in the captured images, since the emission wavelengths for the FISH probes and the NeuN/TUNEL staining show some overlap. Analysis of the frequency of apoptosis among aneuploid brain cells and of aneuploidy among apoptotic brain cells indicates that around 80% of apoptotic cells were aneuploid in both control brains and FTLD-MAPT brains, whereas less than 20% of aneuploid cells were apoptotic in both control brains and FTLD-MAPT brains (F). More than 1000 cells per slide and three slides per sample were scored for statistical analysis. Statistical significances were calculated using a Student’s t test. Error bars indicated SEM. *p < 0.05, **p < 0.005, and ***p < 0.0005.
Expression of human MAPT genes harboring FTLD-causing mutations induces aneuploidy and subsequent apoptosis in karyotypically normal human cells
To rule out the possibility that pathological processes that arise in human FTLD-MAPT and in mutant MAPT mouse models are indirectly responsible for generating aneuploid neurons and glia, we transiently expressed human MAPT genes harboring one of two different FTLD-causing mutations in the karyotypically normal, nonneuronal hTERT-HME1 human cell line and assessed the levels of induced aneuploidy and apoptosis. As shown in Figure 6A and Supplemental Figure 1, A–C, and illustrated in Figure 6B and Supplemental Figure 1B, expression of FTLD-causing mutant forms of MAPT (P301L or V337M) induced a significant increase in aneuploidy within 48 h posttransfection, compared with control cells transfected with the vector alone. Levels of aneuploidy in cells transfected with wild-type MAPT were intermediate between those of cells transfected with the vector alone and cells transfected with the FTLD-causing MAPT mutants. At this 48-h time point, levels of apoptosis were essentially undetectable (data not shown).
FIGURE 6:

Induction of trisomy 12 and 21 followed by apoptosis in karyotypically normal human cells expressing MAPT harboring FTLD-causing mutations. Quantitative FISH analysis of hTERT-HME1 cells transfected in the absence of vector (Reagent), with the pcDNA3.1 vector alone (pcDNA), or with the pcDNA3.1 vector harboring wild-type MAPT (WT-MAPT), MAPT with the P301L mutation (P301L), or MAPT with the V337M mutation (V337M) revealed an increase in trisomy 12 (green) and trisomy 21 (orange) in cells expressing MAPT with the FTLD-causing mutations (P301L or V337M) at 48 h compared with cells transfected with the vector alone (A). Levels of aneuploidy in cells transfected with wild-type MAPT were intermediate between cells transfected with the vector alone and cells transfected with the FTLD-causing MAPT mutants. A representative cell with trisomy for chromosomes 12 (green) and 21 (red) showing three individual signals for each chromosome at 48 h is shown in B. Costaining for TUNEL (green) and FISH for chromosomes 12 and 21 at 72 h revealed a significant increase in apoptosis, indicated by TUNEL(+) staining, in cells expressing MAPT with the P301L or V337M mutation relative to negative control cells (Reagent and pcDNA) (C). Quantitative analysis showed that the majority of the cells that were TUNEL(+) at 72 h were aneuploid, regardless of whether they were negative controls or were expressing wild-type or mutant MAPT (D). A representative apoptotic (green, from Reagent) cell with trisomy 12 at 72 h is shown (E); FISH for chromosomes 12 and 21 (I), FISH and DAPI (II), NeuN staining (III), and merged imaged (IV). Analysis of the frequency of apoptosis among aneuploid cells and of aneuploidy among apoptotic cells at 72 h indicates that around 70–80% of apoptotic cells were aneuploid, but only around 15–30% of aneuploid cells were apoptotic in both negative control cells and in cells expressing wild-type or mutant MAPT (F). The graphs show mean differences between treatments using ANOVA with Dunnett t test (A) and post hoc test (C, D, and F) and pcDNA3.1 as comparison group (one-tailed; effect size η2 = 0.60 for trisomy 21 and η2 = 0.58 for trisomy 12). On average, ∼500 cells/slide and three slides/sample for each of three replicates were scored. Significance was calculated using an independent t test. Error bars indicate SEM. *p < 0.05, **p < 0.005, and ***p < 0.0005.
TUNEL staining at a later time point of 72 h posttransfection revealed significantly higher levels of apoptosis in cells expressing either MAPT P301L or MAPT V337M, relative to control cells transfected with the vector alone (Figure 6C). Quantitative analysis of cells costained for TUNEL and FISH for chromosomes 12 and 21 showed that the majority of TUNEL(+) cells at 72 h were aneuploid, regardless of whether they were negative controls or were expressing wild-type or mutant MAPT (Figure 6, D and E). Analysis of the frequency of apoptosis among aneuploid cells and of aneuploidy among apoptotic cells at 72 h indicated that ∼80% of apoptotic cells were aneuploid, but less than 20% of aneuploid cells were apoptotic (Figure 6F). This relationship between aneuploidy and apoptosis was true both in negative control cells, which had low levels of aneuploidy and apoptosis, and in cells expressing wild-type or mutant MAPT, which had significantly higher levels of aneuploidy and apoptosis. Notably, although the cells expressing the FLTD-causing mutant MAPT genes showed increased levels of aneuploidy at 48 h posttransfection, they did not exhibit increased levels of apoptosis until 72 h posttransfection. This finding suggests that aneuploidy precedes and most likely plays a causative role in the neuronal cell death that accompanies FTLD-MAPT.
Apoptosis induced by expression of MAPT harboring FTLD-causing mutations is not caused by oxidative stress
Our data thus far show that FTLD-causing forms of human MAPT induce aneuploidy and apoptosis in human brain cells and when expressed in a karyotypically normal human cell line. The temporal order of aneuploidy followed by apoptosis in our cell culture experiments suggests a cause–effect relationship. However, it is theoretically possible that FTLD-associated MAPT mutations cause both aneuploidy and apoptosis through some common intermediate step. Indeed, oxidative stress has been described in transgenic mice expressing MAPT harboring the P301S FTLD mutation (Lopez-Gonzalez et al., 2015). To test the possibility that FTLD-causing forms of MAPT induce oxidative stress, which in turn leads to both aneuploidy and apoptosis, we measured oxidative stress levels, including reactive oxygen species (ROS) and superoxide, in cells expressing the FTLD mutant forms of MAPT. In these experiments, we found no evidence of increased stress relative to controls (Supplemental Figure 2). These findings show that the effects of FTLD mutant MAPT on aneuploidy and apoptosis are not likely to be caused by oxidative stress.
Expression of MAPT harboring FTLD-causing mutations induces mitotic spindle defects in karyotypically normal human cells
To investigate the mechanism by which MAPT dysfunction leads to chromosome mis-segregation, aneuploidy, and subsequent apoptosis, we analyzed mitotic spindle structures in hTERT-HME1 cells transfected with a vector for expressing wild-type MAPT, MAPT P301L, or MAPT V337M or with the vector alone as a negative control (Figure 7). The cells transfected with either of the MAPT mutants exhibited increased levels of various types of mitotic spindle abnormalities, including lagging chromosomes, multiple centrosomes, and microtubule aggregates at 48 h posttransfection (Figure 7, D–J), when aneuploidy is observed but before apoptosis is observed. These data provide strong support for the conclusion that microtubule instability and consequent mitotic spindle dysfunction are a mechanism by which mutant MAPT induces chromosome mis-segregation, aneuploidy, and, ultimately, apoptosis.
FIGURE 7:
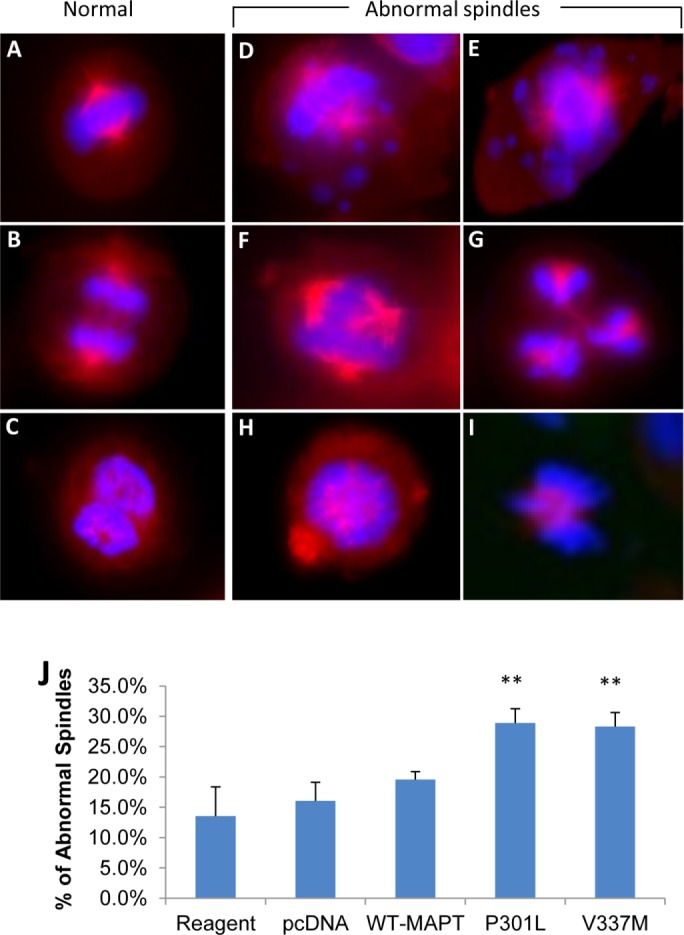
Mitotic spindle defects induced by expression of MAPT harboring FTLD-causing mutations in a karyotypically normal human cell line. The hTERT-HME1 cell line was transfected in the absence of vector (Reagent), with the pcDNA3.1 vector alone (pcDNA), or with the pcDNA3.1 vector harboring wild-type MAPT (WT-MAPT), MAPT with the P301L mutation (P301L), or MAPT with the V337M mutation (V337M), and microscopy images were taken during the metaphase and anaphase stages of mitosis at 48 h posttransfection; microtubules are shown in red, and DNA is shown in blue. Normal spindles: the chromosomes are aligned properly in metaphase and are also undergoing proper segregation in anaphase (A–C). Abnormal spindles: lagging chromosomes, the majority of the chromosomes are aligned properly but some remain unattached and unaligned (D, E); multiple centrosomes: more than two microtubule arrays are present during mitosis (F, G); and microtubule aggregate: the tubulin subunits do not polymerize properly but instead form a cluster inside the nucleus (H, I). Cells expressing the mutant MAPT genes (P301L or V337M) show a significant increase in MT abnormalities in comparison to samples transfected with the pcDNA vector alone or in the absence of vector (J). The graphs show mean difference and significance (*) of the cells transfected with the mutant MAPT genes compared with the pcDNA3.1 vector alone (sham-control) using ANOVA with a Tukey post hoc test. Error bars indicate SEM. **p < 0.005.
Mutant MAPT-induced aneuploidy and apoptosis both depend on cell division
The data presented above are most consistent with the defects in mitosis and consequent chromosome mis-segregation and aneuploidy preceding and thus likely leading to apoptosis (Figure 8). As a more direct test of whether the aneuploidy caused by mutant MAPT expression induces apoptosis or whether the apoptosis caused by mutant MAPT expression occurs independently of the aneuploidy, hTERT-HME1 cells transfected with the pcDNA vector for expressing MAPT P301L or MAPT V337M, or with the pcDNA vector alone as a negative control, were treated for 48 h with Nutlin-3 at 24 h posttransfection to halt cell division and inhibit the generation of aneuploid cells. As expected, cells expressing mutant MAPT (P301L or V337M) exhibited significantly higher levels of aneuploidy (Supplemental Figure 3A) and apoptosis (Supplemental Figure 3B) relative to mock transfected cells, cells transfected with the pcDNA vector, or cells expressing wild-type MAPT; however, Nutlin-3 treatment of cells expressing mutant MAPT (P301L or V337M) prevented them from becoming aneuploid, as measured using a FISH probe for chromosome 21 (Supplemental Figure 3C), and it also prevented them from undergoing apoptosis, as measured by TUNEL staining (Supplemental Figure 3D). If the expression of mutant MAPT induces apoptosis independently of the aneuploidy, then we would have expected to observe increased apoptosis but not increased aneuploidy in the cells expressing mutant MAPT, since the Nutlin-3 treatment would prevent them from dividing and becoming aneuploid. However, the data clearly show that both mutant MAPT-induced aneuploidy and apoptosis were suppressed by Nutlin-3 treatment, indicating that the apoptosis is dependent on the aneuploidy caused by abnormal cell division (Supplemental Figure 3).
FIGURE 8:
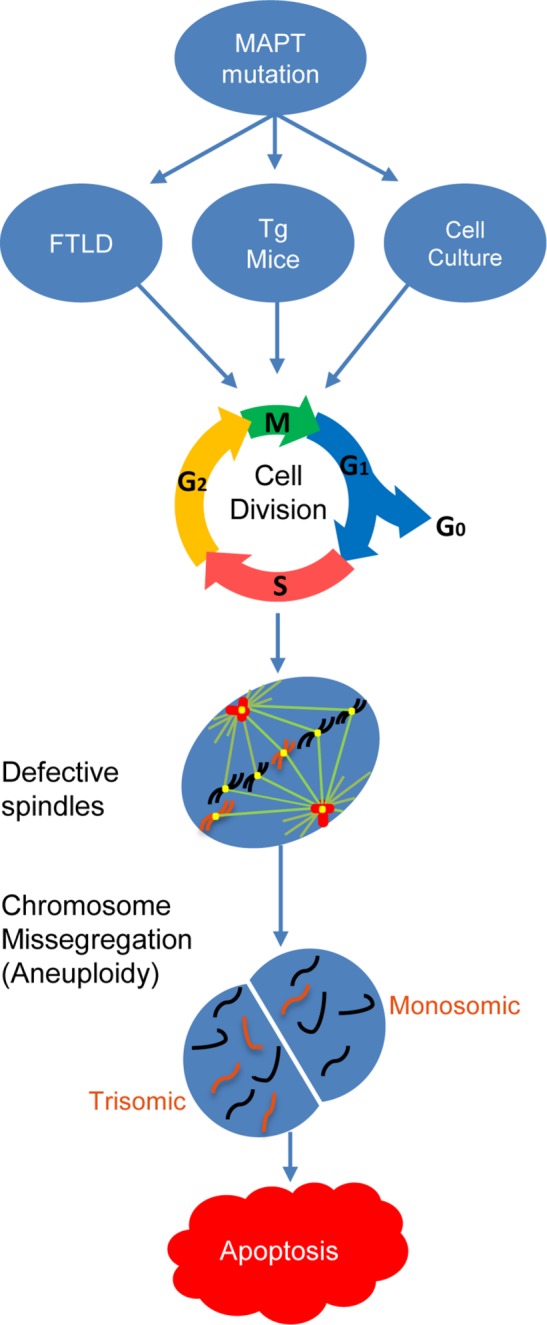
Proposed theoretical mechanistic pathway of aneuploidy and apoptosis in FTLD-MAPT and other tauopathies. On the basis of the evidence presented here, which shows that mutant MAPT-induced aneuploidy and apoptosis require cell division, we hypothesize that the expression of mutant forms of MAPT in the cells of FTLD-MAPT patients (or in patients with other tauopathies) would induce mitotic spindle defects, which would in turn lead to the accumulation of aneuploid cells over time as a result of aberrant cell division. An increase in the aneuploid cell population would not only place a heavy burden on the aneuploid cells to overcome the detrimental effects of altered gene dosage but also render them more vulnerable and prone to undergoing apoptosis.
DISCUSSION
Aneuploidy and apoptosis in various neurodegenerative diseases
Previous studies have shown that aneuploidy occurs in neurons, glia, and other cells in sporadic and all familial forms of AD (Potter, 1991; Potter et al., 1995; Migliore et al., 1997; Geller and Potter, 1999; Kingsbury et al., 2006; Mosch et al., 2007; Thomas and Fenech, 2008; Iourov et al., 2009) and that the specific degeneration of aneuploid neurons that occurs between midstage and advanced AD accounts for 90% of total neuronal cell loss observed at autopsy (Arendt et al., 2010). Although chromosome instability was described previously in peripheral cells from FTLD patients with MAPT mutations (Rossi et al., 2008), aneuploidy levels in the brain were not determined.
Here we show that the brains of familial FTLD patients with MAPT mutations exhibit significantly increased levels of mosaic aneuploidy for chromosomes 12 and 21, together with a significant increase in apoptosis in both neuronal and nonneuronal cells. Furthermore, ∼80% of the neuronal apoptosis occurs in the aneuploid neurons, which parallels the finding described above that 90% of neuronal cell death in advanced AD can be attributed to the specific loss of aneuploid neurons (Arendt et al., 2010).
Our finding that apoptosis occurs preferentially in aneuploid cells from familial FTLD patients with MAPT mutations, including neurons, combined with our observation that a karyotypically normal human cell line expressing MAPT with an FTLD-causing mutation first exhibits aneuploidy without apoptosis at 48 h and then undergoes apoptosis at 72 h suggest that apoptosis arises from aneuploidy and not vice versa. This conclusion was tested directly by experiments with Nutlin-3 showing that the aneuploidy and the apoptosis induced by mutant MAPT expression in cultured cells are not independent of each other, because both require cell division. Our additional finding of mosaic aneuploidy in nonneuronal brain cells from FTLD patients with MAPT mutations reinforces the previous finding of chromosome instability in peripheral cells of FTLD patients with MAPT mutations (Rossi et al., 2008) and indicates that chromosome mis-segregation is likely a general feature of all cells in patients with FTLD-MAPT. The fact that mitotic spindle abnormalities and aneuploidy arise within two mitotic cell cycles (at 48 h posttransfection) in karyotypically normal human cells expressing FTLD-causing mutant forms of MAPT and later lead to apoptosis provides a mechanistic link between microtubule dysfunction and neurodegeneration in FTLD-MAPT. Over time, an individual with FTLD-MAPT who has carried one of these MAPT mutations in all of their cells for their entire life would be expected to also show significant levels of mosaic aneuploidy in cells throughout their body, and their aneuploid neurons would be at significant risk for apoptosis.
The general cell-cycle dysfunction shown in our FTLD-MAPT experiments and the similar findings in AD indicate that multiple chromosomes undergo mis-segregation in these and potentially other neurodegenerative diseases (Potter et al., 1995; Migliore et al., 1997; Geller and Potter, 1999; Kingsbury et al., 2006; Mosch et al., 2007; Boeras et al., 2008; Thomas and Fenech, 2008; Iourov et al., 2009; Arendt et al., 2010, 2015; Arendt, 2012; Granic et al., 2010; Borysov et al., 2011; Yurov et al., 2014). For example, we have found that Niemann-Pick Type C1, another tauopathy, caused by defects in cholesterol metabolism, is characterized by aneuploid neurons and other cells (Granic and Potter, 2013). In addition, a recent study of Lewy body dementia showed an increase in neuronal DNA content (hyperdiploidy) that correlated with neuronal loss in all brain regions examined (Yang et al., 2015). These results together with our studies of AD and FTLD-MAPT support the general conclusion that aneuploid neurons arise by different mechanisms in multiple forms of neurodegenerative disease and that the increasing susceptibility of aneuploid neurons to apoptosis may underlie much of the neuronal loss and cognitive decline that characterize these disorders.
Potential means by which aneuploid neurons arise in MAPT-FTLD
The generation and accumulation of aneuploidy in dividing cell populations might, in principle, arise by both genetic and environmental stressors at any time in life (discussed in Potter, 1991, and Oromendia and Amon, 2014). However, it has long been believed that neurons are postmitotic cells and furthermore that vertebrates are born with a finite number of neurons with little or no neurogenesis occurring in the adult brain (Bhardwaj et al., 2006). These long-held beliefs raise the question of when and how aneuploid neurons arise during age-associated neurodegenerative disease.
Aneuploidy generation during embryogenesis.
One explanation for neuronal aneuploidy in neurodegenerative diseases is that it arises in neuronal precursor cells during embryogenesis and then later leads to neurodegeneration. Certainly, our data show that having an FTLD-causing mutation in MAPT is sufficient to induce abnormal mitotic spindles and subsequent aneuploidy. However, the mechanism by which age may contribute to the process remains unclear. It is possible that aging may promote the development of additional processes that promote the degeneration of aneuploid neurons in these disorders (Potter, 1991; Potter et al., 2016).
Aneuploidy generated during adult neurogenesis and aging.
Although neurogenesis appears to be rare in normal adult brain, three processes may generate the neuronal aneuploidy observed at autopsy in patients with neurodegenerative diseases and may arise in the adult and be promoted by aging. First, data from many studies over the past couple of decades have provided evidence that neurogenesis can be induced in many brain regions in adult mice and rats in response to brain damage (Zhao et al., 2008; Spalding et al., 2013; Zheng et al., 2013; Ibrahim et al., 2016) and attempted self-repair by the brain or as part of an ongoing process in the subventricular/granular zone of the brain (Eriksson et al., 1998; Hallbergson et al., 2003; Sakamoto et al., 2014). Therefore, neuronal damage and the mitotic defect in FTLD-MAPT could produce aneuploid replacement neurons, which would not be fully functional and would be particularly prone to apoptosis and degeneration, as our experiments have shown. Aneuploidy has been shown to promote degeneration in many other experimental systems, possibly by inducing proteotoxic stress (Rajendran et al., 2008; Kai et al., 2009; Arendt et al., 2010; Oromendia and Amon, 2014).
The second potential mechanism for aneuploidy generation during aging and neurodegeneration is cell-cycle reentry. Neurons in the AD brain express phosphoproteins usually detected only during mitosis (McShea et al., 1997; Vincent et al., 1997; Arendt, 2012), and in AD mice, the loss of preexisting neurons induces neuronal reentry into the cell cycle (Lopes et al., 2009; Seward et al., 2013). Indeed, Aβ induces the expression of mitotic proteins and cell-cycle reentry in mature neurons in culture (Majd et al., 2008; Seward et al., 2013). A similar process may be occurring in familial FTLD associated with MAPT mutations.
Finally, the recent discovery that striatal astrocytes can transdifferentiate into new neurons capable of forming functional neuronal circuits with preexisting neurons following ischemic brain injury (Magnusson et al., 2014; Duan et al., 2015) suggests that the aneuploid glia we observed in FTLD brains may have the potential to transdifferentiate into the aneuploid neurons that we also observed.
It seems very likely that the aging process may exacerbate any or all of these processes that have the potential to generate aneuploid neurons and other cells. Indeed, aneuploidy has been proposed and found to increase with age (Arendt et al., 2009; Yurov et al., 2009, 2010; Fischer et al., 2012).
Evidence that MAPT plays a role in chromosome segregation in both FTLD and AD
Our studies showing that MAPT plays a role in chromosome segregation in some forms of FTLD is consistent with previous findings in AD, which showed that mutant forms of APP or PSEN1 or Aβ itself also induce chromosome mis-segregation in a MAPT-dependent manner (Boeras et al., 2008; Granic et al., 2010; Borysov et al., 2011). Additionally, the fact that MAPT and APP phosphorylation and processing are cell-cycle-dependent modifications that increase during mitosis, and that MAPT, APP, and presenilin are localized to mitotic structures, such as centrosomes (Li et al., 1997; Sjoberg et al., 2006; Rossi et al., 2008; Judge et al., 2011), further implicates both MAPT and Aβ function in the process of chromosome segregation.
Other studies linking aneuploidy, neurodegeneration, and MAPT
Evidence linking neuronal aneuploidy, neurodegeneration, and MAPT was reported recently by two other groups in Drosophila. Specifically, a study by Bougé and Parmentier (2016) showed that excess Tau causes mitotic spindle defects, aneuploidy, and apoptosis through inhibition of the microtubule-dependent motor protein Kinesin-5/Eg5. Similar results have been observed by Malmanche et al. (2017), who examined photoreceptors and brain neurons in Drosophila and found that adult-onset neurodegeneration mediated by MAPT overexpression included the generation of aneuploid cells (Malmanche et al., 2017). The former result is of particular interest in light of our prior finding that Aβ induces chromosome mis-segregation and aneuploidy though competitive inhibition of Kinesin-5/Eg5 (Borysov et al., 2011). Thus, causal mutations leading to AD and FTLD-MAPT appear to lead to chromosome mis-segregation, aneuploidy, and apoptosis through inhibition of the same target enzyme: Kinesin-5/Eg5.
Possible explanations for how a general cell-cycle defect can lead to regional neurodegeneration
Both AD and FTLD and indeed all neurodegenerative disease show pathology and cell loss in specific regions of the brain, while sparing other regions, and this regional specificity correlates roughly with the clinical symptoms (e.g., tau pathology in AD). Because APP and MAPT are expressed widely, other factors must control essential steps in the pathogenic pathway that results in regional specificity. In AD, the specificity arises from differences in the expression of modulator proteins, such as the amyloid catalysts alpha-1-antichymotrypsin (ACT) and ApoE, which are essential for the conversion of Aβ to oligomers and filaments. Indeed, only Aβ oligomers cause chromosome mis-segregation and aneuploidy (Granic et al., 2010; Borysov et al., 2011). Both ACT and ApoE are inflammation-associated proteins in the brain that are not expressed highly in the cerebellum in AD, for example, where amyloid pathology also does not arise. Prion disease research has also found that species and regional specificity must involve a specific component, termed “protein X,” that is needed for pathology (Yehiely et al., 1997). Protein X has yet to be definitively identified, but one study showed that it is not ApoE (Tatzelt et al., 1996).
The finding that APP and Presenilin mutations and their product, oligomeric Aβ peptide, cause chromosome mis-segregation in cells, transgenic mice, and humans through disruption of the mitotic spindle and that the consequent aneuploidy may underlie much of the neurodegeneration, as discussed above, also begs the same question of how an effect on a common physiological process such as mitosis can lead to region specific neurodegeneration. As anticipated above, the explanation in AD may lie in the regional expression of ApoE.
Similarly, we suggest that one potential explanation for the regional specificity of FTLD is that another protein that is expressed in the cortex interacts with mutant MAPT to promote its pathological effect on microtubules. It is also possible that such a protein or even the direct effect of mutant MAPT only occurs in dividing cells, as our experiment with Nutlin-3 indicates. In that case, only neurons undergoing cell-cycle reentry or being replaced by neurogenesis would be sensitive to mutant MAPT. Alternatively, other microtubule-binding proteins that can substitute for damaged tau may be normally preferentially expressed in certain brain regions (i.e., those not affected in FTLD), thereby underlying region-specific protection.
Targeting microtubule and mitotic spindle dysfunction to prevent neurodegeneration
Taken together with the results of our previous work and recent reports, the data presented herein provide a novel mechanism for neurodegeneration and neuronal dysfunction in FTLD based on microtubule and mitotic spindle dysfunction (Figure 8). Further, the findings may lead to new general treatments or preventative strategies for a wide range of neurodegenerative disorders, including FTLD, AD, Niemann Pick Type C1, ALS, Lewy body dementia, and Down syndrome. For example, microtubule stabilization, by drug therapy, by inhibition of MAPT phosphorylation, or by rescue of inhibited Kinesin-5/Eg5 activity, may improve chromosome segregation and lead to reduced levels of aneuploidy and subsequent neurodegeneration.
MATERIALS AND METHODS
Cell line
The hTERT-HME1 (Clontech) is a telomerase-immortalized primary human mammary epithelial cell line that divides indefinitely and retains normal function, phenotype, and karyotype (Jiang et al., 1999). The cell line was cultured in supplemented mammary epithelium basal medium (MEBM; Lonza), as described previously (Boeras et al., 2008; Granic et al., 2010). Early cell passages were used for the experiments.
Primary mouse cells
Mouse spleens and brains were harvested and processed for culture or immediately fixed as single-cell suspensions for FISH and FISH/immunocytochemistry, as described previously (Boeras et al., 2008; Granic et al., 2010; Granic and Potter, 2013; Caneus et al., 2017).
Human brain cells
Approximately 1–2 g of frozen human brain cortices from patients diagnosed with familial FTLD associated with MAPT mutations (aged 56 ± 5 yr) and controls (aged 65 ± 9 yr; p = 0.054) was obtained from the Mayo Clinic Brain Bank and the Banner Sun Health Research Institute and Brain and Body Donation Program, respectively. The tissues were dissociated into single-nuclei suspensions, fixed, and processed for FISH in combination with NeuN and/or TUNEL immunocytochemistry, as described previously (Granic et al., 2010; Granic and Potter, 2013).
Mice
Tau-/- knockout mice (obtained from the Jackson Laboratory, B6.129 × 1-MAPTtm1Hnd/J [ Dawson et al., 2001]) and their heterozygous (tau+/-) and wild-type (tau+/+) littermates were 6–7 mo old (background strain: C57BL/6). Transgenic mice expressing a human FTLD-causing mutation in the MAPT gene (P301S) from a prion promoter and their nontransgenic control (NON) were ∼8 mo old and were obtained from the Jackson Laboratory (B6;C3-Tg[Prnp-MAPT*P301S]PS19Vle/J). In addition, brain tissue samples from an FTLD mouse model expressing the human mutant MAPT (P301L) transgene under the control of the prion promoter and control nontransgenic mice (∼8 wk old) were purchased from the Jackson Laboratory (FVB-Tg[tetO-MAPT*P301L]#Kha/JlwsJ and FVB/NJ, respectively).
Plasmids
Plasmids constructed by inserting the wild-type human MAPT cDNA or the human MAPT cDNA harboring an FTLD-causing mutation (P301L or V337M) into the pcDNA3.1 expression vector were gifts from Chad Dickey (University of South Florida, Tampa). The NucleoBond Plasmid Purification Kit (BD Bioscience) was used for nucleic acid extraction from NovaBlue Singles competent cells (Novagen).
Antibodies, kits, and DNA probes
Mouse/rabbit anti-neuronal nuclei (NeuN) AlexaFluor antibodies (Millipore, MAB377X; Abcam, ab190195) and DAPI (4’,6-diamidino-2-phenylindole) II (Abbott, Vysis) or VectaShield with DAPI (Vector Laboratories) were used to label neurons and nuclei, respectively. A BAC probe to mouse chromosome 16 was either purchased (Mouse IDetect Chromosome 16 D16Mit88 Point Probe, RP24–288N19, Empire Genomics) or prepared directly using a Nick Translation Kit (Roche) with Spectrum Green or Red dUTP (Vysis) using a chromosome 16 BAC plasmid (a gift from Bruce Lamb, The Cleveland Clinic), as described previously (Boeras et al., 2008; Granic et al., 2010; Granic and Potter, 2013). A mixture of fluorescently labeled DNA probes for the detection of human chromosomes 12 and 21 (LSI TEL/AML1 ES Dual Color Translocation Probe) or individual probes for the detection of human chromosome 21 (Vysis TelVysion 21q SpectrumOrange Probe, Vysis LSI ERG [Tel] SpectrumGreen Probe, Vysis LSI ERG [Cen] SpectrumRed Probe) were obtained from Abbott, Vysis. TUNEL assay was carried out using the Click-iT Plus TUNEL Assay for in situ apoptosis detection with Alexa Fluor dyes (molecular probes) on brain samples and cultured cells in with or without Nutlin-3 (Selleckchem; Cat. No. S1061). A cellular ROS/Superoxide Detection Assay Kit (Abcam; ab139476) was used to measure oxidative stress levels in cultured cells.
FISH analysis
Single nuclei suspensions were dropped on glass slides (FisherBrand) and air-dried/aged at room temperature overnight. The slides/nuclei were then subjected to hybridization procedures according to the manufacturer (Abbott, Vysis) using established protocols with the mouse and human probes described above and previously, including hybridization in a HyBrite chamber (Vysis). In subsequent steps, the slides were either counterstained with Vectashield with DAPI immediately (cultured cells) or processed for NeuN immunostaining followed by DAPI counterstain (brain samples) (Boeras et al., 2008; Granic et al., 2010; Granic and Potter, 2013; Caneus et al., 2017).
TUNEL staining combined with FISH and immunocytochemistry for brain cells
Single-nuclei suspensions prepared from either frozen human brain tissues or from hTERT cells transfected for 72 h with mutant MAPT genes were fixed, dropped, and air-dried overnight on glass slides and subjected to TUNEL staining, which involved incubation with a recombinant terminal deoxynucleotidyl transferase (TdT) and dUTPs with Alexa Fluor dyes for a combined 1 h, 40 min at 37˚C, according to the manufacturer’s recommended procedure (Click-iT Plus TUNEL Assay; Life Technologies). Following TUNEL staining, the cells were processed for FISH with DNA probes for chromosomes 12 and 21 and immunocytochemistry with anti-NeuN antibodies (Millipore and Abcam), as described previously. On average, more than 500 cells/slide and three slides/sample were counted for the transfected hTERT cells, and ∼1000 cells/slide and two slides were counted for each human subject.
Transient transfection
The hTERT-HME1 cells (∼1.8 × 105 cells/2 ml) were plated into a six-well plate and transfected with an empty pcDNA3.1 vector, with the pcDNA3.1 vector harboring a wild-type or mutant (P301L or V337M) human MAPT cDNA, or with a mixture of reduced Opti-MEM and FuGENE 6 transfection reagent (Promega), according to the manufacturer’s protocol, as described previously (Boeras et al., 2008; Granic et al., 2010). Cells from three parallel transfections were treated with Nutlin-3 at 24 h posttransfection and incubated for an additional 48 h. The samples were then harvested, fixed, and scored for aneuploidy using chromosome 21 FISH and apoptosis using TUNEL at 72 h posttransfection.
Immunocytochemistry of hTERT-HME1 cells for microtubule analyses
Confluent hTERT cells were harvested, plated at a density of ∼1.5 × 105 cells/2 ml, and treated with the FuGENE 6 reagent alone or transfected with a FuGene 6-DNA complex of either the plasmid vector (pcDNA3.1) or the pcDNA3.1 plasmid vector harboring wild-type MAPT, MAPT V337M, or MAPT P301L and cultured on glass chamber slides. The cells were allowed to progress through the cell cycle and harvested at the metaphase/anaphase stages of mitosis, fixed in methanol for 5 min at –20˚C, subjected to indirect immunofluorescence staining with a mouse anti–alpha-tubulin antibody (Abcam; Cat. Ab28439) and Alexa Fluor-conjugated goat anti-mouse antibody, and mounted in Vectashield with DAPI.
Image acquisition and analysis
FISH hybridization signals were analyzed following Abbott/Vysis guidelines as described (Boeras et al., 2008; Granic et al., 2010). Between 800 and 1000 interphases per sample per treatment were scored for aneuploidy, and at least 400 NeuN(+) and NeuN(–) mouse brain cells were analyzed for BAC DNA signals. In addition, the percentage of cells that were positive for TUNEL labeling was recorded and scored for apoptosis analysis. Mitotic spindles and DNA were visualized and scored for quantitative analysis using ZEN 2011 SP1 imaging software. Each of the in vitro experiments was done in triplicate.
Statistics
Normality of data was determined by Shapiro-Wilk test. Independent t tests were used to compare aneuploidy in FTLD and control brain and spleen cells in the experiments using transgenic mice expressing human MAPT P301S. One-way analysis of variance (ANOVA) with Tukey HSD and Dunnett test was used to establish significance in all other studies, as indicated by (*) or presented in the figures. The effect size (η2 for ANOVA and Cohen’s d for t test) was calculated for the main outcomes. The analyses were performed in Microsoft Excel 2013 and IBM SPSS Statistics version 22.
Supplementary Material
Acknowledgments
The chromosome 16 BAC and MAPT expression vectors were kind gifts from Bruce Lamb and Chad Dickey, respectively. We also acknowledge with gratitude the contributions of neurologists, notably Neill Graff-Radford and Zbigniew Wszolek, in caring for the FTLD patients. Human brain tissue was obtained from the Mayo Clinic Brain Bank (Jacksonville, FL) and from the Banner Sun Health Research Institute Brain and Body Donation Program of Sun City (Arizona), which is supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinson’s Disease and Related Disorders), the National Institute on Aging (P30-AG19610, Arizona Alzheimer’s Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer’s Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05–901, and 1001 to the Arizona Parkinson’s Disease Consortium), and the Michael J. Fox Foundation for Parkinson’s Research. We appreciate the opportunity to present early results of this study at the 2013, 2014, and 2015 Alzheimer’s Association International Conferences. We also thank Joaquin Espinosa for his helpful suggestion to use Nutlin-3 to inhibit cell division in our experiments. This study was funded by National Institute of Aging grants (AG-025711 and AG-037942). A.G. is funded by the AGE Research Group to Avan A. Sayer (Institute of Neuroscience, Newcastle Institute for Ageing, National Institute for Health Research Newcastle Biomedical Research Centre, Newcastle University, UK). We also acknowledge the generous support of donors.
Abbreviations used:
- AD
Alzheimer’s disease
- FTD
frontotemporal dementia
- FTLD
frontotemporal lobar degeneration.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-01-0031) on December 27, 2017.
REFERENCES
- Arendt T. (2012). Cell cycle activation and aneuploid neurons in Alzheimer’s disease. Mol Neurobiol , 125-135. [DOI] [PubMed] [Google Scholar]
- Arendt T, Bruckner MK, Losche A. (2015). Regional mosaic genomic heterogeneity in the elderly and in Alzheimer’s disease as a correlate of neuronal vulnerability. Acta Neuropathol , 501-510. [DOI] [PubMed] [Google Scholar]
- Arendt T, Bruckner MK, Mosch B, Losche A. (2010). Selective cell death of hyperploid neurons in Alzheimer’s disease. Am J Pathol , 15-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt T, Mosch B, Morawski M. (2009). Neuronal aneuploidy in health and disease: a cytomic approach to understand the molecular individuality of neurons. Int J Mol Sci , 1609-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj RD, Curtis MA, Spalding KL, Buchholz BA, Fink D, Bjork-Eriksson T, Nordborg C, Gage FH, Druid H, Eriksson PS, Frisen J. (2006). Neocortical neurogenesis in humans is restricted to development. Proc Natl Acad Sci USA , 12564-12568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeras DI, Granic A, Padmanabhan J, Crespo NC, Rojiani AM, Potter H. (2008). Alzheimer’s presenilin 1 causes chromosome missegregation and aneuploidy. Neurobiol Aging , 319-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borysov SI, Granic A, Padmanabhan J, Walczak CE, Potter H. (2011). Alzheimer Abeta disrupts the mitotic spindle and directly inhibits mitotic microtubule motors. Cell Cycle , 1397-1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bougé AL, Parmentier ML. (2016). Tau excess impairs mitosis and kinesin-5 function, leading to aneuploidy and cell death. Dis Model Mech , 307-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caneus J, Granic A, Chial HJ, Potter H. (2017). Using fluorescence in situ hybridization (FISH) analysis to measure chromosome instability and mosaic aneuploidy in neurodegenerative diseases. In: Genomic Mosaicism in Neurons and Other Cell Types, ed. Frade JM, Gage FH, New York: Springer, , 329-359. [Google Scholar]
- Dawson HN, Ferreira A, Eyster MV, Ghoshal N, Binder LI, Vitek MP. (2001). Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J Cell Sci , 1179-1187. [DOI] [PubMed] [Google Scholar]
- Duan CL, Liu CW, Shen SW, Yu Z, Mo JL, Chen XH, Sun FY. (2015). Striatal astrocytes transdifferentiate into functional mature neurons following ischemic brain injury. Glia , 1660-1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, Peterson DA, Gage FH. (1998). Neurogenesis in the adult human hippocampus. Nat Med , 1313-1317. [DOI] [PubMed] [Google Scholar]
- Fischer HG, Morawski M, Bruckner MK, Mittag A, Tarnok A, Arendt T. (2012). Changes in neuronal DNA content variation in the human brain during aging. Aging Cell , 628-633. [DOI] [PubMed] [Google Scholar]
- Geller LN, Potter H. (1999). Chromosome missegregation and trisomy 21 mosaicism in Alzheimer’s disease. Neurobiol Dis , 167-179. [DOI] [PubMed] [Google Scholar]
- Granic A, Padmanabhan J, Norden M, Potter H. (2010). Alzheimer Abeta peptide induces chromosome mis-segregation and aneuploidy, including trisomy 21: requirement for tau and APP. Mol Biol Cell , 511-520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granic A, Potter H. (2013). Mitotic spindle defects and chromosome mis-segregation induced by LDL/cholesterol-implications for Niemann-Pick C1, Alzheimer’s disease, and atherosclerosis. PLoS One , e60718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallbergson AF, Gnatenco C, Peterson DA. (2003). Neurogenesis and brain injury: managing a renewable resource for repair. J Clin Investig , 1128-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim S, Hu W, Wang X, Gao X, He C, Chen J. (2016). Traumatic brain injury causes aberrant migration of adult-born neurons in the hippocampus. Sci Rep , 21793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iourov IY, Vorsanova SG, Liehr T, Yurov YB. (2009). Aneuploidy in the normal, Alzheimer’s disease and ataxia-telangiectasia brain: differential expression and pathological meaning. Neurobiol Dis , 212-220. [DOI] [PubMed] [Google Scholar]
- Jiang XR, Jimenez G, Chang E, Frolkis M, Kusler B, Sage M, Beeche M, Bodnar AG, Wahl GM, Tlsty TD, Chiu CP. (1999). Telomerase expression in human somatic cells does not induce changes associated with a transformed phenotype. Nat Genet , 111-114. [DOI] [PubMed] [Google Scholar]
- Judge M, Hornbeck L, Potter H, Padmanabhan J. (2011). Mitosis-specific phosphorylation of amyloid precursor protein at threonine 668 leads to its altered processing and association with centrosomes. Mol Neurodegener , 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kai Y, Wang CC, Kishigami S, Kazuki Y, Abe S, Takiguchi M, Shirayoshi Y, Inoue T, Ito H, Wakayama T, Oshimura M. (2009). Enhanced apoptosis during early neuronal differentiation in mouse ES cells with autosomal imbalance. Cell Res , 247-258. [DOI] [PubMed] [Google Scholar]
- Kingsbury MA, Yung YC, Peterson SE, Westra JW, Chun J. (2006). Aneuploidy in the normal and diseased brain. Cell Mol Life Sci , 2626-2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulnane LS, Lehman EJ, Hock BJ, Tsuchiya KD, Lamb BT. (2002). Rapid and efficient detection of transgene homozygosity by FISH of mouse fibroblasts. Mamm Genome , 223-226. [DOI] [PubMed] [Google Scholar]
- Li J, Xu M, Zhou H, Ma J, Potter H. (1997). Alzheimer presenilins in the nuclear membrane, interphase kinetochores, and centrosomes suggest a role in chromosome segregation. Cell , 917-927. [DOI] [PubMed] [Google Scholar]
- Lopes JP, Blurton-Jones M, Yamasaki TR, Agostinho P, LaFerla FM. (2009). Activation of cell cycle proteins in transgenic mice in response to neuronal loss but not amyloid-beta and tau pathology. J Alzheimer’s Dis , 541-549. [DOI] [PubMed] [Google Scholar]
- Lopez-Gonzalez I, Aso E, Carmona M, Armand-Ugon M, Blanco R, Naudi A, Cabre R, Portero-Otin M, Pamplona R, Ferrer I. (2015). Neuroinflammatory gene regulation, mitochondrial function, oxidative stress, and brain lipid modifications with disease progression in Tau P301S transgenic mice as a model of frontotemporal lobar degeneration-Tau. J Neuropathol Exp Neurol , 975-999. [DOI] [PubMed] [Google Scholar]
- Magnusson JP, Goritz C, Tatarishvili J, Dias DO, Smith EM, Lindvall O, Kokaia Z, Frisen J. (2014). A latent neurogenic program in astrocytes regulated by Notch signaling in the mouse. Science , 237-241. [DOI] [PubMed] [Google Scholar]
- Majd S, Zarifkar A, Rastegar K, Takhshid MA. (2008). Different fibrillar Abeta 1–42 concentrations induce adult hippocampal neurons to reenter various phases of the cell cycle. Brain Res , 224-229. [DOI] [PubMed] [Google Scholar]
- Malmanche N, Dourlen P, Gistelinck M, Demiautte F, Link N, Dupont C, Vanden Broeck L, Werkmeister E, Amouyel P, Bongiovanni A, et al. (2017). Developmental expression of 4-Repeat-Tau induces neuronal aneuploidy in Drosophila tauopathy models. Sci Rep , 40764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell MJ, Lindberg MR, Brennand KJ, Piper JC, Voet T, Cowing-Zitron C, Shumilina S, Lasken RS, Vermeesch JR, Hall IM, Gage FH. (2013). Mosaic copy number variation in human neurons. Science , 632-637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McShea A, Harris PL, Webster KR, Wahl AF, Smith MA. (1997). Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer’s disease. Am J Pathol , 1933-1939. [PMC free article] [PubMed] [Google Scholar]
- Migliore L, Testa A, Scarpato R, Pavese N, Petrozzi L, Bonuccelli U. (1997). Spontaneous and induced aneuploidy in peripheral blood lymphocytes of patients with Alzheimer’s disease. Hum Genet , 299-305. [DOI] [PubMed] [Google Scholar]
- Mosch B, Morawski M, Mittag A, Lenz D, Tarnok A, Arendt T. (2007). Aneuploidy and DNA replication in the normal human brain and Alzheimer’s disease. J Neurosci , 6859-6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oromendia AB, Amon A. (2014). Aneuploidy: implications for protein homeostasis and disease. Dis Model Mech , 15-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter H. (1991). Review and hypothesis: Alzheimer disease and Down syndrome—chromosome 21 nondisjunction may underlie both disorders. Am J Hum Genet , 1192-1200. [PMC free article] [PubMed] [Google Scholar]
- Potter H, Granic A, Caneus J. (2016). Role of trisomy 21 mosaicism in sporadic and familial Alzheimer’s disease. Curr Alzheimer Res , 7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter H, Ma J, Das S, Geller LN, Benjamin M, Kayyali US, Dressler D. (1995). Beyond β-protein: new steps in the pathogenic pathway to Alzheimer’s disease. In: Research Advances in Alzheimer’s Disease and Related Disorders, ed. Iqbal JAMK, Winblad B, Wisniewski HM, New York: John Wiley & Sons Ltd, 643-654. [Google Scholar]
- Rademakers R, Neumann M, Mackenzie IR. (2012). Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol , 423-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran RS, Wellbrock UM, Zupanc GK. (2008). Apoptotic cell death, long-term persistence, and neuronal differentiation of aneuploid cells generated in the adult brain of teleost fish. Dev Neurobiol , 1257-1268. [DOI] [PubMed] [Google Scholar]
- Rossi G, Conconi D, Panzeri E, Paoletta L, Piccoli E, Ferretti MG, Mangieri M, Ruggerone M, Dalpra L, Tagliavini F. (2014). Mutations in MAPT give rise to aneuploidy in animal models of tauopathy. Neurogenetics , 31-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi G, Conconi D, Panzeri E, Redaelli S, Piccoli E, Paoletta L, Dalpra L, Tagliavini F. (2013). Mutations in MAPT gene cause chromosome instability and introduce copy number variations widely in the genome. J Alzheimer’s Dis , 969-982. [DOI] [PubMed] [Google Scholar]
- Rossi G, Dalpra L, Crosti F, Lissoni S, Sciacca FL, Catania M, Di Fede G, Mangieri M, Giaccone G, Croci D, Tagliavini F. (2008). A new function of microtubule-associated protein tau: involvement in chromosome stability. Cell Cycle , 1788-1794. [DOI] [PubMed] [Google Scholar]
- Sakamoto M, Ieki N, Miyoshi G, Mochimaru D, Miyachi H, Imura T, Yamaguchi M, Fishell G, Mori K, Kageyama R, Imayoshi I. (2014). Continuous postnatal neurogenesis contributes to formation of the olfactory bulb neural circuits and flexible olfactory associative learning. J Neurosci , 5788-5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seward ME, Swanson E, Norambuena A, Reimann A, Cochran JN, Li R, Roberson ED, Bloom GS. (2013). Amyloid-beta signals through tau to drive ectopic neuronal cell cycle re-entry in Alzheimer’s disease. J Cell Sci , 1278-1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoberg MK, Shestakova E, Mansuroglu Z, Maccioni RB, Bonnefoy E. (2006). Tau protein binds to pericentromeric DNA: a putative role for nuclear tau in nucleolar organization. J Cell Sci , 2025-2034. [DOI] [PubMed] [Google Scholar]
- Spalding KL, Bergmann O, Alkass K, Bernard S, Salehpour M, Huttner HB, Bostrom E, Westerlund I, Vial C, Buchholz BA, et al. (2013). Dynamics of hippocampal neurogenesis in adult humans. Cell , 1219-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatzelt J, Maeda N, Pekny M, Yang SL, Betsholtz C, Eliasson C, Cayetano J, Camerino AP, DeArmond SJ, Prusiner SB. (1996). Scrapie in mice deficient in apolipoprotein E or glial fibrillary acidic protein. Neurology , 449-453. [DOI] [PubMed] [Google Scholar]
- Thomas P, Fenech M. (2008). Chromosome 17 and 21 aneuploidy in buccal cells is increased with ageing and in Alzheimer’s disease. Mutagenesis , 57-65. [DOI] [PubMed] [Google Scholar]
- Vincent I, Jicha G, Rosado M, Dickson DW. (1997). Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer’s disease brain. J Neurosci , 3588-3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Shepherd C, Halliday G. (2015). Aneuploidy in Lewy body diseases. Neurobiol Aging , 1253-1260. [DOI] [PubMed] [Google Scholar]
- Yehiely F, Bamborough P, Da Costa M, Perry BJ, Thinakaran G, Cohen FE, Carlson GA, Prusiner SB. (1997). Identification of candidate proteins binding to prion protein. Neurobiol Dis , 339-355. [DOI] [PubMed] [Google Scholar]
- Yurov YB, Vorsanova SG, Iourov IY. (2009). GIN’n’CIN hypothesis of brain aging: deciphering the role of somatic genetic instabilities and neural aneuploidy during ontogeny. Mol Cytogenet , 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurov YB, Vorsanova SG, Iourov IY. (2010). Ontogenetic variation of the human genome. Curr Genomics , 420-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurov YB, Vorsanova SG, Liehr T, Kolotii AD, Iourov IY. (2014). X chromosome aneuploidy in the Alzheimer’s disease brain. Mol Cytogenet , 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Deng W, Gage FH. (2008). Mechanisms and functional implications of adult neurogenesis. Cell , 645-660. [DOI] [PubMed] [Google Scholar]
- Zheng W, ZhuGe Q, Zhong M, Chen G, Shao B, Wang H, Mao X, Xie L, Jin K. (2013). Neurogenesis in adult human brain after traumatic brain injury. J Neurotrauma , 1872-1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.