

The N-methyl-d-aspartate (NMDA)-type glutamate receptor is one of three major classes of receptors for glutamate, the principle excitatory neurotransmitter in the central nervous system. It plays a key role in learning and in the formation of memories by acting as a “coincidence detector” that initiates changes in synaptic strength that lead to the formation of new neural networks (1). It is also an important mediator of several forms of pathological neuronal toxicity. The NMDA receptor responds at a synapse only when the presynaptic terminal releases glutamate at the same time that the postsynaptic neuron is strongly depolarized by the sum of activating influences impinging on it. In effect, it initiates the strengthening of all synapses that depolarize the same postsynaptic neuron at the same time and thus triggers formation of a new, more stable circuit. When the NMDA-receptor channel opens, it allows passage of calcium ions, as well as sodium and potassium, into the cell. The calcium ions trigger a cascade of biochemical signaling reactions catalyzed by enzymes located just underneath the postsynaptic membrane. These reactions modify other membrane channels in the synapse, ultimately leading to a change in the strength of the electrical signal produced when the synapse is activated again.
The processes of thinking, learning, and remembering are subtle and complex. Therefore, it isn't surprising that the magnitude and timing of calcium flux through NMDA receptors are tightly regulated by phosphorylation by several second-messenger-regulated protein kinases. The paper in this issue of PNAS by Li et al. entitled “Regulation of NMDA receptors by cyclin-dependent kinase-5” now provides evidence that a cyclin-dependent kinase that is highly expressed in brain also may be an important regulator of NMDA-receptor function (2). The cyclin-dependent kinases are a large family of 10 or more members that are known mostly for their involvement in orchestrating transitions in the cell cycle (3, 4). Cdk1 is the mammalian orthologue of the yeast cyclin-dependent protein kinase responsible for initiating the transition into mitosis (cdc2/cdc28). Other members of the family control critical functions, such as DNA replication, during the cell cycle. Most cdks are regulated by a family of small proteins called cyclins that are synthesized and degraded in a tightly controlled fashion as the cell cycle progresses. Only a few members of the family have functions outside the cell cycle; cdk5 is one of them. Its highest level of expression is in postmitotic neurons in developing and adult brain. The cdk5 catalytic subunit alone has no catalytic activity; however, it is not regulated by cyclins. Instead, it is activated by association with brain-specific regulatory proteins that have little sequence similarity to cyclins but seem to assume a cyclin-like fold (3). Cdk5 has many known functions in the brain, including an essential role in neuronal migration during development of the cortex (5, 6), regulation of cadherin adhesion, regulation of actin, neurofilament, and tubulin cytoskeletons, and regulation of dopamine signaling (3). In addition, cdk5 may mediate hyperphosphorylation of tau protein that leads to the formation of paired helical filaments in Alzheimer's disease (7). The work by Li et al. shows that cdk5 is concentrated together with the NMDA receptor at glutamatergic postsynaptic sites and can phosphorylate the cytosolic tail of a subunit of the NMDA receptor. The authors also show that roscovitine, an inhibitor of cdk5, reduces current through the NMDA receptor and blocks induction of long-term potentiation (LTP) in hippocampal slices. Their results suggest that cdk5 is part of a pathway that, directly or indirectly, regulates the amplitude of current through NMDA receptors.
Like most ligand-gated channels, the NMDA receptor is an oligomer of four to five individual subunits that come together in the membrane to form the channel (1). One of these subunits, NR1, is necessary for channel function and is similar in structure to the subunits of other ligand-gated channels (8). However, the four other principal subunits, termed NR2A–D, are unique among ligand-gated channels in having long extensions of their carboxyl-terminal tails that protrude into the cytoplasm beneath the membrane and serve as anchoring points for signal transducing enzymes (see Fig. 1; refs. 9 and 10). In the hippocampus and cortex, NR2A and NR2B are the most prominent. The ≈630-residue cytosolic tail of NR2A and the ≈650-residue tail of NR2B are homologous but are only ≈30% identical in sequence. The most similar regions are clustered in small segments of the tails (see Fig. 1). Thus, the two subunits may interact with some of the same, and some distinct, proteins. The tails of both subunits bind to the scaffold protein PSD-95 (11, 12) and also bind Ca2+/calmodulin-dependent protein kinase II (CaMKII; refs. 13 and 14), but the nature and consequences of that interaction may differ (15).
Figure 1.
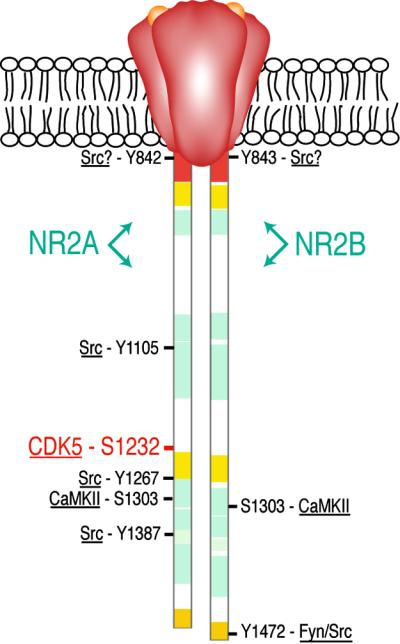
Phosphorylation sites on the cytosolic tails of NMDA-receptor subunits NR2A and NR2B. The domain diagrams compare the cytosolic tail of NR2A (residues 838-1464) and that of NR2B (residues 839-1482). Regions shown in red are 70% identical; in gold, 50%; in yellow, 40%; and in light blue, 20–30%. White regions show no similarity. The positions of phosphorylation sites discussed in the text are labeled, and the protein kinase believed to phosphorylate each is noted. Phosphorylation of Ser-1232 (in red) by Cdk5 is described by Li et al. in this issue (2).
Although a number of sites on the tails of these two subunits have been shown to be phosphorylated by specific protein kinases (14, 16–21), evidence about the functional significance of these modifications is still inconclusive. Perhaps the best documented example of a functionally significant phosphorylation is the up-regulation of activity of NMDA receptors through phosphorylation by the src/fyn family of cytosolic protein Tyr kinases (for review, see ref. 22). The introduction of src or a peptide that activates src-family kinases into heterologous cells expressing NMDA receptors increases NMDA-evoked currents. Similar treatment of neurons increases the NMDA-receptor component of synaptically evoked currents. The same effects can be seen after application of these reagents to inside-out membrane patches containing NMDA receptors. However, the functional significance of these effects and the mechanisms underlying them are uncertain. For example, enhancement of recombinant NR1/NR2A currents is blocked by mutating specific Tyr residues in the cytosolic tail to Phe (Y1105, Y1267, Y1387; ref. 17), supporting the notion that the enhancement is caused by Tyr phosphorylation. However, the increased current is caused by a reduction of tonic, high-affinity, voltageindependent blockade of the channels by extracellular Zn2+, an effect that is not observed when recording from NMDA receptors in intact neurons (23). To complicate the interpretation further, the principal site phosphorylated in NR2B in vivo by the fyn Tyr kinase, a close relative of src, does not correspond to the residues that, when mutated in NR2A, block up-regulation by src (Y1472; ref. 20). This site, Tyr-1472, becomes more heavily phosphorylated in hippocampal slices after induction of LTP and after transient global ischemia (20). The sequence surrounding Tyr-1472 in NR2A is a consensus clathrin adaptor-2 (AP2)-binding domain that can mediate NMDA-receptor internalization if it is not masked by binding to the PDZ domain of PSD-95 (24). The increase in phosphorylation of the Tyr in the AP2-binding domain after induction of LTP suggests that phosphorylation of this site may help to regulate turnover of the NMDA receptor. In the same vein, Vissel et al. (25) have reported recently that dephosphorylation of a ring of Tyr residues in NMDA-receptor subunits NR1 and NR2A results in down-regulation of recombinant NR1/NR2A receptors, apparently by clathrin-mediated endocytosis. These sites can be phosphorylated and down-regulation can be blocked by the Src and Fyn protein Tyr kinases.
It is apparent that the NMDA receptor can be modified by a complex, integrative network of phosphorylation and dephosphorylation.
Thus, although it is not yet clear which phosphorylation sites are important for physiological regulation of the NMDA receptor or for assembly of its associated signaling complexes, it is apparent that the receptor can be modified by a complex, integrative network of phosphorylation and dephosphorylation. The organization and functions of this network will be the focus of much research over the next few years. In particular, it will be interesting to see whether the Ser site in NR2A that is phosphorylated by cdk5 in heterologous cells also is phosphorylated in neurons, and whether phosphorylation of the site is required for normal current through the NMDA receptor.
Footnotes
See companion article on page 12742.
References
- 1.Dingledine R, Borges K, Bowie D, Traynelis S F. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- 2.Li B-S, Sun M-K, Zhang L, Takahashi S, Ma W, Vinade L, Kulkarni A B, Brady R O, Pant H C. Proc Natl Acad Sci USA. 2001;98:12742–12747. doi: 10.1073/pnas.211428098. . (First Published October 2, 2001; 10.1073/pnas.211428098) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith D S, Greer P L, Tsai L H. Cell Growth Differ. 2001;12:277–283. [PubMed] [Google Scholar]
- 4.Maccioni R B, Otth C, Concha I I, Munoz J P. Eur J Biochem. 2001;268:1518–1527. doi: 10.1046/j.1432-1033.2001.02024.x. [DOI] [PubMed] [Google Scholar]
- 5.Ohshima T, Ward J M, Huh C G, Longenecker G, Veeranna, Pant H C, Brady R O, Martin L J, Kulkarni A B. Proc Natl Acad Sci USA. 1996;93:11173–11178. doi: 10.1073/pnas.93.20.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chae T, Kwon Y T, Bronson R, Dikkes P, Li E, Tsai L H. Neuron. 1997;18:29–42. doi: 10.1016/s0896-6273(01)80044-1. [DOI] [PubMed] [Google Scholar]
- 7.Ahlijanian M D, Barrezueta N X, Williams R D, Joaklowski A, Kowsz K P, McCarthy S, Coskran T, Carlo A, Seymour P A, Burkhardt J E, et al. Proc Natl Acad Sci USA. 2000;97:2910–2915. doi: 10.1073/pnas.040577797. . (First Published March 7, 2000; 10.1073/pnas.040577797) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moriyoshi K, Masu M, Ishii T, Shigemoto R, Mizuno N, Nakanishi S. Nature (London) 1991;354:31–37. doi: 10.1038/354031a0. [DOI] [PubMed] [Google Scholar]
- 9.Monyer H, Sprengel R, Schoepfer R, Herb A, Higuchi M, Lomeli H, Burnashev N, Sakmann B, Seeburg P H. Science. 1992;256:1217–1221. doi: 10.1126/science.256.5060.1217. [DOI] [PubMed] [Google Scholar]
- 10.Kennedy M B. Science. 2000;290:750–754. doi: 10.1126/science.290.5492.750. [DOI] [PubMed] [Google Scholar]
- 11.Cho K-O, Hunt C A, Kennedy M B. Neuron. 1992;9:929–942. doi: 10.1016/0896-6273(92)90245-9. [DOI] [PubMed] [Google Scholar]
- 12.Kornau H-C, Schenker L T, Kennedy M B, Seeburg P H. Science. 1995;269:1737–1740. doi: 10.1126/science.7569905. [DOI] [PubMed] [Google Scholar]
- 13.Strack S, Colbran R J. J Biol Chem. 1998;273:20689–20692. doi: 10.1074/jbc.273.33.20689. [DOI] [PubMed] [Google Scholar]
- 14.Gardoni F, Bellone C, Cattabeni F, Di Luca M. J Biol Chem. 2001;276:7609–7613. doi: 10.1074/jbc.M009922200. [DOI] [PubMed] [Google Scholar]
- 15.Bayer K U, De Koninck P, Leonard A S, Hell J W, Schulman H. Nature (London) 2001;411:801–805. doi: 10.1038/35081080. [DOI] [PubMed] [Google Scholar]
- 16.Omkumar R V, Kiely M J, Rosenstein A J, Min K-T, Kennedy M B. J Biol Chem. 1996;271:31670–31678. doi: 10.1074/jbc.271.49.31670. [DOI] [PubMed] [Google Scholar]
- 17.Zheng F, Gingrich M B, Traynelis S F, Conn P J. Nat Neurosci. 1998;1:185–191. doi: 10.1038/634. [DOI] [PubMed] [Google Scholar]
- 18.Cheung H H, Gurd J W. J Neurochem. 2001;78:524–534. doi: 10.1046/j.1471-4159.2001.00433.x. [DOI] [PubMed] [Google Scholar]
- 19.Liao G Y, Wagner D A, Hsu M H, Leonard J P. Mol Pharmacol. 2001;59:960–964. doi: 10.1124/mol.59.5.960. [DOI] [PubMed] [Google Scholar]
- 20.Nakazawa T, Komai S, Tezuka T, Hisatsune C, Umemori H, Semba K, Mishina M, Manabe T, Yamamoto T. J Biol Chem. 2001;276:693–699. doi: 10.1074/jbc.M008085200. [DOI] [PubMed] [Google Scholar]
- 21.Yang M, Leonard J P. J Neurochem. 2001;77:580–588. doi: 10.1046/j.1471-4159.2001.00255.x. [DOI] [PubMed] [Google Scholar]
- 22.Ali D W, Salter M W. Curr Opin Neurobiol. 2001;11:336–342. doi: 10.1016/s0959-4388(00)00216-6. [DOI] [PubMed] [Google Scholar]
- 23.Xiong Z G, Pelkey K A, Lu W Y, Lu Y M, Roder J C, MacDonald J F, Salter M W. J Neurosci. 1999;19:RC37. doi: 10.1523/JNEUROSCI.19-21-j0003.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roche K W, Standley S, McCallum J, Dune Ly C, Ehlers M D, Wenthold R J. Nat Neurosci. 2001;4:794–802. doi: 10.1038/90498. [DOI] [PubMed] [Google Scholar]
- 25.Vissel B, Krupp J J, Heinemann S F, Westbrook G L. Nat Neurosci. 2001;4:587–596. doi: 10.1038/88404. [DOI] [PubMed] [Google Scholar]