

Abstract
Bordetella pertussis causes whooping cough, a highly contagious respiratory disease that is reemerging in many world regions. The spread of antigen-deficient strains may threaten acellular vaccine efficacy. Dynamics of strain transmission are poorly defined because of shortcomings in current strain genotyping methods. Our objective was to develop a whole-genome genotyping strategy with sufficient resolution for local epidemiologic questions and sufficient reproducibility to enable international comparisons of clinical isolates. We defined a core genome multilocus sequence typing scheme comprising 2,038 loci and demonstrated its congruence with whole-genome single-nucleotide polymorphism variation. Most cases of intrafamilial groups of isolates or of multiple isolates recovered from the same patient were distinguished from temporally and geographically cocirculating isolates. However, epidemiologically unrelated isolates were sometimes nearly undistinguishable. We set up a publicly accessible core genome multilocus sequence typing database to enable global comparisons of B. pertussis isolates, opening the way for internationally coordinated surveillance.
Keywords: Bordetella pertussis, whole-genome sequencing, multilocus sequence typing, nomenclature database, phylogeny, epidemiology, bacteria, pertussis, whooping cough, surveillance
Whooping cough (or pertussis) is a vaccine-preventable disease caused mainly by the bacterium Bordetella pertussis, and to a lesser extent by B. parapertussis. The infection is most severe in infants who are too young to be vaccinated or are not yet fully vaccinated. The burden of disease is still high: 24 million pertussis cases and 160,700 deaths from pertussis in children <5 years of age in 2014 (1). The introduction of vaccination using whole-cell vaccines in the 1950s, and the switch to acellular vaccines targeting only some antigens in the 1980–1990s, have played a central role in the control of whooping cough. However, increasing incidence of the disease and large outbreaks have been reported recently in many countries (2–5). The observed resurgence of whooping cough underlines the need for reinforced surveillance of strain evolution, local spread, and global transmission. For example, the relative contributions of intercountry spread compared with local, independent evolution of strains that do not express pertactin (6–8), one of the components of acellular vaccines, are unknown. This gap limits our ability to interpret the local prevalence of pertactin-negative isolates and to define the effects of country-specific vaccine strategies on the emergence of antigen-deficient isolates.
Until now, strain genotyping for surveillance and epidemiology has been based mostly on pulsed-field gel electrophoresis (PFGE), antigen and virulence factor genotyping, 7-gene multilocus sequence typing (MLST), or multilocus variable-number tandem-repeat analysis (MLVA) (9–11). PFGE achieves some level of resolution given the high structural dynamics of B. pertussis genomes, driven by insertion sequence elements dynamics (12), and is more discriminatory than MLVA or MLST. However, B. pertussis clinical isolates exhibit strong genetic homogeneity (11,13). Therefore, these traditional typing methods have largely failed to define local chains of transmission.
Whole-genome sequencing (WGS) provides the highest possible resolution of genetic differences among individual isolates. Working with WGS of an international collection of B. pertussis isolates collected through 2010, Bart et al. (13) provided a global phylogenetic structure of B. pertussis and analyzed genome evolutionary dynamics across the prevaccine and vaccine eras. This pioneering study found genotype mixing across countries at shallow phylogenetic depth, revealing frequent long-distance spread of B. pertussis isolates and underlining the importance of defining standard genotyping methods that would allow tracing international transmission. Core genome MLST (cgMLST), using the set of genes conserved among isolates of a given bacterial group, represents an approach that combines the high resolution of genome-level variation and the high reproducibility and portability of MLST (14). cgMLST genotyping strategies were recently implemented for international coordinated surveillance of several pathogenic bacterial species (15–20).
We report on the development and evaluation of a cgMLST scheme for genotyping of B. pertussis clinical isolates. We demonstrate the resolution power of this approach to recognize groups of intrafamilial isolates or multiple isolates recovered from the same patient. We also show that, in some cases, temporally or geographically unrelated isolates can be nearly undistinguishable, illustrating the rapid diffusion of isolates through hidden chains of transmission. We made the cgMLST strategy for B. pertussis isolate characterization publicly available through a Web-accessible genotyping platform (http://bigsdb.pasteur.fr/bordetella), providing a novel tool for tracking the international spread of B. pertussis variants.
Materials and Methods
Isolates and DNA Preparation
We sequenced a set of 55 isolates (Technical Appendix 1 Table 1). Of these, 24 isolates corresponded to 11 related groups of isolates: 8 isolates originated from 4 different pairs of intrafamilial transmission cases and 16 isolates corresponded to multiple isolates collected from 7 patients (6 pairs and 1 quadruplet); 30 corresponded to a random selection of temporally cocirculating isolates. We used as reference the Tohama isolate (GenBank accession no. NC_002929).
We grew isolates at 36°C for 72 hours on Bordet-Gengou agar (Becton Dickinson, Le Pont de Claix, France) supplemented with 15% defibrinated horse blood (BioMérieux, Marcy l’Étoile, France) and subcultured them in the same medium for 24 hours. We suspended the bacteria in physiologic salt to reach an optical density at 650 nm of 1, and pelleted 400 μL. We suspended the pellets in 100 μL of 1× phosphate-buffered saline, 100 μL of lysis buffer (Roche), and 40 μL of proteinase K; heated them at 65°C for 10 minutes and then at 95°C for 10 minutes; and used them for DNA extraction.
PFGE
We obtained PFGE profiles using the XbaI enzyme, as described previously (9,21). We conduated analyses by using BioNumerics version 6.6 (Applied-Maths, Sint-Martens-Latem, Belgium).
MLVA Analysis
We identified variable-number tandem-repeat (VNTR) sequences (22) on each whole-genome sequence using blastn (https://blast.ncbi.nlm.nih.gov/Blast) with Tohama alleles as query. To define Tohama alleles, we located the loci using the primer sequences defined for each locus (VNTR-1, -3, -4, -5, and -6) in the Protocols and Tables section of the Netherlands’ National Institute for Public Health and the Environment’s MLVA website (http://www.mlva.net/bpertussis/default.asp). We defined alleles by counting the number of repeats in the retrieved sequences. We then determined MLVA types using the Single Profile Query section at the same website.
WGS, Definition of the Core Genome, and Data Analysis
We describe WGS, our definition of the core genome, and data analysis in Technical Appendix 1. The study accession number in the European Nucleotide Archive is PRJEB21744, including samples ERS1869830–ERS1869884 and their corresponding sequence data.
Results
Constitution of the cgMLST Scheme
We identified protein-coding genes of B. pertussis that were found in >95% of a set of 300 genomes of B. pertussis gathered from publicly available data and from our sequencing of isolates from France. We subjected these genes to several filters designed to ensure robustness of genotyping data (Technical Appendix 1). We then chased artifactual variation of allele calls using assemblies available for 3 reference strains obtained from different sequencing methods and assemblies of 17 isolates from France sequenced with different Illumina (San Diego, CA, USA) sequencing systems (HiSeq and NextSeq; Technical Appendix 1 Table 2). We also assessed the dependency of allele calls to assembly coverage depth, by using randomly selected read pairs from raw sequencing data of 1 isolate (FR6072), and eliminated the loci that showed variation above 20× coverage depth. These steps led to a final set of 2,038 gene loci, together constituting a B. pertussis cgMLST scheme that should minimize artifactual variation caused by the use of different sequencing platforms or sequencing depths. The set of 2,038 core genes had a total length of 1,751,253 bp, covering 42.9% of the Tohama reference genome. The median gene length was ≈1,000 bp (Technical Appendix 1 Figure 1). Eleven loci were >3,000 bp long and corresponded to genes encoding large proteins such as BrkA, DnaE, RpoB or CyaA (Technical Appendix 2). Most core genes had <10 alleles within our selection of 300 B. pertussis genomes used to define the cgMLST scheme, consistent with previous estimates of sequence variation within this homogeneous pathogenic species (11,13). Core genes belonged to diverse gene classification categories (Technical Appendix 1 Figure 2).
Phylogenetic Analysis of cgMLST
Phylogenetic analysis of the 55 isolates of the study based on concatenated alignments of the 2,038 gene sequences showed 2 early diverging branches comprising the reference strain Tohama, which belongs to the early-diverging ptxP1 clade (13), and ptxP1 strain FR6022 (Figure 1). Of the clinical isolates, 49 belonged to clade ptxP3 and 4 belonged to the previously described clade ptxP21, which is derived from ptxP3 (23) (Technical Appendix 1 Table 1). These 53 non-ptxP1 isolates were separated according to their fim3 allele, either fim3-1 or fim3-2. These results are congruent with previous phylogenetic analyses (13). Cluster analysis of the cgMLST allelic profiles led to a very similar grouping of isolates (Technical Appendix 1 Figure 3), indicating that this method can be used for rapid classification purposes.
Figure 1.
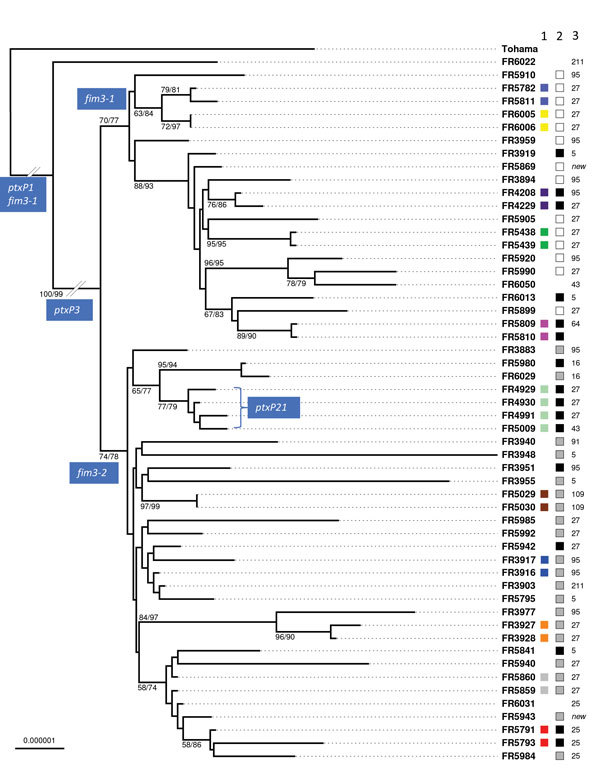
Maximum-likelihood phylogenetic tree for Bordetella pertussis isolates based on the concatenated multiple sequence alignments of 2,038 core genome multilocus sequence typing loci. The tree was rooted on the Tohama reference isolate (GenBank accession no. NC_002929). Only branch support values >50 are labeled (bootstrap/aLRT-SH). Column 1 to the right of the isolates’ names shows colors indicating the intrafamilial groups or groups of multiple isolates from single patients (corresponding to colors shown in inner circle of Figure 2). Column 2 shows pulsed-field gel electrophoresis types (French nomenclature; white, IVα; gray, IVβ; black, IVγ). Column 3 shows multilocus variable-number tandem-repeat analysis types. Scale bar indicates nucleotide substitutions per site.
Groups of intrafamilial or of multiple isolates from the same patient were largely distributed across the phylogenetic tree (Figure 1). Although most groups formed a distinct branch (Figure 1), 3 sets of isolates (FR3916 and FR3917, FR5859 and FR5860, and FR5791 and FR5793), were intermixed in the tree with isolates with no documented epidemiologic link.
Numbers of Allelic Differences among Epidemiologically Related or Nonrelated Isolates
When considering the 55 isolates of the study, we found only 1 or 2 uncalled allele(s) among the 2,038 loci of the cgMLST scheme: 53.6% of the isolates had 2,038 core genes tagged, 42.8% had 1 missing allele, and 3 (5.4%) isolates had 2 missing alleles. We evaluated the pairwise comparisons of allelic profiles and recorded the number of mismatches, defined as allelic differences at loci where both isolates had an allele called. We found the highest numbers of allelic mismatches (close to 66 allelic differences) for the comparisons of non-PtxP1 isolates with the Tohama reference strain, consistent with this strain belonging to a distant lineage. In turn, comparisons of the ptxP1 isolate FR6022 with the non-ptxP1 clinical isolates showed 15–25 allelic differences. Among non-ptxP1 clinical isolates, the number of allelic mismatches varied from 0 to 15 (median 9). All pairwise comparisons between pairs of intrafamilial isolates or pairs of multiple isolates collected from the same patient showed a median value of 1 allelic mismatch (maximum 2). However, 2.8% (39 of 1,415) pairs of nonrelated isolates also showed either 1 or 2 mismatches only.
Comparison of cgMLST with PFGE and MLVA
Based on PFGE, all clinical isolates belonged to PFGE group IV. This group is highly predominant in France among contemporaneous (post-2000) isolates, and is subdivided into 3 different subgroups, IVα, IVβ, and IVγ (24,25), which were all represented in our selection. Analysis of the distribution of PFGE profiles along the cgMLST-based phylogenetic tree (Figure 1) revealed that subgroups IVα and IVβ were separated into 2 clades, corresponding with fim3-1 (associated with IVα) and fim3-2 (associated with IVβ). In contrast, PFGE subgroup IVγ was found interspersed in these 2 clades, indicating that it does not represent a natural (monophyletic) grouping of B. pertussis isolates. As expected, the same PFGE subgroups were shared by nonrelated and related isolates (Figure 1).
We extracted MLVA profiles from whole-genome sequence assemblies. The main MLVA types were MLVA-27 (38.9%), MLVA-95 (18.5%), MLVA-5 (11.1%), and MLVA-25 (7.4%). The first 3 genotypes were distributed widely across the phylogenetic tree (Figure 1), indicating that, similar to PFGE subgroups, they do not represent proper phylogenetic clades.
Comparison of cgMLST with a Whole-Genome Single Nucleotide Polymorphism–Based Approach
We used a mapping approach against the Tohama strain genome as reference (GenBank accession no. NC_002929), and compared the derived single-nucleotide polymorphism (SNP)–based phylogenetic tree to the one inferred from the concatenated multiple sequence alignments obtained from the cgMLST loci (Technical Appendix 1 Figure 4). Both approaches were highly congruent, grouping the isolates in nearly identical clades. The SNP-based approach led to the identification of 721 variable positions. The highest numbers of SNPs were found for the comparisons of recent isolates (ptxP3 and ptxP21) with the Tohama reference strain (266 ± 10 SNPs) and with the ptxP1 isolate FR6022 (98 ± 5 SNPs). Among unrelated ptxP3 clinical isolates, the number of SNPs was 34 ± 9. In the cgMLST gene sequences, there were 206 variable positions, 83.5% of which were included in those identified in the SNP-based analysis. The genome-wide SNP approach might thus be useful as a complementary approach when very high resolution is needed. Comparisons among the 11 related cases showed a very low number of SNPs (no SNP in 9 out of 11 comparisons, 1 SNP in 1 comparison, and 2 SNPs in the remaining comparison), consistent with the cgMLST results. When considering the 3 pairs of isolates not fully resolved using cgMLST, we noticed that FR3916 and FR3917 displayed no SNP between each other, and no SNP with cocirculating isolate FR3903; FR5859 and FR5860 displayed no SNP between each other but >18 SNPs compared with FR5940 and 12 SNPs compared with FR5841; and FR5791 and FR5793 displayed no SNP between each other but 3 or 4 SNPs with cocirculating isolate FR5984. These observations show that, except for the first case, whole-genome SNPs discriminate the related pairs from epidemiologically nonrelated isolates better than cgMLST does. Altogether, these results emphasize that, for the highly monomorphic B. pertussis, genotyping data will need to be complemented with epidemiologic data to unravel transmission chains.
Application of cgMLST to Study of Outbreaks from Different Countries
We analyzed publicly available whole-genome sequences corresponding to 3 outbreaks that occurred in California and Vermont (26,27), USA, and in the United Kingdom (4) (Technical Appendix 1 Table 3). Figure 2 illustrates the phylogenetic relationships of these isolates compared with those from France, based on cgMLST gene sequences. We observed that all pairs of intrafamilial isolates from France and all pairs of multiple isolates recovered from the same patient remained grouped. Isolates from each of the 3 US and UK outbreaks were found in different branches of the phylogenetic tree, consistent with previous results showing that they did not result from the spread of a unique strain (4). This finding confirms that the outbreaks of pertussis disease we analyzed corresponded to the simultaneous emergence of multiple strains, consistent with the hypothesis of the silent maintenance of a genetically heterogeneous pool of B. pertussis strains in the human population (4,27).
Figure 2.
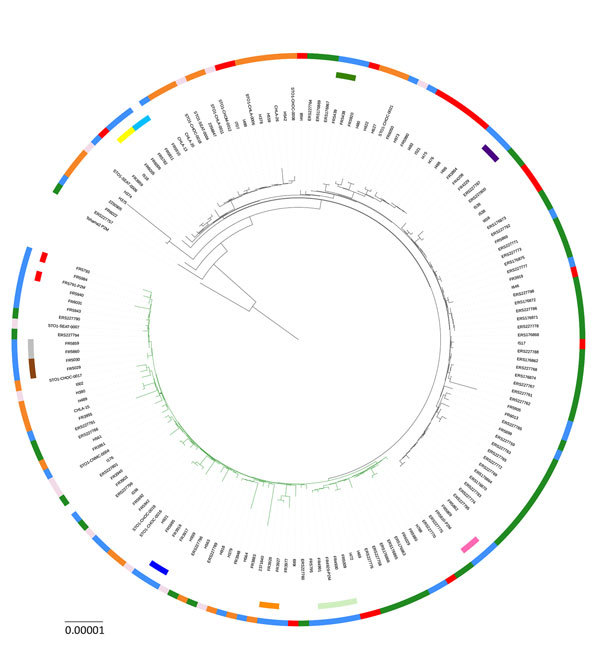
Maximum-likelihood phylogenetic tree for Bordetella pertussis based on the concatenated alignments of the 2,038 cgMLST loci sequences of isolates from France (this study) and isolates from outbreaks in the United States and the United Kingdom. The tree was rooted on the Tohama reference isolate (GenBank accession no. NC_002929). Black tree branches indicate fim3-1 clade and green tree branches indicate fim3-2 clade. Intrafamilial groups of isolates and multiple groups of isolates recovered from the same patient are represented by sectors of the internal circle surrounding the tree (corresponding to colors in column 1 of Figure 1). The external circle indicates the geographic origin of isolates (blue, France; red, Vermont, USA; orange, California, USA; light pink, other United States; green, United Kingdom). Scale bar indicates nucleotide substitutions per site.
Discussion
We developed a cgMLST scheme for B. pertussis, one of the most monomorphic bacterial pathogens (11,13,28). Resolving groups of related isolates (such as intrafamilial cases or multiple isolates recovered from the same patient) from nonrelated cocirculating isolates is challenging, given that the B. pertussis population is very homogeneous. Consistent with expectations, few of the 2,038 gene loci of the genotyping scheme were variable among non-ptxP1 isolates. Despite this low level of genetic diversity, the cgMLST scheme grouped most sets of isolates from direct transmission chains and distinguished them most of the time from cocirculating unrelated isolates. Therefore, this genotyping approach will help define chains of transmission of B. pertussis. Nevertheless, close genetic relatedness does not demonstrate direct epidemiologic relatedness. Conversely, isolates from different outbreaks (as defined by the notable increase of clinical cases in defined geographic areas) were genetically heterogeneous, demonstrating a diversity of isolates circulating during pertussis epidemics.
Given our ambition to develop a universally applicable cgMLST B. pertussis genotyping scheme, we selected the core genes using 300 B. pertussis genomes of international origins. Although we cannot exclude that some sublineages of B. pertussis, not represented among the 300 isolates, may have lost several of the core loci selected here, we regard this possibility as unlikely and believe that most cgMLST gene loci selected here will be present in most B. pertussis isolates.
Remarkably, cgMLST was much more discriminant than PFGE or MLVA, 2 reference epidemiologic typing methods, which appeared unable to distinguish related intrafamilial isolates or isolates collected from the same patient from cocirculating unrelated ones. PFGE and MLVA are widely used and will likely continue to be used until WGS is largely implemented (29–32), especially in settings in which WGS cannot be achieved because of cost considerations. Our work provides a correspondence between whole-genome–based phylogenetic data and both PFGE and MLVA genotypes and thus provides essential information for the accurate interpretation of typing data from these legacy typing methods.
Even though standardization efforts have been made, it remains difficult to compare PFGE data from different countries. Using isolates from France and the French nomenclature for PFGE profiles (9), we demonstrated that some PFGE subtypes, such as type IV-γ, conflate phylogenetically distinct B. pertussis sublineages. MLVA typing is more comparable across laboratories but even less discriminatory than PFGE. In contrast to these 2 legacy typing methods, the phylogenetic tree based on cgMLST genes was highly congruent with that obtained from whole-genome SNPs. Therefore, the cgMLST loci, used in combination, represent powerful phylogenetic markers and will enable identification of meaningful groupings of B. pertussis isolates. The cgMLST scheme developed here may therefore be a powerful approach for identifying emerging B. pertussis sublineages. Because the cgMLST scheme covers only 43% of the genome, a complementary full-genome analysis will be required to define the particular biologic features, such as the loss of vaccine antigen expression, of emerging sublineages.
The cgMLST scheme developed here forms the basis of a unified allele nomenclature database, which was made openly accessible online at http://bigsdb.pasteur.fr/bordetella. This novel gene-by-gene genotyping strategy (14) opens the prospects of an internationally unified surveillance, whereby emerging genotypes and sublineages can be recognized in real time by surveillance laboratories. The ability of different national reference centers, microbiology laboratories, and public health agencies to compare B. pertussis genotypes will facilitate understanding of transmission dynamics. Further, the harmonization of epidemiologic typing practice by the use of the same genotyping approach will facilitate sharing of experiences among national surveillance systems and has the potential to promote collaboration. Finally, phylogenetic comparisons of isolates from different countries and world regions will facilitate the much-needed studies of the impact of whole-cell or acellular vaccines and the various vaccination strategies in use (33,34) on the transmission success of particular B. pertussis lineages, such as those that evolve toward a lack of expression of vaccine antigens.
Description of Bordetella pertussis whole-genome sequencing and analysis processes.
Bordetella pertussis core gene characteristics.
Acknowledgments
We thank the Collection of the Institut Pasteur for the gift of the CIP 8132 isolate and the Renacoq Network for collecting and sending B. pertussis isolates. We thank 3 anonymous reviewers for their critical reading of earlier versions of the manuscript, which led to important improvements. We also thank the Mutualized Platform for Microbiology of Institut Pasteur for genome sequencing using Illumina NextSeq technology and the Genomics Platform of Institut Pasteur for genome sequencing on Illumina HiSeq.
This work was performed in the Institut Pasteur research unit Biodiversity and Epidemiology of Bacterial Pathogens, which hosts the French National Reference Center for Whooping Cough and other Bordetelloses. This work was supported by the Institut Pasteur, Santé Publique France, and the charitable association Clara Belliveau.
Biography
Dr. Bouchez is a scientist at Institut Pasteur, Paris, France. Her research focuses on Bordetella spp. diversity and evolution in the context of vaccination.
Footnotes
Suggested citation for this article: Bouchez V, Guglielmini J, Dazas M, Landier A, Toubiana J, Guillot S, et al. Genomic sequencing of Bordetella pertussis for epidemiology and global surveillance of whooping cough. Emerg Infect Dis. 2018 June [date cited]. https://doi.org/10.3201/eid2406.171464
References
- 1.Yeung KHT, Duclos P, Nelson EAS, Hutubessy RCW. An update of the global burden of pertussis in children younger than 5 years: a modelling study. Lancet Infect Dis. 2017;17:974–80. 10.1016/S1473-3099(17)30390-0 [DOI] [PubMed] [Google Scholar]
- 2.Crespo I, Broner S, Soldevila N, Martínez A, Godoy P, Sala-Farré MR, et al. Characteristics of pertussis outbreaks in Catalonia, Spain, 1997 to 2010. Hum Vaccin Immunother. 2015;11:231–5. 10.4161/hv.36156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Safarchi A, Octavia S, Wu SZ, Kaur S, Sintchenko V, Gilbert GL, et al. Genomic dissection of Australian Bordetella pertussis isolates from the 2008-2012 epidemic. J Infect. 2016;72:468–77. 10.1016/j.jinf.2016.01.005 [DOI] [PubMed] [Google Scholar]
- 4.Sealey KL, Harris SR, Fry NK, Hurst LD, Gorringe AR, Parkhill J, et al. Genomic analysis of isolates from the United Kingdom 2012 pertussis outbreak reveals that vaccine antigen genes are unusually fast evolving. J Infect Dis. 2015;212:294–301. 10.1093/infdis/jiu665 [DOI] [PubMed] [Google Scholar]
- 5.Winter K, Glaser C, Watt J, Harriman K; Centers for Disease Control and Prevention (CDC). Pertussis epidemic—California, 2014. MMWR Morb Mortal Wkly Rep. 2014;63:1129–32. [PMC free article] [PubMed] [Google Scholar]
- 6.Bouchez V, Brun D, Cantinelli T, Dore G, Njamkepo E, Guiso N. First report and detailed characterization of B. pertussis isolates not expressing Pertussis Toxin or Pertactin. Vaccine. 2009;27:6034–41. 10.1016/j.vaccine.2009.07.074 [DOI] [PubMed] [Google Scholar]
- 7.Zeddeman A, Witteveen S, Bart MJ, van Gent M, van der Heide HG, Heuvelman KJ, et al. Studying Bordetella pertussis populations by use of SNPeX, a simple high-throughput single nucleotide polymorphism typing method. J Clin Microbiol. 2015;53:838–46. 10.1128/JCM.02995-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pawloski LC, Queenan AM, Cassiday PK, Lynch AS, Harrison MJ, Shang W, et al. Prevalence and molecular characterization of pertactin-deficient Bordetella pertussis in the United States. Clin Vaccine Immunol. 2014;21:119–25. 10.1128/CVI.00717-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Advani A, Hallander HO, Dalby T, Krogfelt KA, Guiso N, Njamkepo E, et al. Pulsed-field gel electrophoresis analysis of Bordetella pertussis isolates circulating in Europe from 1998 to 2009. J Clin Microbiol. 2013;51:422–8. 10.1128/JCM.02036-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Gent M, Heuvelman CJ, van der Heide HG, Hallander HO, Advani A, Guiso N, et al. Analysis of Bordetella pertussis clinical isolates circulating in European countries during the period 1998–2012. Eur J Clin Microbiol Infect Dis. 2015;34:821–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diavatopoulos DA, Cummings CA, Schouls LM, Brinig MM, Relman DA, Mooi FR. Bordetella pertussis, the causative agent of whooping cough, evolved from a distinct, human-associated lineage of B. bronchiseptica. PLoS Pathog. 2005;1:e45. 10.1371/journal.ppat.0010045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weigand MR, Peng Y, Loparev V, Batra D, Bowden KE, Burroughs M, et al. The history of Bordetella pertussis genome evolution includes structural rearrangement. J Bacteriol. 2017;199:e00806-16. 10.1128/JB.00806-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bart MJ, Harris SR, Advani A, Arakawa Y, Bottero D, Bouchez V, et al. Global population structure and evolution of Bordetella pertussis and their relationship with vaccination. MBio. 2014;5:e01074. 10.1128/mBio.01074-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maiden MC, Jansen van Rensburg MJ, Bray JE, Earle SG, Ford SA, Jolley KA, et al. MLST revisited: the gene-by-gene approach to bacterial genomics. Nat Rev Microbiol. 2013;11:728–36. 10.1038/nrmicro3093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moura A, Tourdjman M, Leclercq A, Hamelin E, Laurent E, Fredriksen N, et al. Real-time whole-genome sequencing for surveillance of Listeria monocytogenes, France. Emerg Infect Dis. 2017;23:1462–70. 10.3201/eid2309.170336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moura A, Criscuolo A, Pouseele H, Maury MM, Leclercq A, Tarr C, et al. Whole genome-based population biology and epidemiological surveillance of Listeria monocytogenes. Nat Microbiol. 2016;2:16185. 10.1038/nmicrobiol.2016.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bennett JS, Jolley KA, Maiden MC. Genome sequence analyses show that Neisseria oralis is the same species as ‘Neisseria mucosa var. heidelbergensis’. Int J Syst Evol Microbiol. 2013;63:3920–6. 10.1099/ijs.0.052431-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cody AJ, Bray JE, Jolley KA, McCarthy ND, Maiden MCJ. Core genome multilocus sequence typing scheme for stable, comparative analyses of Campylobacter jejuni and C. coli human disease isolates. J Clin Microbiol. 2017;55:2086–97. 10.1128/JCM.00080-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bialek-Davenet S, Criscuolo A, Ailloud F, Passet V, Jones L, Delannoy-Vieillard AS, et al. Genomic definition of hypervirulent and multidrug-resistant Klebsiella pneumoniae clonal groups. Emerg Infect Dis. 2014;20:1812–20. 10.3201/eid2011.140206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Been M, Pinholt M, Top J, Bletz S, Mellmann A, van Schaik W, et al. Core genome multilocus sequence typing scheme for high-resolution typing of Enterococcus faecium. J Clin Microbiol. 2015;53:3788–97. 10.1128/JCM.01946-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weber C, Boursaux-Eude C, Coralie G, Caro V, Guiso N. Polymorphism of Bordetella pertussis isolates circulating for the last 10 years in France, where a single effective whole-cell vaccine has been used for more than 30 years. J Clin Microbiol. 2001;39:4396–403. 10.1128/JCM.39.12.4396-4403.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schouls LM, van der Heide HG, Vauterin L, Vauterin P, Mooi FR. Multiple-locus variable-number tandem repeat analysis of Dutch Bordetella pertussis strains reveals rapid genetic changes with clonal expansion during the late 1990s. J Bacteriol. 2004;186:5496–505. 10.1128/JB.186.16.5496-5505.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bouchez V, Hegerle N, Strati F, Njamkepo E, Guiso N. New data on vaccine antigen deficient Bordetella pertussis isolates. Vaccines (Basel). 2015;3:751–70. 10.3390/vaccines3030751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hegerle N, Paris AS, Brun D, Dore G, Njamkepo E, Guillot S, et al. Evolution of French Bordetella pertussis and Bordetella parapertussis isolates: increase of Bordetellae not expressing pertactin. Clin Microbiol Infect. 2012;18:E340–6. [DOI] [PubMed] [Google Scholar]
- 25.Hallander H, Advani A, Riffelmann M, von König CH, Caro V, Guiso N, et al. Bordetella pertussis strains circulating in Europe in 1999 to 2004 as determined by pulsed-field gel electrophoresis. J Clin Microbiol. 2007;45:3257–62. 10.1128/JCM.00864-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harvill ET, Goodfield LL, Ivanov Y, Meyer JA, Newth C, Cassiday P, et al. Genome sequences of 28 Bordetella pertussis U.S. outbreak strains dating from 2010 to 2012. Genome Announc. 2013;1:e01075-13. 10.1128/genomeA.01075-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bowden KE, Weigand MR, Peng Y, Cassiday PK, Sammons S, Knipe K, et al. Genome structural diversity among 31 Bordetella pertussis isolates from two recent U.S. whooping cough statewide epidemics. mSphere. 2016;1:e00036–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Achtman M. Evolution, population structure, and phylogeography of genetically monomorphic bacterial pathogens. Annu Rev Microbiol. 2008;62:53–70. 10.1146/annurev.micro.62.081307.162832 [DOI] [PubMed] [Google Scholar]
- 29.Bailon H, León-Janampa N, Padilla C, Hozbor D. Increase in pertussis cases along with high prevalence of two emerging genotypes of Bordetella pertussis in Perú, 2012. BMC Infect Dis. 2016;16:422. 10.1186/s12879-016-1700-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haghighi F, Shahcheraghi F, Abbasi E, Eshraghi SS, Zeraati H, Mousavi SA, et al. Genetic profile variation in vaccine strains and clinical isolates of Bordetella pertussis recovered from Iranian patients. Avicenna J Med Biotechnol. 2014;6:178–84. [PMC free article] [PubMed] [Google Scholar]
- 31.Rocha EL, Leite D, Camargo CH, Martins LM, Silva RSN, Martins VP, et al. The characterization of Bordetella pertussis strains isolated in the Central-Western region of Brazil suggests the selection of a specific genetic profile during 2012-2014 outbreaks. Epidemiol Infect. 2017;145:1392–7. 10.1017/S0950268816003332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cassiday PK, Skoff TH, Jawahir S, Tondella ML. Changes in predominance of pulsed-field gel electrophoresis profiles of Bordetella pertussis isolates, United States, 2000–2012. Emerg Infect Dis. 2016;22:442–8. 10.3201/eid2203.151136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carbonetti NH. Bordetella pertussis: new concepts in pathogenesis and treatment. Curr Opin Infect Dis. 2016;29:287–94. 10.1097/QCO.0000000000000264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carbonetti NH, Wirsing von König CH, Lan R, Jacob-Dubuisson F, Cotter PA, Deora R, et al. Highlights of the 11th International Bordetella Symposium: from basic biology to vaccine development. Clin Vaccine Immunol. 2016;23:842–50. 10.1128/CVI.00388-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Bordetella pertussis whole-genome sequencing and analysis processes.
Bordetella pertussis core gene characteristics.