

Abstract
Growth retardation is a constant feature of Noonan syndrome (NS) but its physiopathology remains poorly understood. We previously reported that hyperactive NS-causing SHP2 mutants impair the systemic production of insulin‐like growth factor 1 (IGF1) through hyperactivation of the RAS/extracellular signal-regulated kinases (ERK) signalling pathway. Besides endocrine defects, a direct effect of these mutants on growth plate has not been explored, although recent studies have revealed an important physiological role for SHP2 in endochondral bone growth. We demonstrated that growth plate length was reduced in NS mice, mostly due to a shortening of the hypertrophic zone and to a lesser extent of the proliferating zone. These histological features were correlated with decreased expression of early chondrocyte differentiation markers, and with reduced alkaline phosphatase staining and activity, in NS murine primary chondrocytes. Although IGF1 treatment improved growth of NS mice, it did not fully reverse growth plate abnormalities, notably the decreased hypertrophic zone. In contrast, we documented a role of RAS/ERK hyperactivation at the growth plate level since 1) NS-causing SHP2 mutants enhance RAS/ERK activation in chondrocytes in vivo (NS mice) and in vitro (ATDC5 cells) and 2) inhibition of RAS/ERK hyperactivation by U0126 treatment alleviated growth plate abnormalities and enhanced chondrocyte differentiation. Similar effects were obtained by chronic treatment of NS mice with statins. In conclusion, we demonstrated that hyperactive NS-causing SHP2 mutants impair chondrocyte differentiation during endochondral bone growth through a local hyperactivation of the RAS/ERK signalling pathway, and that statin treatment may be a possible therapeutic approach in NS.
Introduction
Noonan syndrome (NS; MIM # 163950) is a relatively common genetic disorder characterized by growth retardation, heart defects (i.e. pulmonary valve stenosis and hypertrophic cardiomyopathy), developmental delay/learning disability, and predisposition to myeloproliferative disorders (1–3). These features are shared with related syndromes, notably NS with multiple lentigines (NS-ML; MIM # 151100), NS with loose anagen hair (NS-LAH; MIM # 607721), cardiofaciocutaneous syndrome (CFCS; MIM # 115150), Costello syndrome (CS; MIM # 218040), neurofibromatosis type 1 (NF1; MIM # 162200), and Legius syndrome (LS; MIM # 611431). All of these syndromes are caused by germline mutations in genes encoding components or regulators of the RAS/extracellular signal-regulated kinases (ERK) cascade. This signalling pathway plays key roles in development and homeostasis by regulating various cellular processes (e.g. proliferation, differentiation, and survival) to adapt cell fate in response to its environment (e.g. sensing of growth factors and hormones) (3,4). This group of disorders is now termed RASopathies (4) and constitutes one of the largest groups of multiple congenital anomaly diseases known.
About half of NS patients have a missense mutation in the protein-tyrosine phosphatase non-receptor type 11 (PTPN11) gene, which encodes the Src-homology 2 domain-containing tyrosine phosphatase 2 (SHP2) (5). SHP2 is a widely expressed tyrosine phosphatase that is involved in organism development and homeostasis by controlling major growth factors/hormone-triggered signalling pathways, notably the RAS/ERK pathway. NS-causing PTPN11 mutations cause hyperactivation of SHP2 catalytic activity and are thus gain-of-function mutations, leading to enhanced RAS/ERK activation. Several studies in NS animal models have demonstrated that this hyperactivation underlies the development of heart, craniofacial defects, and cognitive deficits observed in NS (6–10).
Short stature, found in 80% of patients, is a major concern in NS, but its pathophysiology is still poorly understood (1–3). In vertebrates, the longitudinal growth of long bones is determined through endochondral ossification at the growth plate level (11). During this process, chondrocytes, derived from the condensation of undifferentiated mesenchymal cells, proceed through a series of proliferation and differentiation steps. First, small chondrocytes rapidly divide and form columns in the proliferating zone. Subsequently, in the pre- and early-hypertrophic zone, chondrocytes undergo hypertrophic differentiation, characterized by their enlarged cell size and the synthesis of collagen type 10 (COL10), which is then calcified by enzymes such as alkaline phosphatase (ALP). In the late-hypertrophic zone, terminally differentiated chondrocytes undergo apoptosis, and the cartilaginous calcified matrix is degraded, through the simultaneous action of matrix metalloproteinase (MMPs) and the invasion of blood vessels, along with osteoclasts, and replaced by bone. Endochondral ossification is highly regulated by multiple hormones, paracrine factors, and extracellular matrix molecules. The growth hormone (GH)/insulin‐like growth factor 1 (IGF1) axis is one of the main endocrine system that regulates endochondral bone growth. GH acts on the growth plate both indirectly, by regulating hepatic IGF1 production and serum levels of IGF1, and also directly, in part through local IGF1 production (12). Several clinical surveys have revealed that NS patients display GH insensitivity, associating high GH levels and low IGF1 levels (13,14). Consistent with these findings, we provided evidence that NS-causing SHP2 mutants impair the systemic production of IGF1 through hyperactivation of the RAS/ERK signalling pathway. Interestingly, inhibition of RAS/ERK activation by a specific inhibitor promotes an increase of IGF1 levels in vitro and in vivo, which is associated with a significant growth improvement in NS mice (15).
Besides endocrine defects, a direct effect of NS-causing SHP2 mutants on the growth plate has not been explored to date, although recent studies have revealed an important physiological role for SHP2 in endochondral bone formation. Thus, conditional Ptpn11-deficient mice develop severe growth plate and bone abnormalities (16–21). Moreover, in human, germline inactivating mutations of PTPN11 have been described in metachondromatosis, a rare inherited disorder associating multiple exostoses and enchondromas (22).
The present study aimed to evaluate, for the first time, the impact of hyperactive NS-associated SHP2 mutants on endochondral ossification and to determine the respective contribution of IGF1 deficiency and RAS/ERK hyperactivation.
Results
NS mice display homogeneous postnatal growth retardation without bone deformity
A mouse model of NS carrying a missense mutation of Ptpn11 (Ptpn11D61G/+, NS mice) and their wild-type littermates (Ptpn11+/+, WT mice) were compared. To avoid potential interference of gender, only male mice were investigated. First, we measured the weight and the anal-nasal length of NS mice and their WT littermates from 1 to 4 weeks of age. Compared with WT mice, NS mice exhibited decreased weight and body length by 2 weeks of age, which continued for at least 4 weeks of age (Fig. 1A), in agreement with previous reports (15). The difference in length between WT and NS mice worsened with age, being 6% at 2 weeks of age and reaching 10% at 4 weeks of age. Gross images and Faxitron X-ray images showed smaller skull length and homogenous growth retardation affecting the axial as well the appendicular skeleton, without obvious bone deformity (Fig. 1B and C).
Figure 1.

Homogeneous postnatal growth retardation in NS mice. (A) Growth curves of male Ptpn11+/+ (WT) (n=12) and Ptpn11D61G/+ (NS) (n=9) mice are shown. Compared with WT mice, NS mice were significantly shorter by 2 weeks of age (2-way repeated-measures ANOVA plus Bonferroni post-test; ***P<0.001, **P<0.01). (B and C) Representative gross images (B) and Faxitron X-ray composites images of entire skeleton, lower legs, and skull (C) of 4-week-old WT and NS mice. NS mice displayed shortened axial and appendicular skeletons, mid-facial hypoplasia, and rounded cranium, without obvious bone deformity.
Growth plate length is reduced in NS mice mostly due to a shortening of the hypertrophic zone
To investigate the mechanisms underlying growth retardation in NS mice, we next focused our analyses on epiphyseal growth plates. The histology of growth plates of proximal tibiae from 4-week-old NS mice and their WT littermates was examined after Alcian Blue and COL10 staining.
Histological analysis revealed a normal organization of the growth plate with columnar structures in the proliferating zone (PZ) and enlarged chondrocytes in the hypertrophic zone (HZ) in NS mice (Fig. 2A and B). However, measurements revealed that the length of the growth plate was significantly shorter in NS mice compared with their WT littermates, mostly due to a decrease of the length of the hypertrophic zone, and to a lesser extent of the proliferating zone (Fig. 2C).
Figure 2.

Decreased growth plate width in NS mice. Histological sections from tibiae of 4-week-old WT (n=12) and NS (n=9) mice. (A and B) Alcian blue (A) and COL10 (B) staining of growth plate. (C) Measurements of different chondrocyte zones, showing a decrease of the widths of the hypertrophic zone, and to a lesser extent of the proliferating zone, in NS mice. (D) PCNA immunostaining to evaluate chondrocyte proliferation. (E) Quantification of the number of cells positive for PCNA per surface in the proliferating zone of the growth plate. There was no difference between WT and NS mice. (F) Colorimetric TUNEL assay to evaluate chondrocyte apoptosis. Values are means ± SEM. Significant statistical differences between groups: **P<0.01, *P<0.05 (two-tailed Student’s t-test).
The decrease in the length of the hypertrophic zone observed in NS mice could be the result of a reduced pool of proliferating chondrocytes, impaired chondrocyte differentiation, or increased chondrocyte apoptosis. We therefore analyzed these different processes.
Chondrocyte proliferation, assessed by immunohistochemistry using an antibody against proliferating cell nuclear antigen (PCNA), was similar between WT and NS mice (Fig. 2D and E). Similarly, chondrocyte apoptosis, assessed by TUNEL analysis, was extremely limited and did not differ between WT and NS mice (Fig. 2F).
Chondrocyte differentiation is impaired in primary chondrocytes from NS mice
To explore chondrocyte proliferation and differentiation in vitro, we next evaluated the transcriptional changes that occur when chondrocytes mature from proliferating to hypertrophic stages (17). For this, quantitative RT-PCR analysis was performed on RNA from primary murine chondrocytes, isolated from WT and NS mice, at baseline and after 7 days of differentiation with ascorbic acid.
As expected, during the differentiation process of WT primary chondrocytes, expression of markers associated with the proliferating stage (Sox9, Col2a1, and Acan) decreased (Fig. 3A–C) whereas those of the pre- (Ihh, Runx2, Pthr1) (Fig. 3D–F) and early- (Alpl) (Fig. 3G and H) hypertrophic stages increased. In NS primary chondrocytes, transcripts for structural protein associated with the proliferating stage (Col2a1 and Acan) also decreased during the differentiation and were decreased to a slightly greater extent compared with WT primary chondrocytes; however, there was no significant difference in the expression of the transcription factor Sox9. In contrast, expression of transcripts associated with the pre- and early-hypertrophic stages was significantly lower in NS chondrocytes at baseline and during differentiation (Fig. 3D–H). In the late hypertrophic-stage, we evaluated the expression of Mmp13, a major metalloproteinase responsible for matrix degradation, and the expression of vascular endothelial growth factor-A (Vegfa), which promotes capillary invasion into the cartilaginous matrix. A trend of decreased expression of Mmp 13 and increased expression of Vegfa in NS primary chondrocytes was observed, but this did not reach statistical significance (Fig. 3I and J). Similarly, the expression of Rankl, which stimulates the formation of osteoclasts, tended to be increased in NS primary chondrocytes but without reaching statistical significance (Fig. 3K).
Figure 3.

NS-causing PTPN11 mutants impair chondrocyte differentiation. Expression of chondrocyte differentiation markers in WT (n=7) and NS (n=7) primary murine chondrocytes, as determined by quantitative RT-PCR at baseline (T0) and after 7 days (T7) of differentiation with ascorbic acid. (A–K) Markers of the proliferating (A–C), pre- (D–F), early- (G and H), and late- (I–K) hypertrophic stages. In NS primary chondrocytes, transcripts associated with pre- and early-hypertrophic stages were very low and did not change during differentiation, suggesting impaired differentiation. Results are expressed as a fold change in mRNA expression, compared with WT mice at baseline. Values are means±SEM. The statistical significance of fold-changes was determined by using a two-way ANOVA for comparison between T0 and T7, and a two-tailed Student’s t-test for comparison between WT and NS primary chondrocytes. Significant statistical differences between groups: ***P<0.001, **P<0.01, *P<0.05.
To further evaluate the impact of NS-causing SHP2 mutant on chondrocytes, we next performed functional assays in vitro. Firstly, to investigate chondrocyte proliferation, primary chondrocytes from WT and NS mice were cultured in the presence of fibroblast growth factor 2 (FGF2), a paracrine factor that plays an important role in endochondral ossification, and the proliferation rate was evaluated by the counting of the total cell number on the one hand and by the incorporation of 5-ethynil-2′-deoxyuridine (EdU, a cell proliferation marker incorporated in cells during the S-phase of the cell cycle) into DNA in the other hand. We observed a similar growth rate between WT and NS primary chondrocytes (Fig. 4B).
Figure 4.

Alkaline phosphatase staining and activity are decreased in NS primary murine chondrocytes. (A and B) Proliferation of WT (n=7) and NS (n=7) primary murine chondrocytes cultured with 10 ng/ml FGF2. (A) Counting of the total cell number at days 1, 2 and 3 showing that the cell number doubled the first 2 days and then reached a plateau without difference between NS and WT chondrocytes. (B) Incorporation of EdU, a cell proliferation marker incorporated in cells during the S-phase of the cell cycle, into DNA between days 1 and 2 and days 2 and 3. The incorporated EdU in DNA coupled to Oregon Green-azide was assessed by the fluorescence detected at an excitation/emission wavelength of 490/585 nm; results were expressed as percentage of WT fluorescence. The proliferation rate was similar between WT and NS chondrocytes. (C and D) Alkaline phosphatase staining and activity in WT (n=7) and NS (n=7) primary murine chondrocytes at baseline (T0) and after 7, 14, and 21 days of differentiation with ascorbic acid. (C) Representative images showing ALP staining. Note significant decreased in NS chondrocytes, compared with WT chondrocytes. (D) ALP activity was determined by fluorometric method using p-nitrophenol-phosphate as a substrate, and normalized to the total DNA content of each sample. ALP activity was significantly decreased in NS chondrocytes compared to WT chondrocytes. Values are means ± SEM. The statistical significance of fold changes was determined using a two-way repeated-measures ANOVA for comparison between T0 and T7 and a two-tailed Student’s t-test for comparison between WT and NS primary chondrocytes. Significant statistical differences between groups: ***P<0.001, **P<0.01, *P<0.05.
Next, to investigate chondrocyte differentiation, we measured ALP staining and activity, which are markers of chondrocyte hypertrophy, in primary murine chondrocytes cultured in differentiation medium with ascorbic acid for periods of time up to 21 days. ALP staining was markedly decreased in NS chondrocytes (Fig. 4C); this was evident as early as day 7 and persisted throughout the overall time-course. Similarly, ALP activity was significantly decreased from baseline to 21 days of differentiation (Fig. 4D).
Taken together, these data suggest that expression of NS-causing SHP2 mutant impairs chondrocyte differentiation during endochondral ossification.
NS-causing SHP2 mutants enhance RAS/ERK activation in chondrocytes in vivo and in vitro
Given the central role of RAS/ERK hyperactivation in the pathophysiology of NS and other RASopathies, we next evaluated if NS-associated SHP2 mutant alters RAS/ERK activation in chondrocytes in vivo and in vitro. First, we assessed RAS/ERK activation in vivo on histologic sections of growth plates by immunohistochemistry using an antibody against phosphoERK1/2. Interestingly, significantly increased ERK1/2 phosphorylation was observed in the hypertrophic chondrocytes of NS mice compared with those of WT littermates (Fig. 5A and B). We then monitored ERK1/2 phosphorylation in vitro in ATDC5 cells, a standard model for chondrocytes, expressing WT SHP2 or the D61Del NS-causing SHP2 mutant, a strong mutant which is closely related to the D61G allele borne by the NS mouse model. These cells were stimulated with FGF2 or GH. We found that the NS-causing SHP2 mutant significantly enhanced FGF2- and GH-induced ERK1/2 phosphorylation in comparison to WT SHP2. This was observed despite the much lower expression of the D61del mutant (Fig. 5C). In contrast, neither STAT5 nor AKT phosphorylation induced by FGF2 or GH was significantly modified in the presence of NS mutant (data not shown).
Figure 5.

NS mutants increase RAS/ERK activation in chondrocytes in vitro and in vivo. (A) Histological sections from tibiae of 4-week-old WT (n=8) and NS mice (n=8) processed for phosphoERK1/2 immunostaining. (B) Quantification of phosphoERK1/2-positive cells per surface in the proliferating and hypertrophic zones of the growth plate. Data are expressed as means ± SEM for two sections per mouse. Significant statistical differences between groups: ***P<0.001, **P<0.01, *P<0.05 (two-tailed Student’s t-test). (C) ATDC5 cells were transduced with adenovectors encoding SHP2-WT and D61del. These cells were either left unstimulated or stimulated with 400 ng/ml GH or 10 ng/ml FGF2 for the indicated times, lysed, and probed by western-blot analysis to determine the level of ERK1/2 phosphorylation with an anti-phospho-ERK1/2 antibody. Note the exposure time was much more important for GH than for FGF2, explaining the detection of bands under unstimulated conditions. Membranes were reprobed with anti-SHP2 and anti-tubulin antibodies to verify SHP2 expression and loading homogeneity. Results from three independent experiments were quantified using ImageJ software and expressed as means ± SEM. Only significant differences versus WT cells for the corresponding time are indicated: ***P<0.001, **P<0.01, *P<0.05 (two-tailed Student’s t-test).
These results suggest that NS-causing SHP2 mutants have a prominent dominant-positive effect on ERK1/2 activation in chondrocytes in vitro and in vivo.
NS-causing SHP2 mutants have a direct IGF1-independent effect on endochondral ossification through RAS/ERK hyperactivation
We previously reported that NS-causing SHP2 mutants impair the systemic production of IGF1 through a hyperactivation of the RAS/ERK signalling pathway and that inhibition of RAS/ERK activation is associated with a significant growth improvement in NS mice (15). We therefore investigated whether IGF1 supplementation or RAS/ERK inhibition could reverse growth plate abnormalities in NS mice. To this aim, we examined the histology of growth plates of proximal tibiae from 4-week-old NS mice and their WT littermates treated from 1 to 4 weeks of age with IGF1 or U0126, a selective inhibitor of MEK (the upstream ERK1/2 kinase), that blocks ERK1/2 phosphorylation and activation.
Interestingly, growth retardation of NS mice was partially corrected by IGF1 and U0126 treatments (Fig. 6A). However, histology of the growth plate revealed that IGF1 treatment led to an increase of the length of the proliferating zone without correcting the decreased hypertrophic zone, suggesting that IGF1 deficiency is not the unique mechanism responsible for growth plate defects in NS (Fig. 6B and Supplementary Material, Fig. S1). In contrast, U0126 treatment restored the length of the growth plate, by simultaneously increasing the length of the proliferating and hypertrophic zones (Fig. 6B and Supplementary Material, Fig. S1).
Figure 6.

RAS/ERK inhibition partially restores growth and growth plate abnormalities in NS mice. (A) Body length of 4-week-old WT and NS mice, treated i.p. from 1 to 4 weeks of age with vehicle (n WT/NS=11/10), IGF1 (4 mg/kg twice daily; n WT/NS=10/7) or U0126 (5 mg/kg daily; n WT/NS=11/6), are shown. IGF1 and U0126 treatments partially restored growth retardation in NS mice. (B) Measurement of growth plate zones widths after Alcian blue staining. IGF1 led to an increase of the length of the proliferating zone without correcting the hypertrophic zone shortening, while U0126 treatment restored the length of the growth plate, by simultaneously increasing the length of the proliferating and hypertrophic zones. (C and D) ALP expression determined by quantitative RT-PCR (C) and activity determined by fluorometric method using p-nitrophenol-phosphate used as a substrate (D), in WT (n=7) and NS (n=7) primary murine chondrocytes at baseline (T0) and after 7 days of differentiation with ascorbic acid, in the presence or absence of U0126 (10 μM) added to the culture medium. U0126 treatment restored ALP expression and activity in NS chondrocytes. Results of Alp expression are expressed as a fold change in mRNA expression compared with WT mice at baseline. ALP activity was normalized to the total DNA content of each sample. Values are means ± SEM. The statistical significance of fold changes was determined using a two-way repeated-measures ANOVA for comparison between T0 and T7 and a two-tailed Student’s t-test for comparison between WT and NS primary chondrocytes. Significant statistical differences between groups: ***P<0.001, **P<0.01, *P<0.05.
To further explore the effect of U0126 treatment on chondrocyte differentiation, we next evaluated ALP expression and activity in primary murine chondrocytes cultured in differentiation medium supplemented or not with U0126. We observed that this treatment restored ALP expression and activity in NS primary chondrocytes (Fig. 6C and D).
These data suggest that NS-causing SHP2 mutants likely have a direct IGF1-independent effect on endochondral ossification through RAS/ERK hyperactivation.
Statin treatment is associated with a significant improvement of impaired chondrocyte differentiation in vivo and in vitro
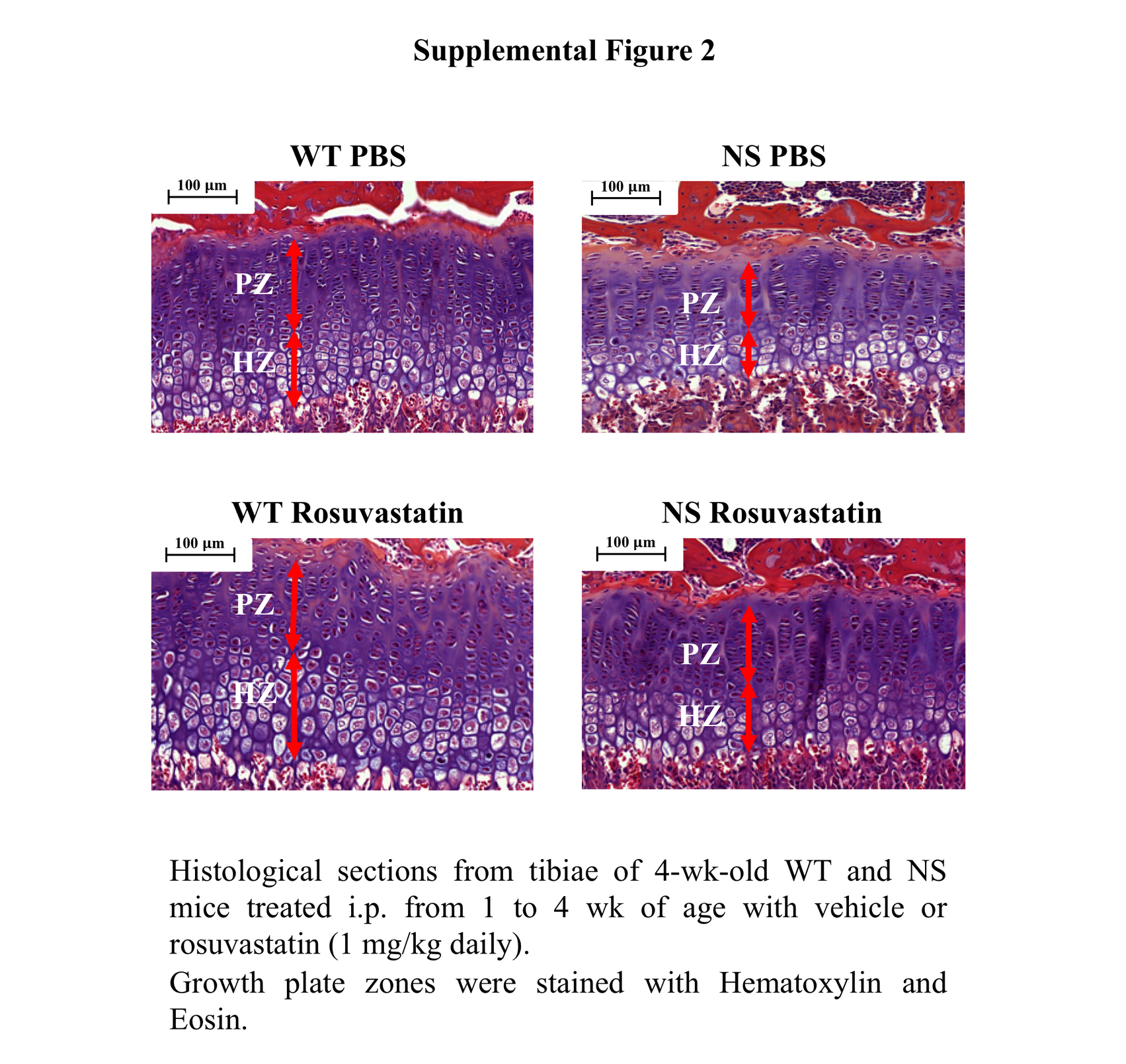
Recently, it has been reported that 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, also known as ‘statins’, can correct growth retardation and growth plate abnormalities in a mouse model of achondroplasia, a skeletal dysplasia associated with hyperactivation of RAS/ERK signalling at the growth plate level (23). We next investigated whether statin treatment may also improve endochondral bone growth in NS mice. To test this hypothesis, NS mice were treated with daily intraperitoneal injections of rosuvastatin from 1 to 4 weeks of age as previously described (23). Quite interestingly, statin treatment significantly improves growth of NS mice (Fig. 7A) and restored the length of the hypertrophic zone growth plate (Fig. 7B and Supplementary Material, Fig. S2).
Figure 7.

Inhibition of RAS/ERK activation by U0126 and statin treatments correct impaired chondrocyte differentiation in vitro. (A) Body length of 4-week-old WT and NS mice, treated i.p. from 1 to 4 weeks of age with vehicle (n WT/NS=12/9) or rosuvastatin (1 mg/kg daily; n WT/NS=12/8), are shown. Statin treatment partially restored growth retardation in NS mice. (B) Measurement of growth plate zone widths after Alcian blue staining. Rosuvastatin treatment restored the length of the growth plate, mostly by increasing the length of the hypertrophic zone. (C and D) ALP expression determined by quantitative RT-PCR (C) and activity determined by fluorometric method using p-nitrophenol-phosphate used as a substrate (D), in WT (n=7) and NS (n=7) primary murine chondrocytes at baseline (T0) and after 7 days of differentiation with ascorbic acid, in the presence or absence of lovastatin (1 μM) added to the culture medium. Lovastatin treatment restored ALP expression and activity in NS chondrocytes. Results of Alp expression are expressed as a fold change in mRNA expression compared with WT mice at baseline. ALP activity was normalized to the total DNA content of each sample. Values are means ± SEM. The statistical significance of fold changes was determined using a two-way repeated-measures ANOVA for comparison between T0 and T7 and a two-tailed Student’s t-test for comparison between WT and NS primary chondrocytes. Significant statistical differences between groups: ***P<0.001, **P<0.01, *P<0.05.
We next assessed the effect of statin treatment on chondrocyte differentiation in vitro by evaluating ALP expression and activity in primary murine chondrocytes cultured in differentiation medium with lovastatin treatment. Interestingly, we observed that this treatment restored ALP expression and activity in NS primary chondrocytes (Fig. 7C and D).
These data suggest that statin treatment may be a possible therapeutic approach in NS.
Discussion
In this study, we demonstrated that hyperactive, NS-causing SHP2 mutants impair ERK-dependent differentiation of chondrocyte during endochondral bone growth, a phenomenon partially restored by MEK inhibitor and statin treatments.
First, we found that growth retardation in NS mice was mostly due to an impairment of chondrocyte hypertrophic differentiation, as suggested by the decrease in the length of the hypertrophic zone on histological studies, the lower expression of transcripts associated with the pre- and early-hypertrophic stages, and the decreased alkaline phosphatase staining and activity during the differentiation of NS primary murine chondrocytes. Concerning the effect of NS-causing SHP2 mutants on chondrocyte proliferation, it was more difficult to conclude, the results depending on the techniques used. Indeed, decreased NS chondrocyte proliferation was suggested by the significant decrease in the length of the proliferating zone and the significant reduction of expression of some markers associated with the proliferative stage (Acan and Col2a1). In contrast, chondrocyte proliferation, assessed by PCNA immunostaining on histological sections and the proliferation rate of primary chondrocytes in vitro, was similar between WT and NS. These apparent discrepancies might be explained by the fact that reduced basal expression of some, but not all, markers of the proliferative stage may not be sufficient to impair chondrocyte proliferation in vitro. Moreover, additional defective mechanisms and/or signals may occur in vivo, resulting in reduced chondrocyte proliferation. In particular, IGF1 deficiency probably explains, at least in part, the reduced proliferative zone in NS, as it is corrected by IGF1 supplementation.
These findings mirror the histologic abnormalities found in different mouse models with PTPN11 inactivation. Indeed, expanded proliferating and hypertrophic zones have been reported in different mouse models with induced global deletion of Ptpn11 (16), inducible deletion of Ptpn11 in chondrocytes (17,19,21), or constitutive deletion of Ptpn11 in mesenchymal stem cells, the immediate precursors of chondrocytes (24). Moreover, our data suggest that hyperactive, NS-causing SHP2 mutant impairs chondrocyte early differentiation during endochondral ossification. This is consistent with previous reports suggesting that SHP2 negatively regulates the early maturation of chondrocytes and positively regulates their terminal differentiation (17,21).As a limitation of this study, we cannot rule out the possibility that hyperactive SHP2 mutants in cells other than chondrocytes also contribute to the aberrant skeletal growth of NS mice. A conditional expression of NS-causing SHP2 mutants specifically in hypertrophic chondrocytes would be helpful in addressing these possibilities.
We previously reported that NS-causing SHP2 mutants lead to impaired IGF1 production, which could contribute to growth retardation (15). Consistent with this notion, IGF1 supplementation improved growth velocity in NS mice. However, in our study, IGF1 treatment of NS mice increased the length of the proliferating zone without modifying the hypertrophic zone abnormalities. This is in accordance with the known action of IGF1 on the growth plate. Thus, mouse mutants lacking GH receptor, IGF1, or both display growth retardation mainly caused by decreased proliferation of chondrocytes (25). In contrast, increased GH signalling, caused by deletion of a negative regulator of GH signalling (SOCS2) leads to an expansion of the proliferating zone and subsequently the hypertrophic zone (26). These data could account for the limited efficacy of GH treatment on growth in NS patients (27). Indeed, GH treatment by increasing IGF1 levels probably stimulates the proliferation of chondrocytes but does not correct the abnormal chondrocyte differentiation.
As previously described in the pathophysiology of other defects associated with NS (i.e. heart, craniofacial defects, and cognitive deficits) (6–10), the hyperactivation of the RAS/ERK pathway appears to be crucial in the development of growth plate abnormalities. Thus, we demonstrated that expression of NS-associated SHP2 mutant results in ERK1/2 hyperactivation in chondrocytes in vitro and in vivo and that inhibition of ERK1/2 activation can alleviate the defective chondrocyte differentiation. In our study, it is important to note that growth retardation in NS mice was partially rescued by U0126 MEK inhibitor. However, this inhibitor, which has been used in previous studies (7,8,15), has a very short half-life and it is likely that MEK inhibitors with longer half-life would have a stronger effect. These findings are consistent with the key role of RAS/ERK signalling pathway in the process of endochondral ossification. Indeed, decreased chondrocyte hypertrophy, related to impaired differentiation, has been reported in different mouse models with overexpression of a constitutively active mutant of MEK in chondrocytes (28) or undifferentiated mesenchymal cells (29), as well as in a mouse model deficient for NF1 (another RASopathy), a negative regulator of RAS/ERK activation (30). In contrast, in a manner similar to SHP2 depletion, the inhibition of RAS/ERK activation leads to an increase of early differentiation and a decrease in terminal differentiation, and is associated with enchondroma-like lesions (17,21,29,31,32). Moreover, the growth plate abnormalities observed in NS mice are reminiscent of, although less severe than, those found in achondroplasia, a common skeletal dysplasia related to gain-of-function mutation in the fibroblast growth factor receptor 3 gene (FGFR3). In achondroplasia, the constitutive activation of the tyrosine kinase activity of the receptor results in a prolonged activation of RAS/ERK signalling, which is involved in the inhibition of chondrocytes proliferation and differentiation in the growth plate (33).
Beside growth retardation, one may wonder whether abnormal endochondral ossification could be responsible for the reduced length of the skull observed in NS mice. In fact, since the base of the skull develops from cartilage structures by endochondral ossification, similar defects could occur, although our study did not address this question. In contrast, development of flat bones of the skulls relies on intramembranous ossification, which is achieved through distinct mechanisms. In intramembranous ossification, mesenchymal cells differentiate directly into osteoblasts whereas in endochondral ossification mesenchymal cells first differentiate into chondrocytes which are secondarily replaced by osteoblasts. However, these two distinct processes share similar regulation, notably through RAS/ERK signalling pathway. Thus, it has been reported that sustained ERK1/2 activation in NS mouse is associated with delayed ossification of frontal bones caused by a lack of osteogenic differentiation (7). Further studies will be necessary to understand the relative contribution of these different mechanisms in NS-associated craniofacial defects.
Given the role of RAS/ERK activation in the pathophysiology of NS and other RASopathies, therapeutic strategies aiming to reduce this activation seem to be very promising. Such treatment could correct growth abnormalities (at the hormonal and also growth plate levels) and also other noncongenital features of NS such as developmental delay and progressive heart defect (i.e. hypertrophic cardiomyopathy). Recently, 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, also known as ‘statins’, which are widely used to treat hypercholesterolemia in humans, have been suggested as a potential therapy for RASopathies. Indeed, RAS activation requires farnesylation which allows RAS to anchor to the plasma membrane where it can be activated by RAS GTPase exchange factor (GEF). Since cholesterol is an obligate precursor of farnesyl, statins, which decrease cholesterol synthesis, may reduce RAS activity. Studies have shown that lovastatin can inhibit RAS farnesylation and activity and restore ERK-dependent learning and attention deficits in a mouse model of NF1 (10,34,35). This treatment has also been assessed in two randomized, double-blind, placebo-controlled trials for the treatment of cognitive deficits and behavioural problems in children with NF1 with no significant or minor effects, but good tolerance (36,37); however, the children were aged 8–16 years, so a therapeutic benefit in younger children cannot be excluded. It has also been reported that statin treatment can correct growth retardation and growth plate abnormalities in a mouse model of achondroplasia, a skeletal dysplasia associated with hyperactivation of RAS/ERK signalling at the growth plate level (23). Interestingly, in a randomized, double-blind, placebo-controlled study conducted to evaluate efficacy and safety of statin therapy in 173 children of average height with familial hypercholesterolemia, the gain in height (albeit non-significant) was greater in patients receiving statins compared to those receiving the placebo (38). Finally, statins may also have a beneficial effect on bone regeneration (39) and a protective effect against cartilage degradation (40,41).
In our study, we demonstrated that statin treatment improves growth of NS mice and chondrocyte differentiation in vivo and in vitro. Further investigations are needed to investigate the effect of statin on RAS/ERK signaling pathway.
Conclusion
Our study helps to better understand how hyperactive, NS-causing SHP2 mutants alter endochondral bone growth, thereby contributing to growth retardation, which could lead to the development of novel therapies for the treatment of growth delay in NS patients, one of those being the use of statins.
Materials and Methods
Mouse studies
A mouse model of NS carrying a missense mutation of Ptpn11 (Ptpn11D61G/+ mice) was used. The heterozygous mice have already been described for developing all the features of the human disease, including growth retardation (15,42). Ptpn11D61G/+ mice were generated and genotyped as previously described (15,42). Colony was maintained on a mixed B6x129Sv background by crossing male Ptpn11D61G/+ with female B6x129Sv F1 hybrids. For all in vivo studies, male Ptpn11D61G/+ mice (NS mice) and their wild-type littermates (Ptpn11+/+, WT mice) were compared.
For IGF1 treatments, animals were injected intraperitoneally (i.p.) with recombinant murine IGF1 (4 mg/kg twice daily; PreProTech) or vehicle (PBS) from 1 to 4 weeks of age. For U0126 treatments, U0126 (5 mg/kg daily; Cell Signalling) or vehicle [PBS, 40% (vol/vol) DMSO] was injected i.p. into nursing females from birth to 2 weeks of age, and then into pups from 2 to 4 weeks of age. For rosuvastatin treatment (1 mg/kg daily; Biovision) or vehicle (PBS) was injected i.p. into pups from 1 to 4 weeks of age.
All animal experiments were performed in compliance with the French Institute of Medical Research (INSERM) guidelines and were approved by the ethical committee on the use and care of animals for animal research (N° 2017040710578733).
X-ray analysis
X-ray images of the entire skeletons, skulls, and lower legs were taken with a Faxitron X-ray system (Faxitron Bioptics, AZ).
Histology and immunohistochemistry
For histological analysis, the left tibiae from 4-week-old mice were dissected and fixed overnight in 4% neutral buffered formalin, decalcified in 10% EDTA, pH 7.4, for 21 days with regular changes, dehydrated, and embedded in paraffin wax using standard procedures.
Paraffin sections (3µm) of the proximal tibiae were stained with hematoxylin and eosin (HE), and alcian blue. For growth plate measurements, the widths of the proliferating (from the start of chondrocyte columns to the first enlargement of flattened cells) and hypertrophic (from the first enlargement of flattened cells to the chondro-osseous junction) zones were measured in a blinded manner at 10 different sites along two stained sections from each growth plate.
Immunohistochemistry was performed according to the manufacturer’s instructions using an antibody against collagen type 10 (monoclonal mouse; Bioscience), proliferating cell nuclear antigen (PCNA, PC10 monoclonal mouse; Dako), and dually phosphorylated forms of extracellular signal-regulated kinases 1 and 2 (ERK-1 and ERK-2, 44 and 42 kDa, respectively, mouse IgG1 isotype; Sigma).
A kit for the detection of apoptotic cells (ApopTagPeroxidase In situ Apoptosis Detection; Millipore) was used according to the manufacturers’ instructions. Immunohistochemistry measurements were performed using the Imagequant Analysis System (3DHistech, Sysmex, Hungary).
Isolation, culture, and differentiation of murine primary chondrocytes
Murine primary chondrocytes were isolated according to previously published protocol (43). Briefly, costal cartilages from 7-day-old mice were dissected, washed in PBS, and digested overnight with collagenase D (Roche). Then, cells aggregates were dispersed and the suspension of isolated cells was filtered through a sterile 48 μm membrane, centrifuged, washed with PBS and resuspended in DMEM with l-glutamine, supplemented with 1% penicillin-streptomycin solution and 10% fetal bovine serum.
Chondrocytes were seeded on a culture dish at a density of 25×103 cells cm−2 and the medium was changed every 2 days. To induce differentiation, after confluence was achieved by days 6–7, cells were cultured with 50 mg/ml ascorbic acid (Sigma) for 21 days. To test the effect of RAS inhibition or statins on chondrocyte differentiation, cells were cultured in the presence or absence of U0126 (10 μM, Cell Signalling) or lovastatin (1 μM, Biovision) added to the culture medium for 7 days.
Cell proliferation assay
Primary murine chondrocytes were seeded at a number of 50×103 cells in 12-well plates and cultured in the presence of 10 ng/ml FGF2 (Sigma–Aldrich). Chondrocyte proliferation rate was determined by the counting of the total cell number and also by the incorporation of EdU (an alternative to bromodeoxyuridine for measuring new DNA synthesis) into DNA, using a Click-iT EdU microplate assay kit (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. The incorporated EdU in DNA was coupled to Oregon Green-azide and then detected using a horseradish peroxidase-conjugated anti-Oregon Green antibody and Amplex UltraRed. The fluorescence [expressed as relative fluorescence units (RFU)] was detected at an excitation/emission wavelength of 490/585 nm and was taken as the cell proliferation rate.
Alkaline phosphatase staining and activity
Primary chondrocyte cultures were stained for alkaline phosphatase (ALP) as previously described (44). Briefly, chondrocyte cultures were washed with PBS and fixed in 10% formalin for 2 h at room temperature. After a 15-min incubation in highly pure water, cells were stained with an ALP substrate containing 0.1 mg/ml naphthol AS-MX phosphate (Sigma Aldrich), 0.5% N,N-dimethylformamide (Sigma Aldrich), and 0.6 mg/ml Red Violet LB salt (Sigma Aldrich) in 0.2 M Tris–HCl (pH 8.3) for 45 min in the dark. Then, cells were washed and air-dried before visualization. All images were captured using the Leica EC3 microscope and Leica Application Suite V3 Software.
ALP activity was determined by fluorometric method with p-nitrophenol-phosphate used as the substrate, as described previously (45). Briefly, primary chondrocyte cultures were washed with PBS and lysed with 0.2% NP40. ALP activity was measured by incubating cell lysates with the p-nitrophenol phosphate (Sigma). The reaction was terminated by the addition of NaOH and absorbance was measured at 410 nm. Values were normalized for total DNA, which was quantified by the picogreen assay (Invitrogen); ALP activity was expressed as nmol of product/min/μg of DNA to correct for cell number differences.
Quantitative RT-PCR
Reverse transcription was performed with 1µg of total RNA using M-MLV (Promega), and 25µM Hexamers (Fermentas), in a 20-µl final reaction volume, according to the manufacturer’s protocol. Quantitative RT-PCR was performed by using LightCycler 480 DNA SYBR Green I Master reaction mix (Roche Lifescience) with primers (Supplementary Material, Table S1) designed by using the QuantPrime (46) or the Universal ProbeLibrary Assay Design Center (Roche Lifescience). Reactions were carried out in duplicate on a 96-well. The amplification program consisted of an initial denaturation at 95°C for 5 min, followed by 40 cycles of denaturation (95°C, 10 s)/elongation (60°C, 40 s) using the LightCycler® 480 System. The reference gene tubulin was used for normalization, and the 2−ΔΔCt method was used for quantification.
Cell culture and stimulation
Chondrogenic ATDC5 cells were cultured at 37°C in a humidified atmosphere of 5% CO2 in DMEM/Ham’s F-12 GlutaMAX medium (Gibco), supplemented with 5% (vol/vol) fetal bovine serum and antibiotics. Cells were seeded in 35-mm dishes, incubated 6 h in serum-free medium, then stimulated or not with 400 ng/ml growth hormone (norditropine; NovoNordisk) or 10 ng/ml FGF2 (Sigma–Aldrich) during indicated times, rinsed with ice cold PBS, and processed for western-blot analysis.
Adenoviral infection
Generation of recombinant adenoviruses has been previously described (47). Briefly, bicistronic adenovectors encoding green fluorescent protein (GFP) and SHP2-WT or –D61Del were obtained by subcloning the corresponding V5-tagged SHP2-encoding construct into the pTrack-CMV vector. Recombinant adenovectors were then obtained using the pAdEasy homologous-recombination system, followed by plaque purification, expansion, and titration in HEK293 cells. For transduction of ATDC5 cells, adenoviruses were mixed with 0.5 μg/ml poly-l-lysine for 90 min in DMEM Ham’s F-12 GlutaMAX medium. Cells were then incubated with this mixture for 2 h at a multiplicity of infection (MOI) of 2500.
Western blots
Lysate aliquots containing identical amounts of protein (Bio-Rad) were diluted in Laemmli’s sample buffer, boiled, and processed for immunoblotting by using a standard procedure. Antibodies used were as follows: polyclonal anti-SHP2 (Santa Cruz) and HRP-conjugated secondary antibodies, and monoclonal anti-phospho ERK1/2, anti-α-tubulin, all from Sigma. Blots were revealed by chemiluminescence on a Chemidoc Apparatus (Bio-Rad).
Statistics
All data are expressed as mean ± SEM. Unless otherwise indicated, statistical significance was determined by using paired or unpaired two-tailed Student’s t-test, with Welch correction in the case of unequal variances, two-way ANOVA with Bonferroni post hoc test, or two-way ANOVA with repeated measures, as appropriate. Throughout the study P < 0.05 was considered significant (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001). Calculations were performed using the GraphPad Prism software, version 6 for Mac OS X (GraphPad Software, Inc., CA).
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgements
We thank the personnel of the ANEXPLO/CREFRE animal facilities for animal handling, of the histopathology unit, and of the non-invasive exploration service, INSERM/UPS—UMS 006/CREFRE.
Conflict of Interest statement. None declared.
Funding
This work was supported by grants from the European Union’s Horizon 2020 research and innovation programme under the ERA-NET Cofund action in the framework of E-Rare-3 (NS-EuroNet) (N° 643578 to A.Y.); the National Institutes of Health (R37 CA41932 to B.G.N.); the ASPIRE Young Investigator Research Awards in Endocrinology supported by Pfizer to T.E.; the INSERM/DHOS translational clinical research (C13–59) to T.E. and J.P.S.; and the French Society of Pediatric Endocrinology and Diabetology (Société Française d’Endocrinologie et de Diabétologie Pédiatrique, SFEDP) and Pfizer to J.P.
References
- 1. Roberts A.E., Allanson J.E., Tartaglia M., Gelb B.D. (2013) Noonan syndrome. Lancet, 381, 333–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Romano A.A., Allanson J.E., Dahlgren J., Gelb B.D., Hall B., Pierpont M.E., Roberts A.E., Robinson W., Takemoto C.M., Noonan J.A. (2010) Noonan syndrome: clinical features, diagnosis, and management guidelines. Pediatrics, 126, 746–759. [DOI] [PubMed] [Google Scholar]
- 3. Tartaglia M., Gelb B.D., Zenker M. (2011) Noonan syndrome and clinically related disorders. Best Pract. Res. Clin. Endocrinol. Metab., 25, 161–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tidyman W.E., Rauen K.A. (2016) Expansion of the RASopathies. Curr. Genet. Med. Rep., 4, 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tartaglia M., Mehler E.L., Goldberg R., Zampino G., Brunner H.G., Kremer H., van der Burgt I., Crosby A.H., Ion A., Jeffery S. (2001) Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet., 29, 465–468. [DOI] [PubMed] [Google Scholar]
- 6. Nakamura T., Colbert M., Krenz M., Molkentin J.D., Hahn H.S., Dorn G.W. II, Robbins J. (2007) Mediating ERK 1/2 signaling rescues congenital heart defects in a mouse model of Noonan syndrome. J. Clin. Invest., 117, 2123–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nakamura T., Gulick J., Pratt R., Robbins J. (2009) Noonan syndrome is associated with enhanced pERK activity, the repression of which can prevent craniofacial malformations. Proc. Natl. Acad. Sci. U. S. A., 106, 15436–15441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Araki T., Chan G., Newbigging S., Morikawa L., Bronson R.T., Neel B.G. (2009) Noonan syndrome cardiac defects are caused by PTPN11 acting in endocardium to enhance endocardial-mesenchymal transformation. Proc. Natl. Acad. Sci. U. S. A., 106, 4736–4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Krenz M., Gulick J., Osinska H.E., Colbert M.C., Molkentin J.D., Robbins J. (2008) Role of ERK1/2 signaling in congenital valve malformations in Noonan syndrome. Proc. Natl. Acad. Sci. U. S. A., 105, 18930–18935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee Y.S., Ehninger D., Zhou M., Oh J.Y., Kang M., Kwak C., Ryu H.H., Butz D., Araki T., Cai Y.. et al. (2014) Mechanism and treatment for learning and memory deficits in mouse models of Noonan syndrome. Nat. Neurosci., 17, 1736–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kronenberg H.M. (2003) Developmental regulation of the growth plate. Nature, 423, 332–336. [DOI] [PubMed] [Google Scholar]
- 12. Nilsson O., Marino R., De Luca F., Phillip M., Baron J. (2005) Endocrine regulation of the growth plate. Horm. Res., 64, 157–165. [DOI] [PubMed] [Google Scholar]
- 13. Binder G., Neuer K., Ranke M.B., Wittekindt N.E. (2005) PTPN11 mutations are associated with mild growth hormone resistance in individuals with Noonan syndrome. J. Clin. Endocrinol. Metab., 90, 5377–5381. [DOI] [PubMed] [Google Scholar]
- 14. Limal J.M., Parfait B., Cabrol S., Bonnet D., Leheup B., Lyonnet S., Vidaud M., Le Bouc Y. (2006) Noonan syndrome: relationships between genotype, growth, and growth factors. J. Clin. Endocrinol. Metab., 91, 300–306. [DOI] [PubMed] [Google Scholar]
- 15. Serra-Nedelec A.D.R., Edouard T., Treguer K., Tajan M., Araki T., Dance M., Mus M., Montagner A., Tauber M., Salles J.-P.. et al. (2012) Noonan syndrome-causing SHP2 mutants inhibit insulin-like growth factor 1 release via growth hormone-induced ERK hyperactivation, which contributes to short stature. Proc. Natl. Acad. Sci. U. S. A., 109, 4257–4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bauler T.J., Kamiya N., Lapinski P.E., Langewisch E., Mishina Y., Wilkinson J.E., Feng G.S., King P.D. (2011) Development of severe skeletal defects in induced SHP-2-deficient adult mice: a model of skeletal malformation in humans with SHP-2 mutations. Dis. Model. Mech., 4, 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bowen M.E., Ayturk U.M., Kurek K.C., Yang W., Warman M.L. (2014) SHP2 regulates chondrocyte terminal differentiation, growth plate architecture and skeletal cell fates. PLoS Genet., 10, e1004364.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang W., Wang J., Moore D.C., Liang H., Dooner M., Wu Q., Terek R., Chen Q., Ehrlich M.G., Quesenberry P.J.. et al. (2013) Ptpn11 deletion in a novel progenitor causes metachondromatosis by inducing hedgehog signalling. Nature, 499, 491–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim H.K., Feng G.S., Chen D., King P.D., Kamiya N. (2014) Targeted disruption of Shp2 in chondrocytes leads to metachondromatosis with multiple cartilaginous protrusions. J. Bone Miner. Res., 29, 761–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim H.K., Aruwajoye O., Sucato D., Richards B.S., Feng G.S., Chen D., King P.D., Kamiya N. (2013) Induction of SHP2 deficiency in chondrocytes causes severe scoliosis and kyphosis in mice. Spine (Phila Pa 1976), 38, E1307–E1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang L., Huang J., Moore D.C., Zuo C., Wu Q., Xie L., von der Mark K., Yuan X., Chen D., Warman M.L.. et al. (2017) SHP2 Regulates the Osteogenic Fate of Growth Plate Hypertrophic Chondrocytes. Sci. Rep., 7, 12699.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bowen M.E., Boyden E.D., Holm I.A., Campos-Xavier B., Bonafe L., Superti-Furga A., Ikegawa S., Cormier-Daire V., Bovee J.V., Pansuriya T.C.. et al. (2011) Loss-of-function mutations in PTPN11 cause metachondromatosis, but not Ollier disease or Maffucci syndrome. PLoS Genet., 7, e1002050.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yamashita A., Morioka M., Kishi H., Kimura T., Yahara Y., Okada M., Fujita K., Sawai H., Ikegawa S., Tsumaki N. (2014) Statin treatment rescues FGFR3 skeletal dysplasia phenotypes. Nature, 513, 507–511. [DOI] [PubMed] [Google Scholar]
- 24. Lapinski P.E., Meyer M.F., Feng G.S., Kamiya N., King P.D. (2013) Deletion of SHP-2 in mesenchymal stem cells causes growth retardation, limb and chest deformity, and calvarial defects in mice. Dis. Model. Mech., 6, 1448–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lupu F., Terwilliger J.D., Lee K., Segre G.V., Efstratiadis A. (2001) Roles of growth hormone and insulin-like growth factor 1 in mouse postnatal growth. Dev. Biol., 229, 141–162. [DOI] [PubMed] [Google Scholar]
- 26. Pass C., MacRae V.E., Huesa C., Faisal Ahmed S., Farquharson C. (2012) SOCS2 is the critical regulator of GH action in murine growth plate chondrogenesis. J. Bone Miner. Res., 27, 1055–1066. [DOI] [PubMed] [Google Scholar]
- 27. Giacomozzi C., Deodati A., Shaikh M.G., Ahmed S.F., Cianfarani S. (2015) The impact of growth hormone therapy on adult height in noonan syndrome: a systematic review. Horm Res Paediatr., 83, 167–176. [DOI] [PubMed] [Google Scholar]
- 28. Murakami S., Balmes G., McKinney S., Zhang Z., Givol D., de Crombrugghe B. (2004) Constitutive activation of MEK1 in chondrocytes causes Stat1-independent achondroplasia-like dwarfism and rescues the Fgfr3-deficient mouse phenotype. Genes Dev., 18, 290–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Matsushita T., Chan Y.Y., Kawanami A., Balmes G., Landreth G.E., Murakami S. (2009) Extracellular signal-regulated kinase 1 (ERK1) and ERK2 play essential roles in osteoblast differentiation and in supporting osteoclastogenesis. Mol. Cell. Biol., 29, 5843–5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ono K., Karolak M.R., Ndong J. d l C., Wang W., Yang X., Elefteriou F. (2013) The ras-GTPase activity of neurofibromin restrains ERK-dependent FGFR signaling during endochondral bone formation. Hum. Mol. Genet., 22, 3048–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen Z., Yue S.X., Zhou G., Greenfield E.M., Murakami S. (2015) ERK1 and ERK2 regulate chondrocyte terminal differentiation during endochondral bone formation. J. Bone Miner. Res., 30, 765–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sebastian A., Matsushita T., Kawanami A., Mackem S., Landreth G.E., Murakami S. (2011) Genetic inactivation of ERK1 and ERK2 in chondrocytes promotes bone growth and enlarges the spinal canal. J Orthop Res., 29, 375–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Monsonego-Ornan E., Adar R., Feferman T., Segev O., Yayon A. (2000) The transmembrane mutation G380R in fibroblast growth factor receptor 3 uncouples ligand-mediated receptor activation from down-regulation. Mol. Cell. Biol., 20, 516–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li W., Cui Y., Kushner S.A., Brown R.A., Jentsch J.D., Frankland P.W., Cannon T.D., Silva A.J. (2005) The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Curr. Biol., 15, 1961–1967. [DOI] [PubMed] [Google Scholar]
- 35. Sebti S.M., Tkalcevic G.T., Jani J.P. (1991) Lovastatin, a cholesterol biosynthesis inhibitor, inhibits the growth of human H-ras oncogene transformed cells in nude mice. Cancer Commun., 3, 141–147. [DOI] [PubMed] [Google Scholar]
- 36. Krab L.C., de Goede-Bolder A., Aarsen F.K., Pluijm S.M., Bouman M.J., van der Geest J.N., Lequin M., Catsman C.E., Arts W.F., Kushner S.A. (2008) Effect of simvastatin on cognitive functioning in children with neurofibromatosis type 1: a randomized controlled trial. JAMA, 300, 287–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. van der Vaart T., Plasschaert E., Rietman A.B., Renard M., Oostenbrink R., Vogels A., de Wit M.C., Descheemaeker M.J., Vergouwe Y., Catsman-Berrevoets C.E.. et al. (2013) Simvastatin for cognitive deficits and behavioural problems in patients with neurofibromatosis type 1 (NF1-SIMCODA): a randomised, placebo-controlled trial. Lancet Neurol., 12, 1076–1083. [DOI] [PubMed] [Google Scholar]
- 38. de Jongh S., Ose L., Szamosi T., Gagne C., Lambert M., Scott R., Perron P., Dobbelaere D., Saborio M., Tuohy M.B.. et al. (2002) Efficacy and safety of statin therapy in children with familial hypercholesterolemia: a randomized, double-blind, placebo-controlled trial with simvastatin. Circulation, 106, 2231–2237. [DOI] [PubMed] [Google Scholar]
- 39. Shah S.R., Werlang C.A., Kasper F.K., Mikos A.G. (2015) Novel applications of statins for bone regeneration. Natl. Sci. Rev., 2, 85–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Simopoulou T., Malizos K.N., Poultsides L., Tsezou A. (2010) Protective effect of atorvastatin in cultured osteoarthritic chondrocytes. J Orthop Res., 28, 110–115. [DOI] [PubMed] [Google Scholar]
- 41. Yudoh K., Karasawa R. (2010) Statin prevents chondrocyte aging and degeneration of articular cartilage in osteoarthritis (OA). Aging (Albany NY), 2, 990–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Araki T., Mohi M.G., Ismat F.A., Bronson R.T., Williams I.R., Kutok J.L., Yang W., Pao L.I., Gilliland D.G., Epstein J.A.. et al. (2004) Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nat. Med., 10, 849–857. [DOI] [PubMed] [Google Scholar]
- 43. Gosset M., Berenbaum F., Thirion S., Jacques C. (2008) Primary culture and phenotyping of murine chondrocytes. Nat. Protoc., 3, 1253–1260. [DOI] [PubMed] [Google Scholar]
- 44. Sun M.M., Beier F. (2014) Liver X Receptor activation delays chondrocyte hypertrophy during endochondral bone growth. Osteoarthritis Cartilage, 22, 996–1006. [DOI] [PubMed] [Google Scholar]
- 45. Katagiri T., Yamaguchi A., Komaki M., Abe E., Takahashi N., Ikeda T., Rosen V., Wozney J.M., Fujisawa-Sehara A., Suda T. (1994) Bone morphogenetic protein-2 converts the differentiation pathway of C2C12 myoblasts into the osteoblast lineage. J. Cell. Biol., 127, 1755–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Arvidsson S., Kwasniewski M., Riano-Pachon D.M., Mueller-Roeber B. (2008) QuantPrime – a flexible tool for reliable high-throughput primer design for quantitative PCR. BMC Bioinformatics, 9, 465.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Edouard T., Combier J.P., Nedelec A., Bel-Vialar S., Metrich M., Conte-Auriol F., Lyonnet S., Parfait B., Tauber M., Salles J.P.. et al. (2010) Functional effects of PTPN11 (SHP2) mutations causing LEOPARD syndrome on epidermal growth factor-induced phosphoinositide 3-kinase/AKT/glycogen synthase kinase 3beta signaling. Mol. Cell. Biol., 30, 2498–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.