

Abstract
Neurological disorders affecting individuals in infancy to old age elude interventions for meaningful protection against neurodegeneration, and preclinical work has not translated to humans. We studied human neuron responses to injury and death stimuli compared to those of animal neurons in culture under similar settings of insult (excitotoxicity, oxidative stress, and DNA damage). Human neurons were differentiated from a cortical neuron cell line and the embryonic stem cell-derived H9 line. Mouse neurons were differentiated from forebrain neural stem cells and embryonic cerebral cortex; pig neurons were derived from forebrain neural stem cells. Mitochondrial morphology was different in human and mouse neurons. Human and mouse neurons challenged with DNA-damaging agent camptothecin showed different chromatin condensation, cell death, and DNA damage sensor activation. DNA damage accumulation and repair kinetics differed among human, mouse, and pig neurons. Promoter CpG island methylation microarrays showed significant differential DNA methylation in human and mouse neurons after injury. Therefore, DNA damage response, DNA repair, DNA methylation, and autonomous cell death mechanisms in human neurons and experimental animal neurons are different.
Keywords: Alzheimer disease, Amyotrophic lateral sclerosis, Apoptosis, Cerebral ischemia, Excitotoxicity, Neuronal cell death
INTRODUCTION
There are many unmet needs for effective therapeutic interventions for all human neurological disorders ranging from infancy to old age. In fact, translational development and application of disease- and injury-modifying treatments for human acute and chronic neurological disorders in newborns, children, and adults have been very disappointing, despite extraordinary financial and labor investments. For example, spinal muscular atrophy clinical trials aimed at restoring levels of survival motor neuron protein in infants have failed (1). Therapeutic hypothermia is used for neonatal hypoxic-ischemic encephalopathy (HIE), but clinical trials show partial, limited efficacy (2), and no efficacy for out-of-hospital and in-hospital pediatric cardiac arrest (3, 4). Drugs and biologics that show positive outcomes in preclinical animal models of adult brain and spinal cord injury and neurodegenerative disease fail repeatedly in clinical trials. The cerebral ischemia field has witnessed clinical failures with >120 different glutamate receptor and ion channel antagonists, free radical scavengers/antioxidants, and other drugs (5, 6). The available treatments for Alzheimer disease (AD) offer symptomatic benefits with marginal clinical significance; cholinesterase inhibitors of the donepezil-type have been used for ∼30 years and, more recently, the N-methyl-d-aspartate (NMDA) receptor antagonist memantine offers little therapeutically to AD patients (7, 8). Antiamyloid trials for AD have also failed (9). The failures in clinical trials for amyotrophic lateral sclerosis (ALS) continue to grow (10–13). The cost of each CNS disorder-related phase III clinical trial is ∼$36,000/person/year (14), so preclinical studies have to be human disease-relevant and truly reflect the reality of human disease.
The biology of neuronal injury, repair, and death is complex and nuanced, especially in the context of the cell death matrix concept (15, 16), and clinical trial design has many limitations and caveats (17). There is an insufficient understanding of all forms of human CNS pathobiology. In addition, the absence of genetic, epigenetic, nutritional, and lifestyle uniformity in human patient populations is a complex confounder. The failure to identify human-relevant cell and molecular mechanism-based targets in human neurons for drug discovery and effective therapeutics could also be related to the relevancy of preclinical animal or cell models that are predicated on the major unvalidated assumption that mechanisms of neuronal injury and death are the same in experimental animal systems and in humans. Neuroanatomical and molecular studies fuel concern about the relevancy of preclinical experimental systems for translational medicine. Cortical neuron dendritic morphology varies markedly among different anthropoid species (18), human and macaque monkeys (19), and human and mouse (20). Transcriptome analyses also show species-specific gene expression patterns between human and mouse (21). However, it is unknown whether these structural and gene expression differences have meaning in the context of biochemical and molecular mechanisms of neuronal repair, degeneration, and cell death. Species postreproductive life span and longevity are also important considerations for disease modeling (22). With the advent of human stem cell-derived neurons, human-relevant mechanisms of cell injury and human neuron-relevant therapeutics can be investigated directly and decisively. Moreover, fundamental mechanisms of disease can be explored in human differentiated neurons. Our study was undertaken to test the assumption that human neurons and experimental animal neurons behave similarly to injury and degenerate similarly. We found distinct differences among human neurons and animal neurons in response to pathological insults such as DNA damage, excitotoxicity, and oxidative stress and conclude that human neurons and rodent neurons degenerate and die differently.
MATERIALS AND METHODS
Cell Culture
Human Cortical Neuron (HCN) Cell Line
The HCN-1A cell line (American Type Culture Collection) was used as a source of human neurons. The HCN cell line was derived from cerebral cortex resected from an 18-month-old female patient with intractable seizures (23). HCN cells can be induced to differentiate when cultured in media containing nerve growth factor, dibutyryl cyclic adenosine monophosphate (cAMP), and 1-isobutyl-3-methylxanthine. Reports show that HCN cells can differentiate into cells immunophenotyped with neuronal markers such a neurofilament protein, neuron-specific enolase, glutamate, and a variety of neuropeptides (23, 24). This cell line has a long history of use (25–28). We propagated the cells under atmospheric oxygen/5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) and 10% fetal bovine serum (FBS).
Human Neural Stem Cell (NSC)-Derived Neurons
We used human neural stem cells (Invitrogen-Gibco, N7800-200), derived from the H9 human embryonic stem cell line isolated from a normal female (29), to generate differentiated human forebrain neurons. Neuronal differentiation was induced, as described in detail (30), by plating the neurosphere-derived neural precursors on polyornithine/laminin-coated plates with neurobasal complete medium in the absence of mitogens or with the differentiation medium (DMEM/F12 supplement, 1% N2 supplement, 1% B27 supplement, 1 mM cAMP, 20 ng/mL brain-derived neurotrophic factor, and 20 ng/mL glial cell line-derived neurotrophic factor). Human neurons were cultured under atmospheric oxygen/5% CO2, differentiated, and maintained for ∼30–40 days.
To establish that human differentiated neurons are functional neurons, we did whole-cell patch-clamp recordings (31). Glass pipettes were pulled on a P-97 electrode puller (Sutter Instruments, Novato, CA). The resistance of the pipettes was 3–5 MΩ when filled with an intracellular solution (135 mM KCl, 2 mM MgCl2, 10 mM HEPES, 5 mM EGTA, 2 mM Mg-ATP, 0.3 mM NaGTP, and 10 mM phosphocreatine, pH 7.25 adjusted with KOH). Cultured neurons on coverslips were superfused at a rate of 2 mL/minute with an external bath solution containing the following: 150 mM NaCl, 2.5 mM KCl, 10 mM HEPES, 10 mM d-glucose, 2 mM CaCl2, and 1 mM MgCl2, pH 7.3–7.4. We performed experiments at room temperature (22–24°C). Currents from cells were monitored with an Axopatch 200B amplifier (Molecular Devices, San Jose, CA) and acquired through Digidata 1440 A (Molecular Devices) onto a computer using pClamp 10 software (Molecular Devices). All chemicals used for electrophysiological recordings were purchased from Sigma. All drugs and solutions were made fresh from stock solutions.
Mouse Forebrain NSC-Derived Neurons
We used mouse NSCs isolated from the subventricular zone and olfactory bulb as described (32–34). Neurons were cultivated from neurospheres that were thawed and expanded in vitro. After one passage, they were collected by centrifugation, washed once with Hank’s salt balanced solution (Ca2+ and Mg2+ free), and dissociated with TryLE Express (Gibco, Waltham, MA) for 10 minutes at 37°C and triturated into single cell suspensions. Single cells were regrown under atmospheric oxygen/5% CO2 as neurospheres and were then dissociated and seeded on poly-d-lysine-coated 35-mm petri dishes at densities of 6×105/mL with DMEM/F12, 1% N2, B27, 10% FBS, DNase I, and antibiotics (100 U/mL penicillin and 100 μg/mL streptomycin). Mouse neurons were cultured, differentiated, and maintained for ∼20–30 days.
Mouse Primary Cortical Neurons
Timed-pregnant wildtype mice (C57BL/6 strain, The Jackson Laboratory, Bar Harbor, ME) were used. Animal care was provided in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Mouse cortical neuron cultures were prepared as described (35–38). Cortical tissue (both hemispheres) was digested in trypsin and washed. Cell suspensions were counted. Cells (∼106) were plated onto 35-mm tissue culture dishes coated with 33 μg/mL poly-d-lysine (Sigma, St. Louis, MO) and human fibronectin (Sigma) or onto poly-d-lysine- and laminin-coated 12-mm glass coverslips (BD Biosciences, Bedford, MA). The cells were plated in Neurobasal medium (Invitrogen, Carlsbad, CA) or DMEM supplemented with B27 (Invitrogen), 25 μM β-mercaptoethanol (Invitrogen), and streptomycin/penicillin (Invitrogen). Three days after plating, 50% of the medium was changed and then subsequently changed every 5 days. This cell culture is nearly neuron-pure because glial contamination is very low (35). Mouse primary neurons were cultured under atmospheric oxygen/5% CO2, differentiated, and maintained for ∼20–30 days.
Pig Forebrain NSC-Derived Neurons
We used pig NSCs isolated from the anterior subventricular zone (SVZ) and olfactory bulb SVZ as described (39). Clonal neurospheres that were cryopreserved in DMEM containing DMSO and FBS were thawed and expanded in vitro again. After one passage, they were collected by spinning, washed once in HBSS and dissociated with TryLE Express (Gibco) for 10 minutes at 37°C and triturated into single cell suspensions and counted. Neurosphere-derived cells were seeded at densities of 6×105/mL into poly-d-lysine-coated 35-mm well plates in DMEM/F12, 1% N2, B27, and antibiotics. Pig forebrain neurons were cultured under atmospheric oxygen/5% CO2, differentiated, and maintained for ∼20–30 days.
Mitochondria Tracking
Transient transfections of human and mouse neural cells were done with a Thy1-mitoDsRed2 expression plasmid, engineered as described (38), or with mitoDsRed2 plasmid with a CMV promoter (Clontech, Mountain View, CA) using Lipofectamine 2000 (Invitrogen). Live cells were imaged using epifluorescence microscopy 48 hours later and were then fixed for 20 minutes in 4% paraformaldehyde/4% sucrose. Digital images were used to measure mitochondrial length, diameter, and cluster size. The length and diameter of individual mitochondria were measured usually in the cell body periphery and proximal dendrites where single mitochondria could be discerned. The major diameter of mitochondrial clusters comprised of apparently 2 or more mitochondria were measured at a perikaryal location.
Camptothecin, Excitotoxin, and Oxidative Stress Challenges to Forebrain Neuron Cell Cultures
We studied the responses of human, mouse, and pig forebrain neurons to toxic stress. One cytotoxic insult was exposure to the plant alkaloid camptothecin (CPT) which has been derivatized to make standard-of-care cancer chemotherapeutic agents topotecan and irinotecan (40). CPT inhibits DNA topoisomerase-I (Topo-I) (41). Topo-I is a nuclear enzyme that catalyzes DNA single-strand cleavage and reunion of the phosphodiester backbone to allow relaxation of supercoiling in response to a torsional stress generated by replication proteins at replication forks during DNA synthesis or by transcriptional proteins during RNA synthesis (41). CPT poisoning of nonneural cells is believed to involve stabilization of Topo-I-DNA complexes, and the trapping Topo-I in an intermediate state to disallow religation of the DNA backbone and thus causing the formation of DNA strand breaks (41, 42). Topo-I is the only known target of CPT. CPT (Sigma, St. Louis, MO) was dissolved in double-distilled water supplemented with sodium hydroxide and heated at 55° C for 30 minutes to dissolve completely the drug at a final concentration of 50 mM. CPT was used at 10 μM for 2–24 hours.
We subjected human, mouse, and pig neurons (grown in DMEM) to excitotoxic stress by changing medium to DMEM without B27 and adding the highly specific NMDA glutamate receptor agonist QA (quinolinic acid) or NMDA to the media at a final concentration of 10–100 µM. Neurons were exposed to excitotoxin for 4 hours and then were changed to complete DMEM growth medium.
We subjected human, mouse, and pig neurons (grown in DMEM) to chemical oxidative stress by changing medium to DMEM without B27 and adding hydrogen peroxide (H2O2), nitric oxide (NO) donor, or peroxynitrite (ONOO−). H2O2 was used at a final concentration of 1 mM. Two different NO donors were used: sodium nitroprusside (SNP, Sigma) and N-(2-aminoethyl)-N-(2-hydroxyl-nitrosohydrazino)-1, 2-ethylenediamine (spermine-NONOate, OXIS International, Portland, OR). Spermine-NONOate was used because the in vitro generation of NO is slower than SNP and is steady state (43). Cells were exposed to SNP (10 µM) or spermine-NONOate (10 µM) for 60 minutes. Neurons were also exposed directly to ONOO− (Alexis, San Diego, CA) at concentrations of 10 µM for 15 minutes. ONOO− is a potent and relatively long-lived reactive oxygen species (ROS) formed by a reaction between superoxide and NO (44). For controls, neurons were incubated in medium for the same time in the absence of H2O2 or SNP, with spermine tetrahydrochloride/sodium nitrite () as a control for NONOate, or with decomposed ONOO− in alkaline solution. After exposure, neurons in the different treatment groups were collected and gently pelleted at 4°C for 5 minutes. Each cell pellet was resuspended and analyzed with the comet assay (45).
Cell Survival and Cell Death Analysis in Neuron Cultures
For living neuron viewing, cells were viewed under phase contrast optics using a Nikon Eclipse TS100 microscope (40× objective) equipped with an Infinity camera to capture digital images. For neuronal degeneration observations, fixed cultures were rinsed in phosphate-buffered saline (PBS) and stained with nuclear dye (Hoechst 33258); digital images were captured from nonoverlapping microscopic fields to assess classic apoptotic nuclei or brightly stained condensed nuclei as before (36).
Immunofluorescence
Immunofluorescence was used to identify cell types and define the maturation of the neuronal cultures. Staining for several neuronal markers (cytoskeletal, synaptic, and transcription factor proteins) was done: microtubule-associated protein-2 (MAP2), synaptophysin, α-synuclein (αSyn), β-synuclein (βSyn), and T-box brain 1 (TBR1). Staining for glial fibrillary acidic protein (GFAP) was done to assess astrocyte presence in the cell cultures (see Supplementary Data Methods). Preparations were viewed with a Nikon TS100 microscope and digital images were captured with a Nikon Infinity CCD camera or they were viewed using a Zeiss LSM confocal microscope. Counting of immunopositive cells was done from digital images acquired in raster scanning patterns covering the culture.
Western Blotting for Caspases, Bcl-2 Family Proteins, and DNA Damage Response Proteins and for Validation of DNA Methylation
Differentiated human and mouse neurons were used for Western blotting of cell death proteins and DNA damage response (DDR) proteins after CPT treatment (see Supplementary Data Methods).
Caspase Activity Assays
Human and mouse neurons were lysed in cell lysis buffer and evaluated for the activity of caspases that recognize the DEVD caspase-3 cleavage motif (Chemicon, Temecula, CA) or the VEID caspase-6 cleavage motif (BioVision, Milpitas, CA) using colorimetric assay kits. Human recombinant active caspase-3 (Chemicon) and active caspase-6 (BioVision) were used as positive controls. Samples were treated with caspase-3 inhibitor (Ac-DEVD-CHO) or the caspase-6 inhibitor (Z-VEID-FMK) were negative controls.
Comet Assay for DNA Damage and DNA Repair
Cells were detached from culture plates by incubation with 0.25% trypsin/EDTA, and then were collected, centrifuged, and washed with PBS to remove trypsin, and resuspended in PBS. A commercial comet assay kit was used (Trevigen, Gaithersburg, MD, 4252-040-K) according to manufacturer instructions. Comets were observed using SYBR gold or ethidium bromide DNA dye. Images were acquired and analyzed using Slidebook or Spot software. Comets were manually counted and comet moments were calculated as described (45, 46).
Genome-Wide Promoter CpG DNA Methylation Profiling of Human Neurons and Mouse Neurons
Promoter microarrays provide an efficient, high-throughput genome-wide screen of CpG islands with differential methylation (47). Differentiated human neurons and mouse neurons were treated with CPT (10 μM) or vehicle and 6 hours later were lysed for genomic DNA isolation and screening on CpG island promoter microarrays. Genomic DNA was isolated by phenol-chloroform extraction and was sonicated to generate random fragments. All DNA restriction enzyme cutting, methylated DNA immunoprecipitation (MeDIP), sample labeling, hybridization, and processing were performed under standardized and strict operating procedures.
For human neuron CpG island DNA methylation, we used University Health Network (UHN) 12 K human CpG arrays (UHN Microarray Center, Toronto Canada). These arrays have 12,192 spotted clones that represent a large percentage of the CpG islands found in the human genome (48). The sequences on this array are derived from a CpG island library in which 75% of all clones are unique sequences and 25% correspond to repetitive elements as well as ribosomal and mitochondrial DNA (48). About half of the unique clones map to the 5′ end of known gene promoters. Human neuron genomic DNA (2 µg) was subjected to MseI cutting, leaving CpG islands intact, and linker ligation followed by BstUI and HpaII digestion. Methylated fragments were amplified by linker ligation-mediated PCR. Amplicons from CPT-treated human neuron DNA samples were labeled with Cy5 by direct incorporation in a random priming reaction using the Klenow fragment. Vehicle-treated control DNA was treated accordingly and labeled with Cy3. Array image acquisition was done using GeneChip Scanner 3000. Arrays were analyzed with Microarray suite software. Features with poor signal-to-noise ratios or saturated pixels were excluded from the analysis. After normalization, the microarray data were imported into Affymetrix microarray suite software for analysis. The individual spot sequence identities with differential methylation signals were determined from automated sequencing of the genomic fragment from the clone and comparison to online databases.
For mouse methylated CpG island DNA microarrays, we used NimbleGen RN34 Meth 3x720K mouse CpG plus promoter arrays. This array contains all known annotated CpG islands and well-characterized RefSeq promoter regions covered by ∼720,000 probes. Immunoprecipitation of methylated DNA was performed using BiomagTM magnetic beads coupled to monoclonal antibody to 5-methylcytosine (clone 33D3, Active Motif, Carlsbad, CA). The total input and matched immunoprecipitation DNA were labeled with Cy3- and Cy5-labeled random 9-mers, respectively, and then were hybridized to arrays. An Axon GenePix 4000B microarray scanner was used.
Statistical Analysis
All quantitative data on mitochondrial morphology, cell death, DNA damage/repair, and Western blotting are expressed and mean ± standard deviation. Human, mouse, and pig neurons from 3 independent cultures were used. Samples for each assay were run in triplicate. Statistical analysis among groups was performed with one-way analysis of variance (ANOVA) or two-way ANOVA followed by the Duncan’s multiple range test, with p < 0.05 considered statistically different.
For the analysis of microarrays, raw data were extracted as pair files by NimbleScan software (Roche NimbleGen, Inc.). Median-centering quartile normalization and linear smoothing by Ringo, Limma, and MEDME was performed. From the normalized log2-ratio data, a sliding-window peak-finding algorithm, as part of NimbleScan v2.5 (Roche NimbleGen, Inc.), was used to find the methylated CpG enriched peaks with specified parameters (sliding window width: 750 bp; mini probes per peak: 2; p value minimum cut-off: 2; maximum spacing between nearby probes within peak: 500 bp). A Kolmogorov-Smirnov test was used to assign a -log10 p value enrichment score. The hybridization results of promoter CpG island microarrays are presented as mean log2 ratios between CPT-treated (E, experimental) and vehicle-treated (C, control) human or mouse neurons of 3 independent microarray experiments. To compare promoters with differentially enriched methylation between in the E and C groups, log2-ratio values were averaged for the calculation of the M′ value [M′ = Average(log2 fluorescence intensity signal MeDIPE/fluorescence instnsity signal InputE)−Average(log2 fluorescence intensity signal MeDIPC/fluorescence intensity signal InputC)] for each probe. To find the differential enrichment peaks, a NimbleScan sliding-window peak-finding algorithm was used. The differential enrichment peaks were filtered by: at least 1 of the 2 groups having a median value of log2 MeDIP/Input ≥0.3 and a median value of M′ >0 within each peak region; and at least half of the probes in a peak having a median value of coefficient of variability ≤0.8 in both groups within each peak region. A fold-change of 1.5 was generally the cut-off for significance (p < 0.05).
Figure Generation
Figures were prepared using Microsoft Excel, SigmaStat, CorelDraw, and Adobe Photoshop. Images might have been adjusted for brightness, tone, or contrast without altering any content.
RESULTS
Cell Culture and Differentiation of Human Neurons and Animal Forebrain Neurons
Our human NSC-derived neuron culture has been described (30), but more characterization of the cultured differentiated human neurons is presented here (Fig. 1). Robust and healthy human neurons were differentiated from human neuroprogenitors derived from embryonic H9 stem cells (Fig. 1). H9-NSCs differentiated into pyramidal-like neurons and developed extensive processes and neuritic networks as seen by differential interference microscopy of living neurons (Fig. 1A). These observations were substantiated after cell fixation and immunophenotyping for the neuron-specific cytoskeletal protein MAP2, the synaptic protein synaptophysin (Fig. 1B, C), and the cortical neuron transcription factor TBR1 (Fig. 1C inset). MAP2-positive human neurons had bipolar and multipolar cell bodies and long, branching dendritic arborizations (Fig. 1B, red). Elaborate axonal differentiation was inferred in human neuron cultures by the formation of numerous presynaptic boutons, identified by synaptophysin that formed apparent axosomatic and axodendritic synapses (Fig. 1B, C, green). Despite the presence of apparent robust health and elaborate morphological differentiation, human neurons in culture can often be electrically silent (30), unlike healthy mouse neuron cultures that tend to display robust spontaneous activity and induced action potentials (31). Whole-cell patch-clamp methods demonstrated that these human cells differentiated into functional neurons (Fig. 1G–I). These human neuron cultures showed the capacity to generate action potentials in response to current injections (Fig. 1G). Tetrodotoxin (TTX; 0.5 μm) blocked the action potentials (data not shown). In voltage-clamp mode, large inward and outward currents were induced in human neurons by incremental depolarization steps between −70 mV to +20 mV (10 mV increments) from a holding potential of −110 mV (Fig. 1H). The fast-activating, fast-inactivating inward currents were blocked by sodium channel blocker TTX (0.5 μM) (data not shown). To demonstrate human neurons established functional synaptic networks in culture, as predicted by the synaptic marker immunostaining (Fig. 1B, C), we analyzed continuous whole-cell voltage clamp recordings. At the holding potential of −70 mV, spontaneous postsynaptic currents were detected in human neurons (Fig. 1I). The HCN cell line was much less favorable than the H9-NSCs in generating neurons and had considerable (>90% of total cells) astrocyte-like cell differentiation (Supplementary Data Fig. 1D), whereas in H9-NSC-derived neuron cultures GFAP-positive cells were <2% of the population.
FIGURE 1.

Human, pig, and mouse forebrain neurons in cell culture characterized by immunofluorescence for neuron-specific markers, and human neurons characterized by patch-clamp recording. (A) Phase contrast microscopy of live human neurons generated by culturing and differentiating human NSCs derived from the H9 human embryonic stem cell line. The human neurons are well differentiated and healthy and form a network with elaborate dendrites. (B, C) Immunofluorescence for MAP2 (red) and synaptophysin demonstrates that the cultured human cells are neurons that have differentiated elaborate dendrites (MAP2, red) decorated with a rich network of synaptic boutons (green dots). Some neurons have a pyramidal neuron-like morphology (white arrow, asterisk identifies cell nucleus) and are strongly positive for the cortical neuron marker TRB1 (C, inset). (D, E) Pig forebrain NSC-derived neurons were positive for MAP2 (D) and for MAP2 and β-synuclein (E). Many cells were positive for the cortical neuron marker TRB1 (E inset). (F) Mouse forebrain NSC-derived neuronal cultures were enriched in the neuronal markers synaptophysin and β-synuclein. (G) Electrophysiological properties of human NSC-derived neurons. A current-clamp recording from a human neuron showing action potentials in response to current injection. (H) Whole-cell voltage-clamp recordings showing voltage-dependent sodium and potassium currents. (I) Cultured human neurons showed spontaneous postsynaptic currents. Scale bars: A, B = 25 µm; C, D = 15 µm; E, F = 12 µm; C inset, same for E, F insets = 11 µm.
The differentiation of neurons from mouse and pig forebrain NSCs was similarly characterized by enrichments of neuronal cytoskeletal and synaptic markers and minor GFAP staining. Differentiated pig neurons showed elaborate MAP2-positive cell bodies and dendrites that were decorated with axosomatic and axodendritic synapses positive for βSyn (Fig. 1D, E). NSC-differentiated mouse neurons were enriched in βSyn-positive neuronal cells bodies as well as βSyn- and synaptophysin-positive presynaptic terminals (Fig. 1F) and were similar to embryonic mouse primary cortical neurons (data not shown). Both pig and mouse neurons showed high percentages of cells positive for TBR1 (Fig. 1E, F insets). These findings are consistent with previous descriptions (32, 33, 35–37, 39, 49) and further demonstrate that our neuronal cell cultures from different species have comparable health and viability and can be used appropriately as models for comparative neuronal toxicity and death.
Mitochondria in Living Cultured Human Neurons and Mouse Neurons Appear Different
To identify mitochondria in living human and mouse neurons, cell cultures were transfected with a mitoDsRed plasmid that drives the expression of red fluorescent protein specifically within mitochondria (38, 50). Subsets of human and mouse neurons showed exquisitely visible mitochondria (Supplementary Data Fig. 1A, B). The distributions and morphology of mitochondria within human and mouse neurons appeared different (Supplementary Data Fig. 1A, B). Mitochondria within human neurons assumed larger aggregated or clustered networks compared to mouse neurons, particularly at perinuclear locations (Supplementary Data Fig. 1A, B). Further analysis of individual mitochondria revealed that the lengths of human neuron mitochondria were greater compared to mouse mitochondria, but diameters were similar (Supplementary Data Fig. 1C). Most HCN cells differentiated into astrocyte-like cells with a flat broad morphology seen by DIC (Supplementary Data Fig. 1D). DsRed-tagged mitochondria in astrocyte-like cells appeared in patterns very distinct from human neurons (Supplementary Data Fig. 1E).
Nuclear and Chromatin Changes in Degenerating Human Neurons and Mouse Neurons Appear Different
When human and mouse neurons were differentiated from NSCs and then treated similarly with identical doses of DNA damaging insult, such as CPT, the nuclear alterations in degenerating neurons were distinctly different in human and mouse neurons. Exposure of human and mouse nonneural and neural cells to CPT induces DNA damage, interference with DNA-dependent RNA polymerase, and apoptosis; it is believed that CPT triggers apoptosis in neurons by induction of DNA single-strand breaks and activation of DDR (37, 51–55). In differentiated human neurons, DNA staining showed that the chromatin condensation in CPT-treated cells was generally homogeneous and uniform within the nucleus but did not form discrete round chromatin clumps or crescentic marginations at the nuclear envelope (Fig. 2A), as in typical neuronal apoptosis (56). Throughout the time course of cell death (Fig. 2C), most differentiated degenerating human neurons underwent pyknosis apparently without fragmentation of the nucleus and cytoplasm into round masses (Fig. 2A). This feature of degenerating human neurons also has been seen in vivo in autopsy tissue of adult and newborn spinal cord and brain from motor neuron disease patients (ALS and spinal muscular atrophy) and in newborn brain with HIE using conventional staining and DNA fragmentation labeling in tissue sections (16, 57–59). In contrast, DNA staining showed the morphological signature of apoptosis (56) in mouse neurons with condensation of the chromatin into round masses and the fragmentation of the nucleus and cytoplasm into small round masses (Fig. 2B). This feature is consistent in mouse neurons whether they are derived from NSCs or embryonic cerebral cortex, confirming previous reports with primary neurons (35, 36). This mode of degeneration was uniform and progressive (Fig. 2C) in mouse differentiated neurons.
FIGURE 2.

Degenerating human neurons and mouse neurons have different morphological and biochemical features. (A, B) Differentiated human neurons and mouse neurons were treated with 10 μm DNA damaging agent camptothecin (CPT, a DNA topoisomerase-I inhibitor) or vehicle (control) and then fixed after 24 hours and stained with DNA dye Hoechst 33258. The degenerating human neurons treated with CPT have a uniformly bright nonfragmented nucleus (arrows), while the degenerating mouse neurons treated with CPT have a nucleus with that is fragmented into 2 or 3 round masses indicative of classic apoptosis (arrows). (C) Graph showing the time course of nuclear condensation as seen in DAPI-stained fixed differentiated human neurons and mouse neurons treated with vehicle or CPT for 4, 8, 16, and 24 hours. (D) Western blotting for phospho-cofilin in differentiated human and mouse neurons treated with vehicle or CPT for 4, 8, 16, and 24 hours. Ponceau S-stained (protein-binding dye) membranes are shown as loading controls to verify equivalent protein lysate input per lane. Graph (D, right) shows the densitometric analysis of phosphocofilin immunoreactivity. Mouse neurons: significantly above vehicle control (*p < 0.05) and (**p < 0.01); significantly below control (***p < 0.01). Human neurons: significantly above vehicle control (+p < 0.05, ++p < 0.01, +++p < 0.001). Scale bars: A = 10 µm; B = 15 µm.
Differences in dying human and mouse neurons were also revealed by Western blotting for cofilin (Fig. 2D). Cofilin functions in actin dynamics and participates in cytoplasmic and nuclear events associated with apoptosis (60). In apoptotic mouse neurons treated with CPT, cofilin levels were elevated significantly at 4–16 hours and then sharply declined (Fig. 2D). In contrast, in CPT-treated human neurons cofilin levels progressively increased significantly to high enrichment through 4–24 hours, with 4-fold elevations achieved at 24 hours (Fig. 2D).
Caspase Activation Is Different in Dying Human Neurons and Mouse Neurons
To verify that human and mouse neurons were undergoing cell death, caspase activation was assessed (Fig. 3A–C). Caspase isoform utilization was different in human and mouse neurons exposed to CPT (Fig. 3A–C). Mouse neurons underwent apoptosis in response to a lethal DNA damaging insult with a strong progressive increase in caspase-3 biochemical activity at 2–8 hours of CPT exposure compared to culture-matched, vehicle-treated mouse neurons (Fig. 3A), consistent with numerous other studies (35–37, 61). In contrast, CPT-treated human neurons showed only slight increases in caspase-3 biochemical activity at 8 hours of similar treatment (Fig. 3A). Because of studies showing changes in caspase-6 activity and immunostaining in primary human neurons and in postmortem AD brain (62, 63) we assayed for caspase-6 activity. Human neurons had a strong progressive increase in caspase-6 biochemical activity at 2–8 hours of CPT exposure (Fig. 3B) compared to culture-matched vehicle-treated human neurons, but mouse neurons did not (Fig. 3B). This biochemical analysis was corroborated by Western blotting that showed minor accumulation of cleaved caspase-3 in CPT-treated human neurons, while cleaved caspase-6 accumulated robustly (Fig. 3C).
FIGURE 3.

Degenerating human neurons and mouse neurons use different caspases and use Bcl-2 family members temporally different. (A) Caspase-3 activity, determined by biochemical colorimetric assay, is increased abruptly and robustly in mouse forebrain neurons in a time-related manner after 10 µM CPT exposure. In contrast, human neurons have significantly less activation of caspase-3 activity compared to similarly cultured mouse neurons. Values (in units × 10−1) are mean ± SD (n = 3 different cultures). Significantly greater *p < 0.05, **p < 0.001 than control (same species comparison); +p < 0.01 and ++p < 0.001 (time-matched comparison of mouse neurons vs human neurons). (B) Caspase-6 activity, determined by biochemical colorimetric assay, is increased robustly in differentiated human forebrain neurons in a time-related manner after 10 µM CPT exposure. In contrast, mouse neurons have significantly less activation of caspase-6 activity compared to similarly cultured human neurons. Values (in units × 10−1) are mean ± SD (n = 3 different cultures). (C) Western blots for cleaved caspase-3 and cleaved caspase-6 in cell lysates of differentiated human neurons treated with DNA damaging agent (10 µM CPT). Degenerating human neurons display only a modest increase in cleaved caspase-3, while cleaved caspase-6 accumulates abruptly and robustly by 4–8 hours (h) and the accumulation progresses through 16 hours. Ponceau S-stained blot is shown to verify equivalent protein loading per lane. (D, E) Differentiated human neurons (D) and mouse neurons (E) were treated with vehicle or 10 µM CPT for 4, 8, 16, and 24 hours, and then total cell lysates were harvested for Western blotting for Bax, Noxa, and Puma. GAPDH Western blots show protein loading per lane. Graphs at right summarize Western blot densitometry results. Compared to vehicle-treated human neurons, CPT-treated human neurons showed significant increases in Bax at 16 hours (*p < 0.05), Noxa at 4, 8, and 16 hours (*p < 0.05, **p < 0.01, ***p < 0.001, respectively), and Puma at 8, 16, and 24 hours (*p < 0.05, ***p< 0.001, **p < 0.01, respectively). Compared to vehicle-treated mouse neurons, CPT-treated mouse neurons showed significant increases in Bax at 4 and 8 hours (**p < 0.01, *p < 0.05, respectively) and significant decreases at 16 and 24 hours (+p < 0.05), significant decreases (+p < 0.05 or ++p < 0.01) in Noxa at 4, 8, 16, and 24 hours, and significant decreases (+p < 0.05) in Puma at 16 and 24 hours.
Bcl-2 Family Mitochondrial Death Protein Levels Change Differentially in Dying Human Neurons and Mouse Neurons
The intrinsic mitochondrial cell death pathway (16) was examined in human and mouse neurons treated with CPT (Fig. 3D, E). In human neurons, Bax immunoreactivity was elevated significantly at 16 hours but remained similar to control levels at other times (Fig. 3D). Noxa levels in human neuron lysates progressively increased significantly at 4–16 hours and then were back to control low levels at 24 hours (Fig. 3D). An almost similar pattern to Noxa was seen with Puma (Fig. 3D). In contrast, in lysates of CPT-treated mouse neurons, Bax immunoreactivity was found significantly increased acutely at 4 h and then was lower than control levels at 16–24 hours (Fig. 3E). Noxa levels in apoptotic mouse neurons showed sustained decrements compared to control, while Puma immunoreactivity was found elevated at 4 hours and then levels were maintained similar to control levels (Fig. 3E).
DDR Is Different in Human Neurons and Mouse Neurons
Our experiments so far indicate that downstream cell death pathways are activated differently in human neurons and mouse neurons, so we explored upstream cell death signaling. An upstream DNA damage sensor that has been studied in nonneuronal cells is the meiotic recombination 11 homolog A protein (Mre11)-Rad50-Nijmegen breakage syndrome-1 protein (Nbs1) complex (MRN) (64). Nbs1 (nibrin) is phosphorylated by ataxia telangiectasia mutated protein kinase (ATM) in response to DNA double-strand breaks (65). After CPT treatment, mouse neurons did not robustly activate the MRN complex as detected by serine-343 phosphorylated Nbs1 (Fig. 4A, E). In contrast, human neurons with a CPT-DNA damaging insult rapidly and strongly activated the MRN complex as seen by Nbs1 phosphorylation (Fig. 4A, D).
FIGURE 4.

The DDR is different in human neurons and mouse neurons consistent with species-dependent DNA damage accumulation in response to DNA damaging agent and excitotoxic insults. (A) Differentiated human and mouse neurons treated with 10 μm DNA damaging agent CPT show different DNA damage sensor activation patterns assessed by Western blotting for phosphorylated Nbs1. Western blots for β-actin are shown as loading controls to verify equivalent protein lysate input per lane. (B, C) Representative Western blots for phospho-ATR (pATR), phospho-p53 (pp53), and phospho-p73 (pp73) in vehicle-treated control and CPT-treated differentiated human neurons and mouse neurons. Actin Western blots show protein loading per lane. (D) Graph summarizing Western blot densitometry results for DDR in human neurons. Compared to vehicle-treated human neuron lysates, CPT-treated human neurons showed significant increases (*p < 0.01) in pNbs1 and pATR at all time points. (E) Graph summarizing Western blot densitometry results for DDR in mouse neurons. Compared to vehicle-treated mouse neuron lysates, CPT-treated mouse neurons showed significant decreases in pNbs1 at all-time points (**p < 0.001 or *p < 0.01), significant decreases in pATR at all-time points (**p < 0.01 or *p < 0.05), significant increases (++p < 0.001) in pp53 at all time points, and significant increases (++p < 0.001 or +p < 0.01) in pp73 at all time points. (F) Mouse neurons without (left, control) and with (right, CPT) DNA damage (DNA-SSBs) as visualized by single-cell gel electrophoresis (the comet assay). Living, differentiated mouse cortical neurons were treated with vehicle (left) or 10 μm CPT (right) and subjected to alkaline comet assay and DNA staining with SYBR Green. Normal neurons with intact nuclear DNA appear have round nuclei with no tails (A). In neurons with DNA-SSBs (B), some of the DNA elutes out of the nucleus and appears as a tail that gives the appearance of a “comet.” (G) Human, mouse, and pig neurons respond differently to DNA damaging insults. Cultured differentiated neurons were challenged with 10 μm CPT for 8 hours and then harvested for comet assay. Values are mean ± SD. The percentage of mouse neurons with DNA damage was significantly greater (**p < 0.01) than the percentages for human and pig neurons. Pig neurons were also significantly different compared to human neurons but the level of significance was less (*p < 0.05) than for mouse neurons. (H) Human, mouse, and pig neurons respond differently to excitotoxicity by forming different levels of DNA damage. Cultured differentiated neurons were challenged with 10 µM NMDA for 30 mins and then subjected to comet assay. Values are mean ± SD. The percentage of mouse neurons with DNA damage was significantly greater (*p < 0.01) than the percentages for human and pig neurons. Pig neuron response was not different from the human neuron response.
To further explore the potential differences in the DDR of CPT-treated human and mouse differentiated neurons, Western blotting was done for activated ATM/Rad3-related kinase (ATR), p53, and p73, as detected by their phosphorylated forms. p73 is a p53 family member with transcription factor and tumor suppressor activities (66, 67). Human neurons with CPT treatment showed an acute (4 hours) and sustained (8–24 hours) activation of ATR compared to culture-matched vehicle-treated human neurons (Fig. 4B, D), while p53 and p73 activation significantly above control was only transient at 16 hours (Fig. 4B, D). In contrast, mouse neurons with CPT treatment showed an acute (4 hours) and sustained (8–24 hours) activation of p53 and p73, while ATR activation was suppressed at 4 and 24 hours (Fig. 4C, E).
DNA Damage Accumulation and DNA Repair Are Different in Human, Mouse, and Pig Neurons
We studied upstream mechanisms of cell injury in human and mouse differentiated neurons and also included pig forebrain neurons because this species is used to model neonatal/pediatric ischemic and traumatic brain injury (68–70) and has a neuropathology more similar to the human clinical picture than neonatal rodent models of cerebral ischemia (59, 69). Human, mouse, and pig forebrain neuroprogenitors were cultured to differentiation and then, under similar culture conditions, were exposed to identical insults, including topoisomerase inhibition, excitotoxicity, and oxidative stress (Fig. 4F–H). These insults are thought to contribute to the pathology of a variety of neurological disorders, including AD, Parkinson disease, ALS, and neonatal/adult stroke (16, 71). A readout for cell injury was DNA-SSB formation based on single-cell gel electrophoresis (comet assay) (Fig. 4F).
Mouse neurons showed greater vulnerability to accumulate DNA damage than human and pig neurons in response to different types of physiological challenges (Fig. 4G, H). After exposure to the DNA Topo1 inhibitor CPT, mouse neurons exhibited more DNA damage than human and pig neurons (Fig. 4G). Similarly, mouse neurons showed more DNA damage accumulation than human and pig neurons after exposure to NMDA glutamate receptor excitotoxicity, while human and pig neurons show similar responses (Fig. 4H). DNA damage accumulation following oxidative and nitrative stress can also be measured by the comet assay (45). Human and pig neurons showed similar DNA damage responses to H2O2, NO donors (sodium nitropruside and NONOate), and peroxynitrite (ONOO), while mouse neurons had significantly higher levels of genome damage (Fig. 5A–C).
FIGURE 5.

Human, mouse, and pig neurons respond differently to oxidative and nitrative stress and have different DNA repair kinetics. (A–C) Cultured differentiated neurons were challenged with hydrogen peroxide (H2O2), nitric oxide donors (sodium nitroprusside [SNP] and NONOate) and peroxynitrite (ONOO) for 30 mins and then subjected to comet assay. Values are mean ± SD. Human neurons (A), mouse neurons (B), and pig neurons (C) all developed DNA damage after exposures to ROS and reactive nitrogen species. All types of neurons were most sensitive to ONOO because more DNA damage was found these groups. DNA damage accumulations in human and pig neurons were similar with exposure to H2O2, SNP, and NONOate (*p < 0.05), while mouse neurons had more severe DNA damage accumulation (*p < 0.01) compared to human and pig neurons. (D–F) Human neurons have more efficient DNA single-strand break (SSB) repair compared to mouse and pig neurons in cell culture. Differentiated human neurons (D), mouse neurons (E), and pig neurons (F) neurons were challenged with 10 μm CPT for 1 or 4 hours, washed, allowed to recover for 1 or 4 hours, and then subjected to comet assay to determine levels of DNA single-strand break repair as calculated by the comet moment (DNA intensitytail×tail area). Values are mean ± SD. Differentiated human neurons (D) treated with CPT accumulated significant levels of DNA damage (*p < 0.05; +p < 0.001 compared to vehicle-treated control) and showed efficient time-related repair DNA-SSBs (**p < 0.01). In contrast, CPT-treated mouse neurons (E) accumulated greater amounts of DNA damage compared to CPT-treated human neurons (**p < 0.01; +p < 0.001 compared to control) and were not successful at repairing DNA damage over a 4-hour recovery period. CPT-treated pig neurons (F) also accumulated significant levels of DNA damage (*p < 0.05; +p < 0.001 compared to vehicle-treated control neurons) and were significantly successful at repairing DNA (*p < 0.05).
DNA repair activity in cells can be evaluated using the comet assay (46, 72). Human, mouse, and pig neurons were subjected to a low-dose DNA damaging insult and then permitted to recover (Fig. 5D–F). The comet moment represents a calculation of the amount of DNA-SSBs in the comet tail (72). Human neurons displayed highly efficient DNA repair, as evidenced by lower comet moment (Fig. 5D). Comparatively, injured mouse neurons did not have efficient DNA repair in this paradigm (Fig. 5E). Injured pig neurons had DNA repair efficiency closer to human neurons than to mouse neurons (Fig. 5F).
DNA Methylation Profiles in Injured Human Neurons and Mouse Neurons Are Different
Genome-wide promoter CpG island DNA methylation profiling was done as a high-throughput screen for additional proof-of-concept that injury responses at the chromatin level are different in human neurons and mouse neurons. Gene promoter methylation signatures in neurons in response to CPT treatment were very distinct and species-specific (Tables 1 and 2). Promoter hypomethylation was the major response of CPT-treated human neurons compared to vehicle-treated human neurons. Of the significant differentially methylated genes <1% showed hypermethylation (Table 1). The changes in promoter methylation in human neurons were highly significant (Table 1). Only one gene, Foxred1, registered a significant hypermethylation (Table 1) and, interestingly, it encodes a mitochondrial chaperone protein for complex I (73). Many of the genes found to be hypomethylated in injured human neurons (Table 1) encoded nuclear proteins that function in transcriptional repression, DNA repair, and in the ubiquitin-proteasome pathway (74–78). In contrast to human neurons, CPT-treated mouse neurons showed much greater diversity in their DNA methylation responses (Table 2). Of the mouse gene promoters registering significant differential methylation in CPT-treated neurons compared to vehicle-treated neurons, 42% were hypermethylated and 58% were hypomethylated (Table 2). Consistent with the propensity of mouse neurons to undergo apoptosis, they showed hypermethylation of the DNA repair enzyme Neil1 (79) and hypomethylation of genes involved in cell death, such as Bak and DNases controlling internucleosomal fragmentation of DNA (Table 2). An unanticipated pattern that emerged was the hypomethylation of several genes encoding small nucleolar and nuclear RNAs (Table 2).
TABLE 1.
Differentially Methylated Promoter CpG Islands in Degenerating Human Neurons
| Gene | Entrez Gene | Chromo-some | Aliases/Name | UniProtKB ID | Methylation Change | Significance p Value | Functions |
|---|---|---|---|---|---|---|---|
| FOXRED1 | 55572 | 11 | FAD-dependent oxidoreductase domain-containing protein 1; H17 | Q96CU9 | Hyper | 6.8×10−8 | Mitochondrial chaperone protein for complex I |
| UBE2V2 | 7336 | 8 | Ubiquitin-conjugating enzyme E2 variant 2; MMS2 | Q15819 | Hypo | 1.3×10−7 | Catalyzes noncanonical poly-ubiquitination to mediate transcriptional activation; DDR |
| GTPBP4 | 23560 | 10 | GTP binding protein 4; nucleolar GTP-binding protein 1 (NGB) | Q9BZE4 | Hypo | 7.9×10−6 | Signal transduction; 60S ribosomal subunit biogenesis |
| RHOU | 58480 | 1 | Ras homolog family member U/WRCH-1 | Q7L0Q8 | Hypo | 1.3×10−5 | Rho family GTPase; regulation of cell morphology and cytoskeletal organization |
| CPS1 | 1373 | 2 | Carbamoyl-phosphate synthase 1, mitochondrial | P31327 | Hypo | 1.4×10−8 | Urea cycle enzyme; core mitochondrial nucleoid protein; antiapoptotic |
| ENC1 | 8507 | 5 | Ectodermal-neural cortex; Nuclear matrix protein NRP/B | O14682 | Hypo | 5.9×10−5 | p53-induced gene 10; oxidative stress response and regulation of Nrf2; neuron-specific |
| SIM2 | 6493 | 21 | Single-minded family BHLH transcription factor 2 | Q14190 | Hypo | 1.3×10−7 | Major gene in CNS development in cooperation with Arnt; ubiquitination target of parkin; related to Down syndrome phenotypes |
| MAGEE1 | 57692 | X | Melanoma-associated antigen (MAGE) family member E1; DAMAGE (dystrobrevin associated MAGE protein) | 57692 | Hypo | 2.5×10−6 | Nuclear protein that enhances ubiquitin ligase activity of RING-type zinc finger-containing protein ligases; links to mental retardation |
| TEAD2 | 8463 | 19 | TEA domain transcriptional enhancer factor 4 | 8463 | Hypo | 1.5×10−4 | Involved in gene regulation of neuronal development; mediates expression of YAP1 |
| SORCS3 | 22986 | 10 | Sortilin-related VPS10 domain containing receptor 3 | Q9UPU3 | Hypo | 2.6×10−7 | Vacuolar protein sorting (VPS); variants associated with Alzheimer’s disease |
| ZEB2 | 9839 | 2 | Zinc finger E-box binding homeobox 2; ZFHX1B; SMAD interacting protein 1(SIP1) | O60315 | Hypo | 6.8×10−6 | Nuclear protein that functions as a DNA-binding transcriptional repressor; interacts with activated SMADS; protects against DNA damage-induced apoptosis |
| SIL1 | 64374 | 5 | SIL1 nucleotide exchange factor; NiP-associated protein; SIL1-like protein ER chaperone | Q9H173 | Hypo | 1.5×10−6 | ER resident protein; function in UPR |
| SMURF2 | 64750 | 17 | SMAD specific E3 ubiquitin protein ligase 2 | Q9HAU4 | Hypo | 1.1×10−6 | Accepts ubiquitin from E2 and transfers it to target protein; interacts with SMAD1 and SMAD7 to trigger their degradation; SMURF2 is neuroprotective in stroke |
| TOP2B | 7155 | 3 | DNA topoisomerase IIβ | Q02880 | Hypo | 2.8×10−5 | Nuclear enzyme controlling topological stated of DNA |
TABLE 2.
Differentially Methylated Promoter CpG Islands in Degenerating Mouse Neurons
| Gene | MGI | Chromo-some | Aliases/Name | NCBI Gene | Methylation Change | Significance p Value | Functions |
|---|---|---|---|---|---|---|---|
| Sfn | 1891831 | 4 | Stratifin; 14-2-2 sigma | 55948 | hyper | 0.05 | Intracellular signaling adaptor protein; cell survival; neuroprotection |
| Mtfr1l | 1924074 | 4 | Mitochondrial fission regulator 1-like; Fam54b | 76824 | hypo | 0.05 | Regulates mitochondrial organization, fission, and aerobic respiration |
| Neil1 | 1920024 | 9 | Nei endonuclease VIII-like 1 | 72774 | hyper | 0.05 | DNA glycosylase; DNA base excision repair |
| Snord32a | 1351324 | 7 | Small nucleolar RNA CD/box 32A; Rnu32; U32 | 27209 | hypo | 0.05 | snoRNA, ribosome biogenesis |
| Snord35a | 1351319 | 7 | Small nucleolar RNA CD box 35A; Rnu35a; U35 | 27211 | hypo | 0.05 | snoRNA, ribosome biogenesis |
| Snord33 | 1351323 | 7 | Small nucleolar RNA CD box 33; Rnu33; U33 | 27208 | hypo | 0.05 | snoRNA, ribosome biogenesis |
| Snord34 | 1351325 | 7 | Small nucleolar RNA CD box 34; Rnu34; U34 | 27210 | hypo | 0.05 | snoRNA, ribosome biogenesis |
| Rnu7 | 97992 | 6 | U7 small nuclear RNA | 19866 | hypo | 0.05 | snRNA; component of Cajal bodies |
| Atp6v0a4 | 2153480 | 6 | ATPase, H+ transporting lysosomal V0 subunit A4; v-ATPase α4 | 140494 | hyper | 0.05 | Intracellular compartment acidification |
| Htra1 | 1929076 | 7 | High-temperature requirement A serine peptidase; insulin-like growth factor binding protein 5 protease; L56; Prss11 | 56213 | hypo | 0.05 | Heat-shock-induced envelope-associated serine protease; chaperone protein and protease; recognizes and degrades misfolded proteins |
| Pdcd1 | 104879 | 1 | Programmed cell death 1; PD-1 | 18566 | hyper | 0.05 | Cell surface membrane protein; immunoregulatory receptor and possible death receptor; regulates cell survival and death |
| Cox8b | 105958 | 7 | Cytochrome c oxidase subunit VIIb; Cox8H | 12869 | hypo | 0.05 | Mitochondrial complex IV protein; oxidative phosphorylation |
| Znrf1 | 2177308 | 8 | Zinc and ring finger 1; Rnf42; Zrfp1 | 170737 | hyper | 0.05 | E3 ubiquitin protein ligase; role in presynaptic terminal maintenance |
| Bak1 | 1097161 | 17 | Bcl2-antagonist/killer 1; N-Bak | 12018 | hypo | 0.05 | Intrinsic mitochondrial death pathway; proapoptotic |
| Slc25a47 | 2144766 | 12 | Solute carrier family 25 member 47 | 104910 | hyper | 0.05 | Mitochondrial uncoupling protein; regulates mitochondrial membrane potential |
| 0610038-B21Rik | 2927595 | 8 | RIKEN cDNA 0610038B21 | 70345 | hyper | 0.05 | Antisense long-noncoding RNA |
| Igf2 | 96434 | 7 | Insulin-like growth factor 2 | 16002 | hyper | 0.05 | Neuronal growth factor; operates through Map kinase signaling |
| DNase 1 | 1103157 | 16 | Deoxyribonuclease 1 | 13419 | hypo | 0.05 | DNA fragmentation during cell death |
| DNase 1-like 3 | 1314633 | 14 | Deoxyribonucleae 1-like 3; DNase gamma | 13421 | hypo | 0.05 | Mediates cleavage of DNA into internucleosomal fragments during apoptosis |
For selected genes, changes in promoter methylation of protein-coding genes in human neurons and mouse neurons were validated at the protein level by Western blotting (Fig. 6A, B). In human neuron lysates, Foxred1 was downregulated and UBE2V2 was upregulated with CPT treatment (Fig. 6A). In mouse neuron lysates, Bak was upregulated and Neil1 was downregulated with CPT treatment (Fig. 6B).
FIGURE 6.
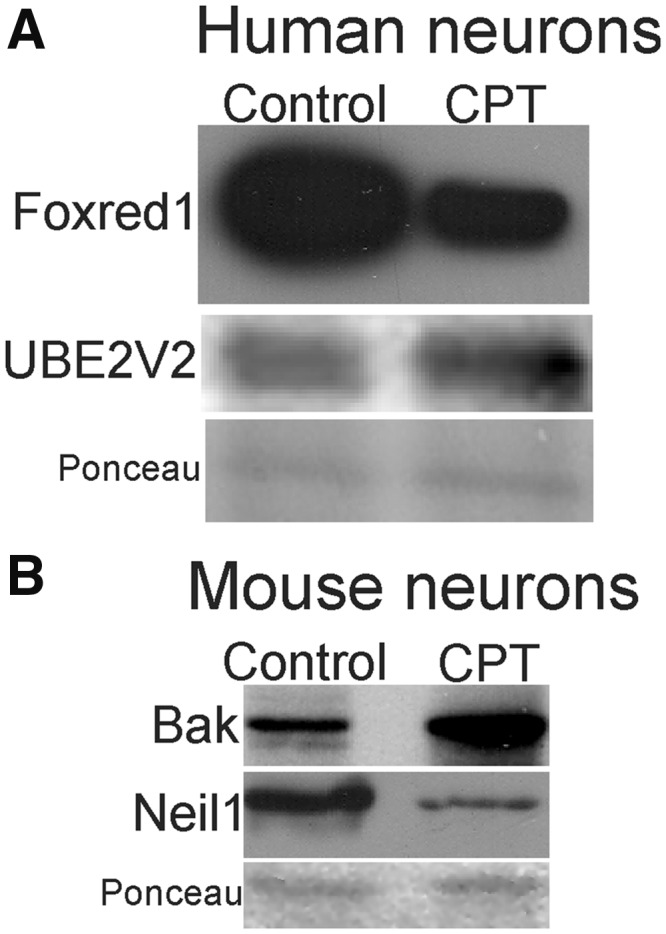
Western blot validation of human (A) and mouse (B) neuron lysates for selected proteins encoded by genes showing significant differential methylation (Tables 1 and 2). Membranes stained for Ponceau S show protein loading.
DISCUSSION
The results of this study are novel and important because they show that human neurons respond to cellular stress and can degenerate and die differently from experimental animal neurons under identical insults. We did several comparisons among human, mouse, and pig differentiated forebrain-derived neurons in cell culture. We found differences in the morphology of mitochondria of human and mouse neurons, differences in the nuclear morphology of dying human and mouse neurons, differences in cell death signaling, and differences in neuronal genome responses to toxic insults such as DNA damage, excitotoxicity, and oxidative/nitrative stress. Notably, the genomic DNA damage response, DNA repair, and cell death mechanisms were different in human neurons compared to mouse and pig neurons. Human and pig neurons had more similar DNA damage/repair responses than did human and mouse neurons. We also found fundamental differences in promoter CpG island DNA methylation patterns between human neurons and mouse neurons. These findings are important because they challenge the long-held assumptions that experimental mammalian animal modeling of nervous system injury and disease is directly reflective of, and relevant to, human neuron biology and should be a launchpad for human CNS disease- and injury-related clinical trials.
Experimental Paradigm
We used cell culture to compare human, mouse, and pig neuronal cell injury and death. The neurons were well differentiated at the time of insult. The neuronal composition and differentiation states of the cell cultures were verified by morphology and immunophenotyping for neuronal and synaptic proteins. The cell cultures under study were robustly positive for neuron-specific cytoskeletal proteins and the presynaptic proteins synaptophysin, αSyn, and βSyn before toxic manipulations. The neurons showed a multipolar morphology and possessed elaborate axodendritic and axosomatic synapses. The human neurons were confirmed as active functionally by patch-clamp recording. An inherent confounder of this experimental design is that, while cultures were maintained similarly and received the same treatments at comparable states of differentiation, they were not identically isochronic due to the differences in the timing of differentiation among human, mouse, and pig cells. While the neurons studied were derived from human, mouse, or pig forebrain NSCs, it is uncertain whether identical subtypes of neurons were represented in the cultures. Another caveat is that, because of the lack of a variable-control O2 incubator, our neurons were cultured under atmospheric O2, which is very high compared to physiologic O2 in tissues. Human and mouse fibroblasts have different sensitivities to O2 (80), and this might be true for neurons. Lastly, the human neurons were female, the mouse neurons had mixed gender, and the pig neurons were male.
This study has an obvious limitation because cultured neurons were examined far-removed from in vivo CNS physiology. However, we reasoned that this strategy, though reductionistic, was a helpful starting point to test the hypothesis that cell autonomous degeneration of CNS neurons is different in different species. It is important to show this fundamental difference in isolated neurons because it is very likely that in vivo tissue modifiers such as vascular physiology, glial microenvironment, immune responses, and other neuropil biology, could render neuronal degeneration even more highly nucanced and species-dependent.
Mitochondrial Morphology Is Different in Human Neurons and Mouse Neurons
We studied the morphology of mitochondria in differentiated human neurons and mouse neurons in culture. Mitochondria were tagged with DsRed (38, 50). We found that human forebrain neuron mitochondria have more complex morphology than mitochondria in mouse forebrain neurons. For example, the length and clustering of human neuron mitochondria was greater than in mouse neuron mitochondria, and human mitochondria appeared as a more interdigitated network within the neuronal cell body and perinuclear vicinity. Moreover, mitochondria in human neurons appeared different from mitochondria in astrocyte-like cells. Our observations are supported by reports showing that mitochondria are heterogeneous in shape (81, 82) and biochemical composition, notably metabolism, cyclophilin D content, and genetics (83–85). Even within a species, the mitochondria of spinal cord and brain show intrinsic differences in metabolic and functional properties (86), sensitivity to Ca2+-induced permeability transition (87, 88), and show sex differences (89, 90).
Nuclear Responses Are Different in Dying Human Neurons and Mouse Neurons
The nuclear changes of differentiated human forebrain neurons during their degeneration were found to be distinct from the nuclear changes seen in mouse differentiated, degenerating forebrain neurons in cell culture. The differences were manifested in the chromatin condensation patterns and the accumulation of cofilin. Cofilin participates in the nuclear program of apoptosis (60). In degenerating human neurons, the nuclear condensation to pyknosis was typically more gradual, subtle, and uniform throughout the nuclear matrix and without the rapid fragmentation of the nucleus into round masses as in mouse neurons. Uncommon nuclear fragmentation during cell death has been shown in Fas-induced death of human embryonic kidney cells (91) and in human embryonal teratoma-derived cells overexpressing truncated fragments of amyloid precursor protein (92). Evident human neuron degeneration with DNA double strand-break accumulation, but without a signature apoptotic-like nuclear change (56), is seen commonly in postmortem human CNS tissue sections of individuals with ALS, Parkinson’s disease, Huntington’s disease, AD, and spinal muscular atrophy (15, 16, 57, 58, 93, 94). In human neonatal HIE, the nuclear features of neocortical neurodegeneration are distinct from rodent neurons (59), and the biochemical signatures of human neuronal cell death in situ also show differences from those seen in ischemic rodent brain (59). We do not have an explanation for the differing nuclear morphology features of degenerating human neurons and degenerating rodent neurons, but it could be related to differences in human nuclear matrix and chromatin proteins (95), DNA methylation (Tables 1 and 2; 47), the posttranslational modification of nuclear proteins such as histone methylation (96), or other human-specific proteome differences (97). A number of major differences exist in chromatin organization and epigenetic modification among mouse, nonhuman primate, and human brain cells (96, 98). The apparent morphological uniqueness of human neuron cell death could also be related to uniquely hominid cell death signaling, as seen in other intracellular signaling pathways (99).
Cell Death Signaling Is Different in Human Neurons and Mouse Neurons
We found that Bcl-2 family and caspase-cell death proteins have different expression and activity patterns in human neurons and mouse neurons during DNA damage-induced cell death. While the levels of some cell death proteins in human and mouse differentiated neurons, such as Bax, differed only temporally after insult, other cell death proteins underwent more striking changes in their levels that are indicative of fundamentally different response patterns intrinsic to human neurodegeneration and mouse neurodegeneration. For example, Noxa and Puma both were elevated in human neurons during their degeneration, but Noxa and Puma levels decreased in mouse neurons during cell death. This contrast suggests different p53-driven gene responses or mobilizations of existing mRNA pools as supported by our promoter CpG island methylation findings. Caspases appeared to differ fundamentally in their utilization in human neurons and mouse neurons. Human differentiated neurons showed robust activation of caspase-6, but not caspase-3, as evidenced by biochemical activity assay and Western blotting, while mouse neurons showed robust activation of caspase-3. Others have found in toxicity assessments that mouse neurons briskly and strongly activate caspase-3, while human neurons do not activate caspase-3 greatly above baseline (100). Another study has shown that caspase-6 appears to have little role in mouse neuronal cell death (101).
DNA Damage Response and Repair Are Different in Human Neurons and Experimental Animal Neurons
Little is known about DNA damage sensor mechanisms in postmitotic cells like neurons compared to knowledge of DDR in nonneuronal cells (102–104). We found that DNA damage in human differentiated neurons was associated with a DDR characterized by activation of Nbs1 and ATR and their downstream targets, including p53 and p73, but was usually unassociated with classic apoptotic morphology. ATR is a protein kinase that is activated in response to DNA damage (103–105). Another DNA damage sensor prominent in nonneuronal cells is the MRN complex (64). This multiprotein Mre11-Rad50-Nbs1 complex is required for full activation of ATM and ATR (64, 106). The observed activation of p53 in cultured neurons is consistent with its role in neurodegeneration and neuronal death in rodents (16, 61, 107–110) and humans (94). However, p53 signaling mechanisms in human neurons appear to be different from those in mouse neurons because of the slight, transient activation (Fig. 4B, D). This modest activation of p53 in cultured human neurons might account for the finding that p53 accumulates in the nucleus of upper and lower motor neurons in human ALS (94) without apparent activation of a classically apoptotic pathway that is seen stereotypically in rodent neurons. Moreover, in human neonatal HIE, activated p53 accumulates in the nucleus of neocortical neurons without a corresponding appearance of a classically apoptotic mode of neurodegeneration (59) that is seen commonly in neonatal rodent neocortex after HIE and excitotoxicity (37, 111). These differences are not surprising in light of the fundamental differences seen with p53-regulated (and many other tumor suppressors) mechanisms in human and mouse cancer biology (112).
Comparative studies of DNA damage and repair in different species in vivo are uncommon due to major differences in aging, nutrition, and lifestyle. An informative approach to address this topic is a cell culture system where the experimental conditions and readouts can be normalized and equivalent. We studied DNA damage accumul ation and DNA repair in human, mouse, and pig neurons after cytotoxic insults using the comet assay. A surprising finding was that the mouse neuron genome was more sensitive to the insults compared to the human and pig neuron genomes under similar culture conditions. Moreover, human neurons generally had more efficient DNA repair than mouse neurons. Pig neurons had responses more similar to human neurons than to mouse neurons. Our findings are consistent with the observation that mouse fibroblasts sustain greater burdens of oxidative stress and DNA damage than human fibroblasts when grown in 20% oxygen (80, 113) and that human and mouse cells undergo senescence differently (113, 114). Moreover, our results are in accord with findings showing that cultured mouse embryonic stem cells are less efficient than human counterparts in rejoining ionizing-radiation-induced DNA-DSBs (115) and that the mRNA expression profiles of several DDR and DNA repair genes in liver are higher in human compared to mice (116). Our observation that MRN activation, shown by the phosphorylation of Nbs1 (nibrin), occurs in human neurons but not in mouse neurons is consistent with this other work.
Precedent for the Concept of Human-Specific Neuronal Cell Injury, Degeneration, and Death Mechanisms
Independent evidence has accrued that supports the premise that human neuron DDR and cell death could be different from neurons in other mammalian species, such as rodents. For example, DNA-binding events for some transcription factors, eg forkhead box (FOX) A1, hepatic nuclear factor (HNF) HNF1A, HNF4A, HNF6, are 41%–89% species-specific for mouse versus human (117). Our DNA methylation results support differences in transcription factor activation in injured human and mouse neurons. Major species differences exist in cell death related genes. Murine and human bax, fas, and caspase-6 gene promoter activities are regulated differently by p53 (118-120). Our DNA methylation experiments also revealed differential activation of cell death-related genes in injured human and mouse neurons. The hypomethylation of the intrinsic mitochondrial death effector bak gene and DNase genes that mediate internucleosomal digestion of DNA in mouse neurons highlights a major difference from human neurons. Even at baseline, human-specific signatures in the cerebral cortex transcriptome exist in vivo (121); indeed, this work is consistent with the fundamental differences we found in the CpG island methylation in human and mouse neurons differentiated from forebrain NSCs. Moreover, we found in human neurons and mouse neurons a variety of differences in the activation of caspases and in the activation of p53 and p73 as seen at activity and protein levels. These caspase-related observations are not too surprising because in rodent cells, the caspase-12 gene product functions in apoptosis induced by endoplasmic reticulum stress, but in human the caspase-12 gene is a pseudogene or produces a truncated, protease-inactive protein (122). Caspase substrates, for example c-Abl, are also known to be species-specific (123). Moreover, human and mouse caspase-3 activation pathways are different (124), consistent with our observations that reliance on caspase-3 activation is different in dying human neurons and mouse neurons. Genome vulnerability to damage and DDR also differ. There is almost no conservation of functional response elements for genes involved in DNA repair and DNA metabolism among human and rodents (125). Here, we found hypermethylation of the Neil1 gene, a DNA base excision repair gene, and Neil1 protein downregulation in injured mouse neurons but not in human neurons. In support of our DDR data, other studies have shown that repair of radiation-induced DNA-SSBs is different in mouse and human cells (115).
Our argument for human-specific neuronal injury and degeneration is strengthened by genetic experiments in mice. Mice with homozygous null mutations in Atm do not develop neuropathologic features consistent with human AT (126, 127) and mice with human XP- and Cockayne syndrome-causing inactivating mutations in nucleotide excision DNA repair genes do not develop neuropathologic features of XP and Cockayne syndrome (128, 129). Mice with hypomorphic mutations in Nbs1 do not show obvious CNS phenotypes that are seen in humans (130). Lastly, mice harboring human gene mutations in tyrosyl-DNA-phosphodiesterase 1 that cause spinocerebellar ataxia with axonal neuropathy do not develop a neurodegenerative disorder as in humans (131).
The concept of human-specific features of neurodegeneration has been articulated before. In the context of age-related neurodegenerative diseases affecting humans, ALS (132) and AD (133, 134) might be unique to human because of evolutionary adaptations in neocortex. For ALS, this possibility was postulated when comparisons were made between the neuropathology of human sporadic and familial ALS and mouse models of familial ALS (135). While the classification of lower motor neuron disease is applicable for the mouse model, the specific phenotypes of the lower motor neuron pathology in most familial ALS mouse models differed dramatically from human (135, 136). Moreover, upper motor neurons in cerebral cortex are mostly unaffected in most current mouse models of ALS, but disease in these neurons is essential for the clinical diagnosis of human ALS (132). Transgenic pigs might model human ALS better than transgenic mice (137). Our findings on the similarities in DNA damage vulnerability and DNA repair in human neurons and pig neurons support this claim.
Implications for Human-Specific Neuronal Cell Injury and Death Mechanisms for Modeling Neurodegeneration
Uniquely hominid neuronal cell injury response, DDR, and cell death mechanisms would be transformative for experimental neuropathology and the modeling of human CNS injury and disease. Descriptive and translational studies would be impacted strongly. Studies of postmortem human brain, particularly studies of early disease events, would be encouraged further. A failure to recognize this species-related neurobiology possibly contributes to the recurring lack of success of costly clinical trials for neurological disorders. Vigilance and caution would be needed in extrapolating neuroprotection outcomes from model organisms to humans. This concept can provide needed incentive to move away from the status quo to make room for a paradigm shift in experimental neuropathology. The design of preclinical studies would need to be changed fundamentally. The preponderant reliance on mouse and rat as preclinical model organisms might have to be reconsidered for more work on large animal models such as pig and macaque monkeys, and other advanced nonhuman primates, or on other species with long postreproductive life spans (22). These genera likely have neuronal injury and neuropathological outcomes more similar to human (68, 138) as well as similar DDR. We found that the DNA repair capacity of pig neurons in the presence of oxidative stress, excitotoxicity, and DNA damage is more similar to that seen in human neurons compared to mouse neurons. One practical extension of our findings is that the screening of drug candidates in the therapeutic pipeline for efficacy in neuroprotection would benefit from high-throughput assays on cultured human, nonhuman primate or pig neurons derived from NSCs and induced pluripotent stem cells, in addition to in vivo testing. Chimeric rodents with major CNS cellular constituents humanized by human NSC-derived neurons and human genes could be developed for in vivo testing. If this concept of human-specific neurodegeneration is correct, then specific cellular mechanism-based therapeutics derived from rodent work is not likely to translate efficiently to human. While these observations relate only to a small part of the comprehensive neuronal injury response, they allow for a critical rethinking of fundamental aspects of how human neuronal injury and neurodegeneration are modeled for human therapeutic relevance.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank: Dr Zhiping Liu for seminal work on mouse NSC isolation, culture, neuronal differentiation, and the comet assay; Dr Christian Lesuisse for early work on mouse primary cortical neuron apoptosis; Dr Alyssa Katzenelson for founding work on pig NSC isolation, culture, cryopreservation, neuronal differentiation; Dr Yi (Gloria) Wang for engineering mitochondrial-targeted DsRed expression vectors; Ms Ann Price for work on human, mouse, and pig cell culture and Western blotting and biochemical assays; and Dr Vasiliki Mahairaki for introducing the H9 cell-derived human neuroprogenitor cell line. We gratefully acknowledge Ms Gabrielle Martin for manuscript editing.
FUNDING
This study was supported by grants from the U.S. Public Health Service NIH-NINDS (R01-NS052098 and R01-NS079348) and the JHU-ADRC (AG05146).
The authors have no actual or potential conflicts of interest to declare.
Supplementary Data can be found at http://www.jnen.oxfordjournals.org.
REFERENCES
- 1. Anderton RA, Mastaglia FL.. Advances and challenges in developing a therapy for spinal muscular atrophy. Expert Rev Neurother 2015;15:895–908 [DOI] [PubMed] [Google Scholar]
- 2. Shankaran S, Pappas A, McDonald SA et al. , . Childhood outcomes after hypothermia for neonatal encephalopathy. N Engl J Med 2012;366:2085–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moler FW, Silverstein FS, Holubkov R et al. , . Therapeutic hypothermia after out-of-hospital cardiac arrest in children. N Engl J Med 2015;372:1898–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Moler FW, Silverstein FS, Holubkov R et al. , . Therapeutic hypothermia after in-hospital cardiac arrest in children. N Engl J Med 2017;376:319–29 [DOI] [PubMed] [Google Scholar]
- 5. Besancon E, Guo S, Lok J et al. , . Beyond NMDA and AMPA glutamate receptors: Emerging mechanisms for ionic imbalance and cell death in stroke. Trends Pharmacol Sci 2008;29:268–75 [DOI] [PubMed] [Google Scholar]
- 6. Kikuchi K, Tanaka E, Murai Y, Tancharoen S.. Clinical trials in acute ischemic stroke. CNS Drugs 2014;28:929–38 [DOI] [PubMed] [Google Scholar]
- 7. Stella F, Radanovic M, Canineu PR et al. , . Anti-dementia medications: Current prescriptions in clinical practice and new agents in progress. Ther Adv Drug Saf 2015;6:151–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huntley J, Howard R.. The importance of high quality trials of cognitive interventions in Alzheimer’s disease. Int Psychogeriatr 2016;28:705–6 [DOI] [PubMed] [Google Scholar]
- 9. Salloway S, Sperling R, Fox NC et al. , . Bapineuzumab 301 and 302 Clinical Trial Investigators. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med 2014;370:322–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gordon PH, Moore DH, Miller RG et al. , . Efficacy of minocycline in patients with amyotrophic lateral sclerosis: A phase III randomized trial. Lancet Neurol 2007;6:1045–53 [DOI] [PubMed] [Google Scholar]
- 11. Lenglet T, Lacomblez L, Abitbol JL et al. , . A phase II-III trial of olesoxime in subjects with amyotrophic lateral sclerosis. Eur J Neurol 2014;21:529–36 [DOI] [PubMed] [Google Scholar]
- 12. Cudkowicz ME, van den Berg LH, Shefner JM et al. , . Dexpramipexole versus placebo for patients with amyotrophic lateral sclerosis (EMPOWER): A randomized, double-blind, phase 3 trial. Lancet Neurol 2013;12:1059–67 [DOI] [PubMed] [Google Scholar]
- 13. Cudkowicz ME, Titus S, Kearney M et al. , . Safety and efficacy of ceftriaxone for amyotrophic lateral sclerosis: A multi-stage, randomised, double-blind, placebo-controlled trial. Lancet Neurol 2014;13:1083–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Battelle Technology Partnership Practice. Biopharmaceutical industry-sponsored clinical trials: Impact on state economy. Battelle Memorial Institute 2015:13
- 15. Martin LJ, Liu Z, Troncoso JC, Price DL. Neuronal cell death in human neurodegenerative diseases and their animal/cell models In: Holcik M, LaCasse E, Korneluk R, MacKenzie A, eds. Apoptosis in Health and Disease. Cambridge: Cambridge University Press; 2005:242–315 [Google Scholar]
- 16. Martin LJ. Mitochondrial and cell death mechanisms in neurodegenerative diseases. Pharmaceuticals (Basel) 2010;3:839–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chiò A, Canosa A, Gallo S et al. , . ALS clinical trials: Do enrolled patients accurately represent the ALS population? Neurology 2011;77:1432–7 [DOI] [PubMed] [Google Scholar]
- 18. Elston GN, Benavides-Piccione R, DeFelipe J.. The pyramidal cell in cognition: A comparative study in human and monkey. J Neurosci 2001;21:RC163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Elston GN, Benavides-Piccione R, Elston A et al. , . Pyramidal cells in prefrontal cortex of primates: Marked differences in neuronal structure among species. Front Neuroanat 2011;5:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mohan H, Verhoog MB, Doreswamy KK et al. , . Dendritic and axonal architecture of individual pyramidal neurons across layers of adult human neocortex. Cereb Cortex 2015;25:4839–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zeng H, Shen EH, Hohmann JG et al. , . Large-scale cellular-resolution gene profiling in human neocortex reveals species-specific molecular signatures. Cell 2012;149:483–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gunn-Moore D, Kaidanovich-Beilin O, Gallego Iradi MC et al. , . Alzheimer’s disease in human and other animals: A consequence of postreproductive life span and longevity rather than aging. Alzheimers Dement 2018;14:195–204 [DOI] [PubMed] [Google Scholar]
- 23. Ronnett GV, Hester LD, Nye JS et al. , . Human cortical neuronal cell line: Establishment from a patient with unilateral megalencephaly. Science 1990;248:603–5 [DOI] [PubMed] [Google Scholar]
- 24. Ronnett GV, Hester LD, Nye JS, Snyder SH.. Human cerebral cortical cell lines from patients with unilateral megalencephaly and Rasmussen’s encephalitis. Neuroscience 1994;63:1081–99 [DOI] [PubMed] [Google Scholar]
- 25. Zhang Z, Drzewiecki GJ, Ho JT et al. , . Human cortical neurons (HCN) cell lines: A model for amyloid β toxicity. Neurosci Lett 1994;177:162–4 [DOI] [PubMed] [Google Scholar]
- 26. Hyslop PA, Zhang Z, Pearson DV, Phebus LA.. Measurement of striatal H2O2 by microdialysis following global ischemia and reperfusion in the rat: Correlation with cytotoxic potential of H2O2 in vitro. Brain Res 1995;671:181–6 [DOI] [PubMed] [Google Scholar]
- 27. Wu S-N, Wang Y-J, Lin M-W.. Potent stimulation of large-conductance Ca2+-activated K+ channels by rottlerin, an inhibitor of protein kinase C-δ, in pituitary tumor (GH3) cells and in cortical neuronal (HCN-1A) cells. J Cell Phys 2007;210:655–66 [DOI] [PubMed] [Google Scholar]
- 28. Brock AJ, Kasus-Jocobi A, Lerner M et al. , . The antimicrobial protein CAP37, is upregulated in pyramidal neurons during Alzheimer’s disease. Histochem Cell Biol 2015;144:293–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thomson JA, Itskovitz-Eldor J, Shapiro SS et al. , . Embryonic stem cell lines derived from human blastocysts. Science 1998;282:1145–7 [DOI] [PubMed] [Google Scholar]
- 30. Mahairaki V, Ryu J, Peters A et al. , . Induced pluripotent stem cells from familial Alzheimer’s disease patients differentiate into mature neurons with amyloidogenic properties. Stem Cells Dev 2014;23:2996–3010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chang Q, Martin LJ.. Motoneuron subtypes show specificity in glycine receptor channel abnormalities in a transgenic mouse model of amyotrophic lateral sclerosis. Channels 2011;5:1–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu Z, Martin LJ.. The olfactory bulb core is a rich source of neural progenitor and stem cells in adult rodent and human. J Comp Neurol 2003;459:368–91 [DOI] [PubMed] [Google Scholar]
- 33. Liu Z, Martin LJ.. Pluripotent fates and tissue regenerative potential of adult olfactory bulb neural stem and progenitor cells. J Neurotrauma 2004;21:1479–99 [DOI] [PubMed] [Google Scholar]
- 34. Liu Z, Martin LJ.. The adult neural stem cell and progenitor cell niche is altered in amyotrophic lateral sclerosis mouse brain. J Comp Neurol 2006;497:468–88 [DOI] [PubMed] [Google Scholar]
- 35. Lesuisse C, Martin LJ.. Long-term culture of mouse cortical neurons as a model for neuronal development, aging, and death. J Neurobiol 2002;51:9–23 [DOI] [PubMed] [Google Scholar]
- 36. Lesuisse C, Martin LJ.. Immature and mature cortical neurons engage different apoptotic mechanisms involving caspase-3 and the MAP kinase pathway. J Cereb Blood Flow Metab 2002;22:935–50 [DOI] [PubMed] [Google Scholar]
- 37. Martin LJ, Liu Z, Chestnut B et al. , . Molecular regulation of DNA damage-induced apoptosis in neurons of cerebral cortex. Cerebral Cortex 2009;19:1273–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang Y, Pan Y, Price A, Martin LJ.. Generation and characterization of transgenic mice expressing mitochondrial targeted red fluorescent protein selectively in neurons: Modeling mitochondriopathy in excitotoxicity and amyotrophic lateral sclerosis. Mol Neurodegen 2011;6:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Martin LJ, Katzenelson A, Koehler PC, Chang Q.. The olfactory bulb in newborn piglet is a reservoir of neural stem and progenitor cells. PLoS One 2013;8:e81105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu YQ, Li WQ, Morris-Natschke SL et al. , . Perspectives on biologically active camptothecin derivatives. Med Res Rev 2015;35:753–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nitiss JL, Wang JC.. Mechanisms of cell killing by drugs that trap covalent complexes between DNA topoisomerases and DNA. Mol Pharmacol 1996;50:1095–102 [PubMed] [Google Scholar]
- 42. Nieves-Neira W, Pommier Y.. Apoptotic response to camptothecin and 7-hydroxystaurosporine (UCN-01) in the 8 human breast cancer cell lines of the NCI anticancer drug screen: Multifactorial relationships with topoisomerase 1, protein kinase C, Bcl-2, p53, MDM-2 and caspase pathways. Int J Cancer 1999;82:396–404 [DOI] [PubMed] [Google Scholar]
- 43. Hrabie JA, Klose JR, Wink DA, Keer LK.. New nitric oxide-releasing zwitterions derived from polyamines. J Org Chem 1993;58:1472–6 [Google Scholar]
- 44. Beckman JS, Carson M, Smith CD, Koppenol WH.. ALS, SOD and peroxynitrite. Nature 1993;364:548. [DOI] [PubMed] [Google Scholar]
- 45. Martin LJ, Liu Z.. DNA damage profiling in motor neurons: A single-cell analysis by comet assay. Neurochem Res 2002;27:1093–104 [DOI] [PubMed] [Google Scholar]
- 46. Fayzullina S, Martin LJ.. DNA damage response and DNA repair in skeletal myocytes from a mouse model of spinal muscular atrophy. J Neuropathol Exp Neurol 2016;75:889–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Diede SJ, Yao Z, Keyes CC et al. , . Fundamental differences in promoter CpG island DNA hypermethylation between human cancer and genetically engineered mouse models of cancer. Epigenetics 2013;8:1254–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cross SH, Charlton JA, Nan X, Bird AP.. Purification of CpG islands using a methylated DNA binding column. Nat Genet 1994;6:236–44 [DOI] [PubMed] [Google Scholar]
- 49. Martin LJ, Liu Z.. Adult olfactory bulb neural neural precursor cell grafts provide temporary protection from motor neuron degeneration, improve motor function, and extend survival in amyotrophic lateral sclerosis mice. J Neuropathol Exp Neurol 2007;66:1002–18 [DOI] [PubMed] [Google Scholar]
- 50. Chestnut BA, Chang Q, Price A et al. , . Epigenetic regulation of motor neuron cell death through DNA methylation. J Neurosci 2011;31:16619–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Recher L, Chan H, Briggs L, Parry N.. Ultrastructural changes inducible with the plant alkaloid camptothecin. Cancer Res 1972;32:2495–501 [PubMed] [Google Scholar]
- 52. Horwitz SB, Horwitz MS.. Effects of camptothecin on the breakage and repair of DNA during the cell cycle. Cancer Res 1973;33:2834–6 [PubMed] [Google Scholar]
- 53. Bertrand R, Solary E, Jenkins J, Pommier Y.. Apoptosis and its modulation in human promyelocytic HL-60 cells treated with DNA topoisomerase I and II inhibitors. Exp Cell Res 1993;207:388–97 [DOI] [PubMed] [Google Scholar]
- 54. Morris EJ, Geller HM.. Induction of neuronal apoptosis by camptothecin, an inhibitor of DNA topoisomerase-I: Evidence for cell cycle-independent toxicity. J Cell Biol 1996;134:757–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Enokido Y, Araki T, Tanaka K et al. , . Involvements of p53 in DNA strand break-induced apoptosis in postmitotic CNS neurons. Eur J Neurosci 1996;8:1812–21 [DOI] [PubMed] [Google Scholar]
- 56. Martin LJ, Al-Abdulla NA, Brambrink AM et al. , . Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: A perspective on the contributions of apoptosis and necrosis. Brain Res Bull 1998;46:281–309 [DOI] [PubMed] [Google Scholar]
- 57. Martin LJ. Neuronal death in amyotrophic lateral sclerosis is apoptosis: Possible contribution of a programmed cell death mechanism. J Neuropathol Exp Neurol 1999;58:459–71 [DOI] [PubMed] [Google Scholar]
- 58. Simic G, Seso-Simic D, Lucassen PJ et al. , . Ultrastructural analysis and TUNEL demonstrate motor neuron apoptosis in Werdnig-Hoffmann disease. J Neuropathol Exp Neurol 2000;59:398–407 [DOI] [PubMed] [Google Scholar]
- 59. Northington F, Chavez-Valdez R, Martin LJ.. Neuronal cell death in neonatal hypoxia-ischemia. Ann Neurol 2011;69:743–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kanellos G, Frame MC.. Cellular functions of the ADF/cofilin family at a glance. J Cell Sci 2016;129:3211–8 [DOI] [PubMed] [Google Scholar]
- 61. Keramaris E, Stefanis L, MacLaurin J et al. , . Involvement of caspase-3 in apoptotic death of cortical neurons evoked by DNA damage. Mol Cell Neurosci 2000;15:368–79 [DOI] [PubMed] [Google Scholar]
- 62. LeBlanc A, Liu H, Goodyer C et al. , . Caspase-6 role in apoptosis of human neurons, amyloidogenesis, and Alzheimer’s disease. J Biol Chem 1999;274:23426–36 [DOI] [PubMed] [Google Scholar]
- 63. Guo H, Albrecht S, Bourdeau M et al. , . Active caspase-6 and caspase-6-cleaved tau in neuropil threads, neuritic plaques, and neurofibrillary tangles of Alzheimer’s disease. Am J Pathol 2004;165:523–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lee J-H, Paull TT.. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 2004;304:93–6 [DOI] [PubMed] [Google Scholar]
- 65. Gatei M, Young D, Cerosaletti KM et al. , . ATM-dependent phosphorylation of nibrin in response to radiation exposure. Nat Genet 2000;25:115–9 [DOI] [PubMed] [Google Scholar]
- 66. Mills AA, Zheng B, Wang XJ et al. , . p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 1999;398:708–13 [DOI] [PubMed] [Google Scholar]
- 67. Yang A, McKeon F.. p63 and p73: p53 mimics, menaces and more. Nat Rev Mol Cell Biol 2000;1:199–207 [DOI] [PubMed] [Google Scholar]
- 68. Martin LJ, Brambrink A, Koehler RC, Traystman RJ.. Primary sensory and forebrain motor systems in the newborn brain are preferentially damaged by hypoxia-ischemia. J Comp Neurol 1997;377:262–85 [DOI] [PubMed] [Google Scholar]
- 69. Martin LJ. Mechanisms of brain damage in animal models of hypoxia-ischemia in newborns In: Stevenson DK, Benitz WE, Sunshine P, eds. Fetal and Neonatal Brain Injury: Mechanisms, Management, and the Risks of Practice. 3rd edition New York: Oxford University Press; 2003:30–57 [Google Scholar]
- 70. Friess SH, Ichord RN, Owens K et al. , . Neurobehavioral functional deficits following closed head injury in the neonatal pig. Exp Neurol 2007;204:234–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ferriero DM. Neonatal brain injury. N Engl J Med 2004;351:1985–95 [DOI] [PubMed] [Google Scholar]
- 72. Collins AR. The comet assay for DNA damage and repair: Principles, applications, and limitations. Mol Biotechnol 2004;26:249–61 [DOI] [PubMed] [Google Scholar]
- 73. Lemire BD. Evolution of FOXRED1, and FAD-dependent oxidoreductase necessary for NADH: Ubiquinone oxidoeductase (Complex I) assembly. Biochimica Biophysica Acta 2015;1847:451–7 [DOI] [PubMed] [Google Scholar]
- 74. McKenna S, Spyracopoulos L, Moraes T et al. , . Noncovalent interaction between ubiquitin and human DNA repair protein Mms2 is required for Ubc13-mediated polyubiquitination. J Biol Chem 2001;276:40120–6 [DOI] [PubMed] [Google Scholar]
- 75. Espantman KC, O’Shea CC.. aMAGEing new players enter the RING to promote ubiquitylation. Mol Cell 2010;39:835–6 [DOI] [PubMed] [Google Scholar]
- 76. Lunardi A, Di Minin G, Provero P et al. , . A genome-scale potein interaction profile of Drosophila p53 uncovers additional nodes of the the human p53 network. Proc Natl Acad Sci U S A 2010;107:6322–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kewley RJ, Whitelaw ML, Chapman-Smith A.. The mammalian basic helix-loop-helix/PAS family of transcriptional regulators. Intl J Biochem Cell Biol 2004;36:189–204 [DOI] [PubMed] [Google Scholar]
- 78. Kim T-A, Jiang S, Seng S et al. , . The BTB domain of the nuclear matrix protein NRP/B is required for neurite outgrowth. J Cell Sci 2005;118:5537–48 [DOI] [PubMed] [Google Scholar]
- 79. Rolseth V, Runden-Pran E, Luna L et al. , . Widespread distribution of DNA glycosylases removing oxidative DNA lesions in human and rodent brains. DNA Repair (Amst) 2008;7:1578–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Parrinello S, Samper E, Krtolica A et al. , . Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol 2003;5:741–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Collins TJ, Bootman MD.. Mitochondria are morphologically heterogeneous within cells. J Exp Biol 2003;206:1993–2000 [DOI] [PubMed] [Google Scholar]
- 82. Jensen RE. Control of mitochondrial shape. Cur Opin Cell Biol 2005;17:384–8 [DOI] [PubMed] [Google Scholar]
- 83. Naga KK, Sullivan PG, Geddes JW.. High cyclophilin D content of synaptic mitochondria results in increased vulnerability to permeability transition. J Neurosci 2007;27:7469–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Martin LJ, Gertz B, Pan Y et al. , . The mitochondrial permeability transition pore in motor neurons: Involvement in the pathobiology of ALS mice. Exp Neurol 2009;218:333–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn of evolutionary medicine. Annu Rev Genet 2005;39:359–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Panov AV, Kubalik N, Zinchenko N et al. , . Metabolic and functional differences between brain and spinal cord mitochondria underlie different predisposition to pathology. Am J Physiol Regul Integr Comp Physiol 2011;300:R844–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Sullivan PG, Rabchevsky AG, Keller JN et al. , . Intrinsic differences in brain and spinal cord mitochondria: Implications for therapeutic interventions. J Comp Neurol 2004;474:524–34 [DOI] [PubMed] [Google Scholar]
- 88. Morota S, Hansson MJ, Ishii N et al. , . Spinal cord mitochondria display lower retention capacity compared with brain mitochondria without inherent differences in sensitivity to cyclophilin D inhibition. J Neurochem 2007;103:2066–76 [DOI] [PubMed] [Google Scholar]
- 89. Martin LJ. An approach to experimental synaptic pathology using green fluorescent protein-transgenic mice and gene knockout mice to show mitochondrial permeability transition pore-driven excitotoxicity in interneurons and motoneurons. Toxicol Pathol 2011;39:220–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Demarest TG, McCarthy MM.. Sex differences in mitochondrial (dys)function: Implications for neuroprotection. J Bioenerg Biomembr 2015;47:173–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ranjan K, Surolia A, Pathak C.. Apoptotic potential of Fas-associated death domain on regulation of cell death regulatory protein cFLIP and death receptor mediated apoptosis in HEK 293T cells. J Cell Commun Signal 2012;6:155–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Nishimura I, Uetsuki T, Kuwako K et al. , . Cell death induced by a caspase-cleaved transmembrane fragment of the Alzheimer amyloid precursor protein. Cell Death Differ 2002;9:199–208 [DOI] [PubMed] [Google Scholar]
- 93. Su JH, Anderson AJ, Cummings BJ, Cotman CW.. Immunohistochemical evidence for apoptosis in Alzheimer’s disease. Neuroreport 1994;5:2529–33 [DOI] [PubMed] [Google Scholar]
- 94. Martin LJ. p53 is abnormally elevated and active in the CNS of patients with amyotrophic lateral sclerosis. Neurobiol Dis 2000;7:613–22 [DOI] [PubMed] [Google Scholar]
- 95. Kimura H. Histone modifications for human epigenome analysis. J Hum Genet 2013;58:439–45 [DOI] [PubMed] [Google Scholar]
- 96. Shulha HP, Crisci JL, Reshetov D et al. , . Human-specific histone methylation signatures at transcription start sites in prefrontal neurons. PLoS Biol 2012;10:e1001427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Uhlén M, Fagerberg L, Hallström BM et al. , . Tissue-based map of the human proteome. Science 2015;347:1260419. [DOI] [PubMed] [Google Scholar]
- 98. Mitchell AC, Bharadwaj R, Whittle C et al. , . The genome in three dimensions: A new frontier in human brain research. Biol Psychiatry 2014;75:961–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Stahl PD, Wainszelbaum MJ.. Human-specific genes may offer a unique window into human cell signaling. Sci Signal 2009;2:pe59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Druwe I, Freudenrich TM, Wallace K et al. , . Sensitivity of neuroprogenitor cells to chemical-induced apoptosis using a multiplexed assay suitable for high-throughput screening. Toxicology 2015;333:14–24 [DOI] [PubMed] [Google Scholar]
- 101. Baburamani AA, Miyakuni Y, Vontell R et al. , . Does caspase-6 have a role in perinatal brain injury? Dev Neurosci 2015;37:321–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Giaccia AJ, Kastan MB.. The complexity of p53 modulation: Emerging patterns from divergent signals. Genes Dev 1998;12:2973–83 [DOI] [PubMed] [Google Scholar]
- 103. Kastan MB, Lim D-S.. The many substrates and functions of ATM. Nat Mol Cell Biol 2000;1:179–86 [DOI] [PubMed] [Google Scholar]
- 104. Brown EJ, Baltimore D.. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Develop 2003;17:615–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Shiloh Y. The ATM-mediated DNA-damage response: Taking shape. Trends Biochem Sci 2006;31:402–10 [DOI] [PubMed] [Google Scholar]
- 106. Dupre A, Boyer-Chatenet L, Gautier J.. Two-step activation of ATM by DNA and the Mre11-Rad50-Nbs1 complex. Nat Struct Mol Biol 2006;13:451–7 [DOI] [PubMed] [Google Scholar]
- 107. Martin LJ, Kaiser A, Yu JW et al. , . Injury-induced apoptosis of neurons in adult brain is mediated by p53-dependent and p53-independent pathways and requires Bax. J Comp Neurol 2001;433:299–311 [DOI] [PubMed] [Google Scholar]
- 108. Martin LJ, Price AC, McClendon K et al. , . Early events in target deprivation/axotomy induced neuronal apoptosis in vivo: Oxidative stress, DNA damage, p53 phosphorylation, and subcellular redistribution of death proteins. J Neurochem 2003;85:234–47 [DOI] [PubMed] [Google Scholar]
- 109. Martin LJ, Liu Z.. Injury-induced spinal motor neuron apoptosis is preceded by DNA single-strand breaks and is p53- and Bax-dependent. J Neurobiol 2002;50:181–97 [DOI] [PubMed] [Google Scholar]
- 110. Morrison RS, Kinoshita Y, Johnson MD et al. , . p53-dependent cell death signaling in neurons. Neurochem Res 2003;28:15–27 [DOI] [PubMed] [Google Scholar]
- 111. Northington FJ, Ferriero DM, Graham EM et al. , . Early neurodegeneration after hypoxia-ischemia in neonatal rat is necrosis while delayed neuronal death is apoptosis. Neurobiol Dis 2011;8:207–19 [DOI] [PubMed] [Google Scholar]
- 112. Kinzler KW, Vogelstein B.. What’s mice got to do with it? Nature 1996;382:672. [DOI] [PubMed] [Google Scholar]
- 113. Coppe J-P, Patil CK, Rodier F et al. , . A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS One 2010;592:e9188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Hornsby PJ. Mouse and human cell versus oxygen. Sci Aging Knowledge Environ 2003;2003:PE21. [DOI] [PubMed] [Google Scholar]
- 115. Banuelos CA, Banath JP, MacPhail SH et al. , . Mouse but not human embryonic stem cells are deficient in rejoining of ionizing radiation-induced DNA double-strand breaks. DNA Repair 2008;7:1471–83 [DOI] [PubMed] [Google Scholar]
- 116. MacRae SL, Croken MM, Calder RB et al. , . DNA repair in species with extreme lifespan differences. Aging (Albany NY) 2015;7:1171–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Odom DT, Dowell RD, Jacobsen ES et al. , . Tissue-specific transcriptional regulation has diverged significantly between human and mouse. Nat Gen 2007;39:730–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Schmidt T, Korner K, Karsunky H et al. , . The activity of the murine Bax promoter is regulated by Sp1/3 and E-box binding proteins but not by p53. Cell Death Differ 1999;6:873–82 [DOI] [PubMed] [Google Scholar]
- 119. Igata E, Inoue T, Ohtani-Fujita N et al. , . Molecular cloning and functional analysis of the murine bax gene promoter. Gene 1999;238:407–15 [DOI] [PubMed] [Google Scholar]
- 120. Horvath MM, Wang X, Resnick MA, Bell DA.. Divergent evolution of human p53 binding sites: Cell cycle versus apoptosis. PLoS Genet 2007;3:e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Somel M, Liu X, Tang L et al. , . MicroRNA-driven developmental remodeling in the brain distinguishes human from other primates. PLoS Biol 2011;9:e1001214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Fischer H, Koenig U, Eckhart L, Tschachler E.. Human caspase 12 has acquired deleterious mutations. Biochem Biophys Res Commun 2002;293:722–6 [DOI] [PubMed] [Google Scholar]
- 123. Ussat S, Werner U-E, Adam-Klages SA.. Species differences in the usage of several caspase substrates. Biochem Biophys Res Commun 2002;297:1186–90 [DOI] [PubMed] [Google Scholar]
- 124. Kerr LE, Birse-Archbold JL, Simon A et al. , . Differential regulation of caspase-3 by pharmacological and developmental stimuli as demonstrated using humanized caspase-3 mice. Apoptosis 2004;9:739–47 [DOI] [PubMed] [Google Scholar]
- 125. Jegga AG, Inga A, Menendez D et al. , . Functional evolution of the p53 regulatory network through its target response elements. Proc Natl Acad Sci U S A 2008;105:944–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Aguilar MJ, Kamoshita S, Landing BH et al. , . Pathological observations in ataxia-telangiectasia. A report of five cases. J Neuropathol Exp Neurol 1968;27:659–76 [PubMed] [Google Scholar]
- 127. Barlow C, Hirotsune S, Paylor R et al. , . Atm-deficient mice: A paradigm of ataxia telangiectasia. Cell 1996;86:159–71 [DOI] [PubMed] [Google Scholar]
- 128. van der Horst GTJ, van Steeg H, Berg RJ et al. , . Defective transcription-coupled repair in Cockayne syndrome B mice is associated with skin cancer. Cell 1997;89:425–35 [DOI] [PubMed] [Google Scholar]
- 129. Jaarsma D, van der Pluijm I, de Waard MC et al. , . Age-associated neuronal degeneration: Complementary roles of nucleotide excision repair and transcription-coupled repair in preventing neuropathology. PLoS Genet 2011;7:e1002405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Kang J, Bronson RT, Xu Y.. Targeted disruption of NBS1 reveals its roles in mouse development and DNA repair. EMBO J 2002;21:1447–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Hirano R, Interthal H, Huang C et al. , . Spinocerebellar ataxia with axonal neuropathy: Consequence of a Tdp1 recessive neomorphic mutation? EMBO J 2007;26:4732–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Eisen A. Amyotrophic lateral sclerosis-evolutionary and other perspectives. Muscle Nerve 2009;40:297–304 [DOI] [PubMed] [Google Scholar]
- 133. Rapoport SI. Hypothesis: Alzheimer’s disease is a phylogenetic disease. Med Hypotheses 1989;29:147–50 [DOI] [PubMed] [Google Scholar]
- 134. Finch CE, Austad SN.. Commentary: Is Alzheimer’s disease uniquely human? Neurobiol Aging 2015;36:553–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Martin LJ, Liu Z.. Opportunities for neuroprotection in ALS using cell death mechanism rationales. Drug Discov Today Models Dis 2004;1:135–43 [Google Scholar]
- 136. Martin LJ, Liu Z, Price A et al. , . Motor neuron degeneration in amyotrophic lateral sclerosis mutant superoxide dismutase-1 mice: Mechanisms of mitochondriopathy and cell death. J Comp Neurol 2007;500:20–46 [DOI] [PubMed] [Google Scholar]
- 137. Yang H, Wang G, Sun H et al. , . Species-dependent neuropathology in transgenic SOD1 pigs. Cell Res 2014;24:464–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Myers RE. Two patterns of perinatal brain damage and their conditions of occurrence. Am J Obstet Gynecol 1972;12:246–76 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.