

Abstract
IDH-mutant astrocytomas are significantly less aggressive than their IDH-wildtype counterparts. We analyzed The Cancer Genome Atlas dataset (TCGA) and identified a small group of IDH-mutant, WHO grade II–III astrocytomas (n = 14) with an unexpectedly poor prognosis characterized by a rapid progression to glioblastoma and death within 3 years of the initial diagnosis. Compared with IDH-mutant tumors with the typical, extended progression-free survival in a control group of age-similar patients, the tumors in the rapidly progressing group were characterized by a markedly increased level of overall copy number alterations ([CNA]; p = 0.006). In contrast, the mutation load was similar, as was the methylation pattern, being consistent with IDH-mutant astrocytoma. Two of the gliomas (14%) in the rapidly progressing, IDH-mutant group but none of the other grade II–III gliomas in the TCGA (n = 283) had pathogenic mutations in genes (FANCB and APC) associated with maintaining chromosomal stability. These results suggest that chromosomal instability can negate the beneficial effect of IDH mutations in WHO II–III astrocytomas. The mechanism of the increased CNA is unknown but in some cases appears to be due to mutations in genes with a role in chromosomal stability. Increased CNA could serve as a biomarker for tumors at risk for rapid progression.
Keywords: Glioblastoma (GBM), Isocitrate dehydrogenase, Prognosis, WHO 2016
INTRODUCTION
Infiltrating gliomas are among the most common primary intracranial tumors. The prognosis depends on the histological grade, age of the patient, and genetic features in particular 1p/19q codeletion and isocitrate dehydrogenase 1/2 (IDH1/2) mutation status (1–3). With the publication of the WHO Classification of Tumors of the Central Nervous System revised fourth edition in 2016, infiltrating gliomas are molecularly subgrouped into IDH-mutant and IDH-wildtype groups (1). Diffuse astrocytomas corresponding to WHO grades II and III with wildtype IDH1/2 have a significantly worse prognosis than their IDH-mutant counterparts and are associated with a more rapid progression to glioblastoma ([GBM]; WHO grade IV astrocytoma) (1). The average overall survival ranges between 81 and 151 months for lower-grade (WHO grade II–III) astrocytomas with IDH mutations and 19–60 months for WHO grade II–III astrocytomas without IDH mutations (4–6).
Much work has been done in recent years to determine additional prognostic biomarkers and potential targetable mutations for these tumors (7–11). Factors such as GBM-like genetic alterations (12, 13) and high nestin expression (14) have been implicated in worse prognosis in some WHO grade II–III astrocytomas. In a previous study, IDH-mutant GBMs showed increased copy number alterations (CNA) compared with IDH-mutant WHO grade II–III astrocytomas (12), suggesting that increased CNA is associated with progression to grade IV in IDH-mutant astrocytomas. However, it was previously unknown if increased CNA levels precede progression to GBM and are therefore associated with a worse prognosis in IDH-mutant grade II–III astrocytomas. We recently described a small cohort of 4 cases IDH-mutant WHO grade II–III astrocytomas from our own institution with rapid progression to GBM and unexpectedly short survival (<3 years) (15). All 4 of these tumors had very high levels of CNA at their initial WHO grade II–III presentation compared with more classically indolent-behaving IDH-mutant lower-grade gliomas, suggesting that increased CNA levels are an adverse prognostic feature.
Herein, we identified and analyzed 14 IDH-mutant WHO grade II–III astrocytomas from The Cancer Genome Atlas (TCGA) online repository (16) and cBio cancer genomics portal (17, 18) with very short intervals to recurrence, progression, and ultimately patient death compared with a group of 16 age-similar IDH-mutant WHO grade II–III astrocytomas from the same database which followed the expected, more indolent clinical course.
MATERIALS AND METHODS
TCGA Case Selection
We performed a search of the 516 lower-grade glioma cases in the TCGA database and identified a total of 14 patients meeting the criteria of an IDH mutation, initial WHO grade of II or III, genetic alterations consistent with astrocytoma, as well as progression to GBM and overall survival <3 years. A control group of 16 patients was selected from the remaining TCGA cases that met the criteria of an IDH mutation and a more conventional clinical course, that is, a disease-free interval >5 years. The original histologic diagnoses reported in TCGA included astrocytomas, oligoastrocytomas, and oligodendrogliomas. All cases were molecularly reclassified according to the WHO 2016 criteria as astrocytomas, WHO grade II–III, based on an intact 1p/19q status and the presence of ATRX and/or TP53 mutations. Mean age at presentation was 40.7 ± 2.7 years and 37.8 ± 2.9 years in the rapidly progressing and control groups, respectively (p = 0.48). No significant difference in tumor grade at initial presentation was identified between these 2 groups (p = 0.52). The study was granted an exemption by the UT Southwestern Institutional Review Board (IRB STU 042014-067).
Genetic and Epigenetic Analysis
The gene expression (Illumina HiSeq, RNASeq) and DNA methylation data (Illumina Human Methylation 450) was downloaded for the selected TCGA low-grade glioma cases and analyzed with TCGAbiolinks (19). The Affymetrix SNP 6.0 micorarray data normalized to germline for copy number analysis for the same TCGA cases was downloaded from Broad GDAC Firehose (http://gdac.broadinstitute.org/runs/stddata__2016_01_28/). The fraction of copy number altered genome was calculated from the above data as the fraction of the genome with log2 of copy number >0.3 following the procedure used in cBioportal (18). The mutation load is the number of nonsynonymous mutations seen in a sample. The differential analysis and visualization of mutations was done using Maftools (Mayakonda A, Koeffle HP, bioRxiv doi:10.1101/052662). The Ideogram for visualization of genome-wide copy number variation results was generated using Genome Decoration Page (https://www.ncbi.nlm.nih.gov/genome/tools/gdp). The pathway and network analyses were conducted using Qiagen’s IPA tool (www.qiagen.com/ingenuity) and R 3.4.1 (http://www.R-project.org/).
GISTIC Analysis
The GISTIC (Genomic Identification of Significant Targets in Cancer) 2.0 algorithm was used to identify regions of the genome that are significantly amplified or deleted across the samples in the rapidly progressing and conventional course groups (20). Each CNA is assigned a G-score that considers the amplitude of the alteration as well as the frequency of its occurrence across samples. The false discovery rate (FDR) was then used to determine the relative significance of each abnormality. Each region predicted to be significantly different between the conventional course and rapidly progressing astrocytomas was screened for tumor suppressor genes, oncogenes, and other genes associated with glioma and malignancy (20, 21). GISTIC 2.0 analysis was run in GenePattern (22).
Mutation Analysis of Genes Involved in Maintenance of Genomic Stability
A group of 43 genes previously known to be involved in maintaining chromosomal stability was identified by a literature review and included the following genes: APC, ATM, ATR, BLM, BRCA1 (FANCS), BRCA2 (FANCD1), BUB1B, CHK1, Claspin, DNA-PK, EME1, FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ (BRIP1), FANCL, FANCM, FANCN (PALB2), FANCO (RAD51C), FANCP (SLX4), FANCQ, FANCR, FANCT (UBE2T), HUS1, Ligase IV, MUS81, NBN, Pol η, Pol κ, RAD51, RAD52, REV3, SMC1, SNM1B, TOP1, TP53, WRN, and XLF (23, 24). Variant annotation was performed using COSMIC (25), dbSNP (26), ClinVar (27), CanProVar 2.0 (28), The 1000 Genomes Project (29), and FATHMM-MKL (30).
Statistical Analysis
Differences in mutation and CNA were evaluated using the Mann-Whitney test in GraphPad Prism version 7.03 (GraphPad, La Jolla, CA). Kaplan-Meier analysis was used for survival analysis. Significance levels of the survival analysis were tested using the Log-rank test.
RESULTS
Clinical Outcome
As expected based on our experimental design, the rapidly progressing glioma patients had both shorter disease-free intervals (p = 2.6e-9; Fig. 1A) and overall survival times (p = 1.7e-9; Fig. 1B).
FIGURE 1.

Kaplan-Meier survival plots demonstrating a significant difference in both (A) disease-free interval (p = 2.6e-9) and (B) total survival (p = 1.7e-9) between the rapidly progressing group (n = 14) and the control group (n = 16).
CNA Analysis
Overall CNA levels were increased in the rapidly progressing IDH-mutant astrocytomas compared with the control IDH-mutant astrocytomas. The total percentage of the genome affected by CNA was significantly higher in the rapidly progressing IDH-mutant astrocytoma group compared with the IDH-mutant control group (16.6% ± 2.8% and 8.1% ± 2.5%, respectively; mean ± STD; p = 0.006; Fig. 2A). Most of the CNA were spread widely throughout the genome, and were considered significant with an amplification or deletion level ≥0.3. (Fig. 3A, B).
FIGURE 2.

(A) Box plot illustrating an increased amount of overall copy number alterations (CNA) in the rapidly progressing group (n = 14) as compared with the control group (n = 16; p = 0.006), and (B) box plot showing no significant difference in the number of overall mutations between the rapidly progressing (n = 5) and control glioma groups (n = 9; p = 0.27).
FIGURE 3.

Overall amplification and deletion levels and chromosomal locations in all of the patients included in the conventional course glioma (A) and rapidly progressing glioma (B) groups. (C) Chromosomal regions with the most significant areas of alteration in both groups including those commonly associated with glioblastoma, 9p, 10q, and 13q.
In addition, multiple specific chromosomal alterations such as areas of deletion frequently present in secondary GBM (13, 31, 32), including on chromosome 9p (reported in 33%–38% of secondary GBMs), chromosome 10q (80%–86%), and chromosome 13q (31%–33%), were identified in some of these lower-grade gliomas before histologic progression to GBM (Fig. 3C). Using GISTIC algorithms (20, 21), we confirmed that the astrocytomas that progressed rapidly to GBM and had short survival intervals had increased frequencies of 9p, 10q, and 13q deletions, as well as less frequent amplifications in cytobands, including 4q12 and 12q14.1, which were not seen in the conventional course group (Figs. 3C and 4). We identified several consistently altered genes within these regions relevant to progression to GBM not seen in the conventional course lower-grade gliomas, including PDGFRA amplification on chromosome 4p12 (which is associated with aggressive and high-grade astrocytomas, including those with H3K27M mutation) (33–36), CDKN2A/B homozygous deletion on chromosome 9p21.3 (7, 8), CDK4 amplification on chromosome 12q14.1 (Fig. 4) (37, 38), as well as less common amplification of BRAF and CDK6 on chromosome 7q (39, 40). These genes are all associated with higher grade and, importantly, have multiple specific inhibitors already under investigation or in use clinically (41–44).
FIGURE 4.
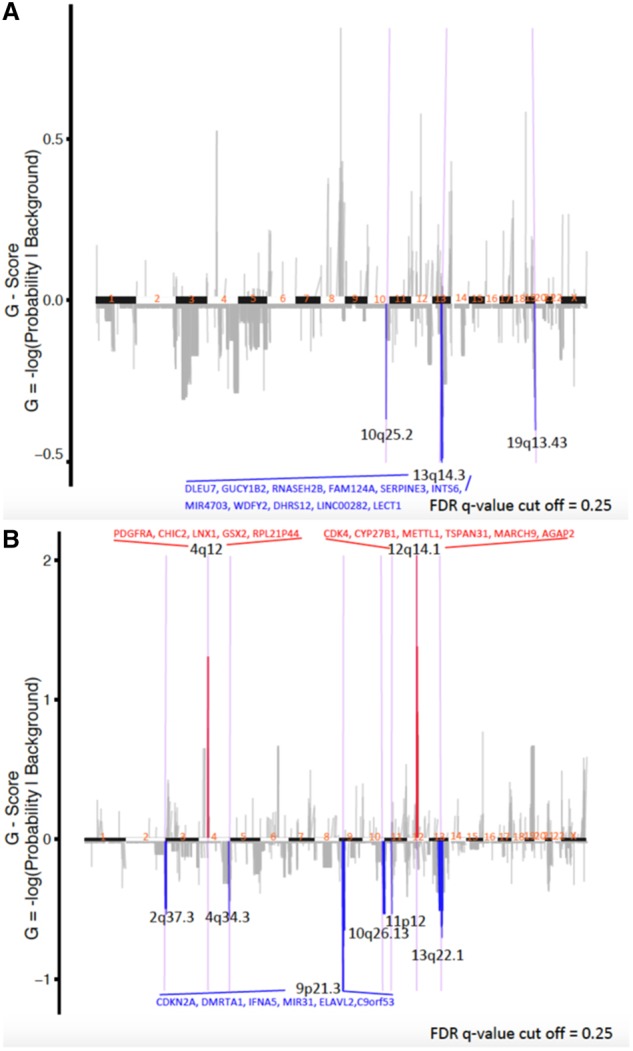
GISTIC analysis showing the most consistent and relevant cytoband/gene alterations in conventional course astrocytomas (A) and rapidly progressing astrocytomas (B). All cytobands shown met the criterion of false discovery rate (FDR) 0.25. The 4 cytobands with gene names annotated met the criterion of FDR 0.05.
MGMT Methylation Analysis
None of MGMT methylation probes demonstrated a significantly different (FDR ≤ 0.05 and difference in mean beta value ≥ 0.25) methylation status between the conventional and rapidly progressing cohorts, indicating that MGMT gene methylation was not significantly different between the 2 groups.
Mutation Load
No significant difference was identified between the overall number of mutations (mutational burden) between a subset of tumors for which full mutation data are available (p = 0.27) in the group of control astrocytomas (25.9 ± 5.7 mutations/case; n = 9) and the group of rapidly progressing astrocytomas (39.0 ± 11.2 mutations/case; n = 5; Fig. 2B). A full list of genes with mutations and the number of cases with mutations in these genes is presented in Supplementary Data Table S1.
Mutation Analysis of Genes Involved in Maintenance of Genomic Stability
Two of the gliomas (14%) among the rapidly progressing IDH-mutant gliomas harbored a pathogenic mutation in genes from a 43 gene panel (FANCB and APC) known to be associated with chromosomal instability (Fig. 5) (45–49). In contrast, none of the gliomas in the IDH-mutant control group and no tumor among the 283 other IDH-mutant and IDH-wildtype WHO grade II–III gliomas with full mutation sequencing data available in the TCGA dataset harbored pathogenic mutations in FANCB, APC, or any of the remaining genes associated with chromosomal instability. The APC (N1808T) and FANCB (P586S) mutations were both missense mutations predicted to be pathogenic with a high specificity (0.98 and 0.96, respectively) based on their locations in highly conserved regions and other factors (FATHMM-MKL algorithm) (30).
FIGURE 5.
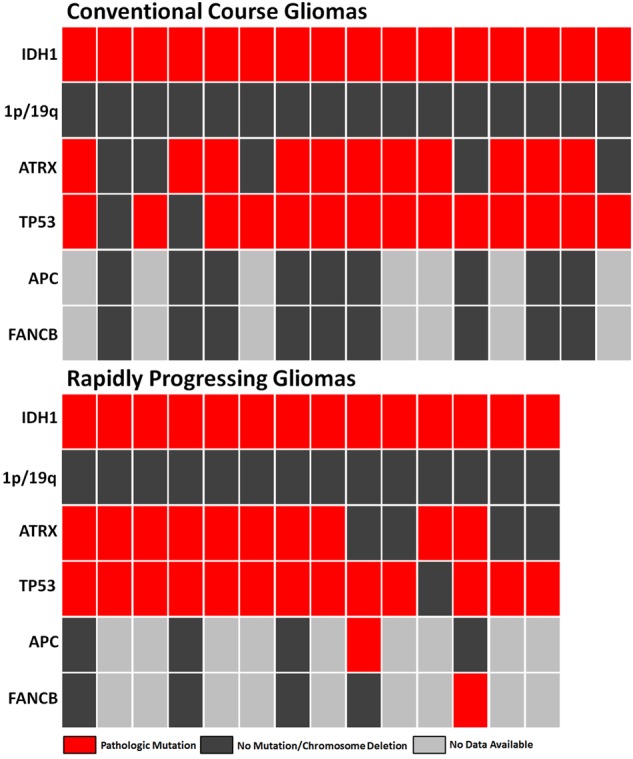
Relevant mutations (IDH1, ATRX, TP53, APC, and FANCB) identified in each case of conventional course (n = 9) and rapidly progressing (n = 5) lower-grade glioma in which partial or complete mutational data were available.
Pathway Analysis
Pathway analysis accounting for both mRNA expression levels and gene amplification/deletion levels was performed (Supplementary Data Fig. S1). The relative deletions and amplifications in rapidly progressing gliomas, as compared with our conventional course glioma control group, shows an overall increase in pathways driving gliomagenesis and tumor progression leading to increased cell survival, growth, and proliferation after accounting for directionality of these alterations.
DISCUSSION
Representing ∼29% of all primary intracranial tumors and 80% of all malignant brain tumors (2), glial tumors are subject to a wide array of ongoing research studies. Recently, IDH1/2 mutational status has emerged as an important prognostic factor in gliomas, particularly lower-grade (WHO grade II–III) astrocytomas. Astrocytomas with mutations in either IDH1 or IDH2 have a significantly better prognosis than IDH-wildtype astrocytomas of comparable grade (1, 5, 6). However, in rare cases, lower-grade IDH-mutant astrocytomas may follow an aggressive clinical course (15).
More recently, new genome-wide analysis methods based on next-generation sequencing and methylation pattern analysis have provided a new approach to the diagnosis and classification of these tumors, resulting in redefinition of many of the diagnostic categories (31, 50, 51). This technology has been used to address the overall state of the genome across IDH-mutant and IDH-wildtype grade II–IV gliomas. These data showed that there were increased levels of overall CNA as well as localized rearrangements (chromothripsis) in IDH-mutant GBMs compared with lower-grade IDH-mutant astrocytomas (12, 16). In contrast, no increase in CNA levels was found with increasing histologic grade in IDH-wildtype astrocytomas (12), an observation that corresponds well with the relatively weak association between increasing histologic grade and worsening clinical outcome in IDH-wildtype astrocytomas (1). If the increased levels of overall chromosomal fracturing and instability occurred prior to or after progression to GBM was not clear from previous studies.
In a small cohort from our own institution, we previously demonstrated that a subgroup of astrocytomas with the IDH1 R132H mutation actually harbored significantly increased levels of CNA prior to progression to secondary GBM. This early chromosomal instability was associated only with tumors that progressed in an unexpectedly rapid manner from lower-grade astrocytoma to GBM and were rapidly fatal, despite the favorable IDH-mutant status (15).
In the present study, we identified 14 cases of IDH-mutant grade II–III astrocytoma with rapid progression to GBM and short patient survival (defined here as 36 months or less) in the TCGA dataset and compared this group to a cohort of age-similar 16 IDH-mutant grade II–III astrocytomas with a clinical course typical for their favorable IDH-mutational status (defined here as patients with a disease-free interval of 60 months or more) from the same dataset (Fig. 1). Of note, several of these patients had original diagnoses of either “oligodendroglioma” or “oligoastrocytoma”; however, with information on the 1p/19q, ATRX, and TP53 status we were able to molecularly reclassify each of these cases as diffuse astrocytomas according to new WHO 2016 guidelines (1). We found that these rapidly progressing gliomas had significantly higher levels of CNA before progression to GBM (Figs. 2A and 3). There were also a number of more specific chromosomal alterations which were previously associated with secondary GBM that were altered in this group of histologically lower-grade, rapidly progressing astrocytomas, including losses at 9p, 10q, 13q, and gains at 4p12 and 12q14.1 containing genes known to be associated with GBM (Figs. 3C and 4) (32, 52). These findings indicate that chromosomal instability, as evidenced by an increased level of CNA, is a prognostic factor in IDH-mutant lower-grade gliomas, predicting a rapid recurrence and progression to GBM. This finding has implications for the management of these patients.
Increased CNA before histologic progression to GBM also suggests a possible mechanism for the poor outcomes in these tumors with otherwise favorable prognostic factors. CNA has been shown to be a factor in increasing malignant potential in tumors through the overall ratio of widespread amplification of DNA regions containing growth factor pathway components and cell cycle proteins, as well as widespread deletions in areas containing tumor suppressor genes (13, 53–56). TP53-inactivating mutations have been associated with increased levels of CNA and chromothripsis, a potential driving force of progression to higher-grade tumors (16, 57). However, since 13/14 of the rapidly progressing tumors and 14/16 of the conventional course control tumors included in this study harbored mutations in TP53, it is unlikely that TP53 mutations are the explanation for the increased CNA levels or clinically aggressive behavior.
In contrast to the increased CNA levels, no consistent specific mutation differences were identified between the 2 groups (Fig. 5), and mutation load (point mutations per megabase) was not significantly altered in the rapidly progressing IDH-mutant astrocytoma group (Fig. 2B), suggesting that chromosomal instability rather than mutation load is the critical factor in the progression to GBM. The methylation pattern was also similar in both groups and consistent with IDH-mutant astrocytoma, as expected, which indicates that the downstream CpG island methylator phenotype (CIMP) caused by IDH-mutations is unchanged in the rapidly progressing group. A survey of 43 tumor suppressor genes with a known protective role in chromosomal stability identified a pathogenic mutation in 2 of the gliomas (14%) in the rapidly progressing IDH-mutant gliomas but none of the control IDH-mutant gliomas; further analysis revealed that none of the other 283 grade II–III gliomas in the TCGA had a pathogenic mutation in these 2 genes, FANCB and APC. The association of FANCB and APC mutations with chromosomal instability has been well established (45–49). Our results show that chromosomal instability can potentially negate the prognostically beneficial effect of IDH mutations in WHO II–III astrocytomas and suggest that increased CNA levels could serve as a biomarker for identifying IDH-mutant lower-grade astrocytomas at risk for rapid progression. The mechanism of the increased chromosomal instability remains unknown in the majority of the rapidly progressing IDH-mutant gliomas but in some cases it may be due to mutations in genes associated with chromosomal instability. While it was previously known that CNA was higher among IDH1/2-wildtype lower-grade gliomas and GBM irrespective of IDH1/2 mutational status when compared with IDH-mutated lower-grade gliomas, the relationship between tumor progression and timing of this genomic instability has been unclear (12). In this report, we show that increased levels of CNA actually precede progression to GBM, at least in some cases with paradoxically rapid progression. This finding may provide an approach for identifying the lower-grade IDH-mutant astrocytomas at risk for a rapid progression and also suggests a mechanism underlying the progression.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank Niccole Williams and Antonio Atkins for excellent administrative professional services. The study was based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/.
FUNDING
This study was supported by the Friedberg Charitable Foundation grant (to MS), Department of Veteran’s Affairs grant I01BX002559 (to AAH), and NIH grant UL1TR001105 (to CX).
Timothy E. Richardson and Adwait Amod Sathe are cofirst authors. Chao Xing and Kimmo J. Hatanpaa are cosenior authors.
The authors have no duality or conflicts of interest to declare.
Supplementary Data can be found at http://www.jnen.oxfordjournals.org.
REFERENCES
- 1. Louis DN, Perry A, Reifenberger G et al. , . The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol 2016;131:803–20 [DOI] [PubMed] [Google Scholar]
- 2. Dolecek TA, Propp JM, Stroup NE et al. , . CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2005–2009. Neuro Oncol 2012;14(Suppl 5):v1–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ostrom QT, Gittleman H, Farah P et al. , . CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro Oncol 2013;15(Suppl 2):ii1–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sanson M, Marie Y, Paris S et al. , . Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol 2009;27:4150–4 [DOI] [PubMed] [Google Scholar]
- 5. Yan H, Bigner DD, Velculescu V et al. , . Mutant metabolic enzymes are at the origin of gliomas. Cancer Res 2009;69:9157–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yan H, Parsons DW, Jin G et al. , . IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009;360:765–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Appin CL, Brat DJ.. Biomarker-driven diagnosis of diffuse gliomas. Mol Aspects Med 2015;45:87–96 [DOI] [PubMed] [Google Scholar]
- 8. Appin CL, Brat DJ.. Molecular pathways in gliomagenesis and their relevance to neuropathologic diagnosis. Adv Anat Pathol 2015;22:50–8 [DOI] [PubMed] [Google Scholar]
- 9. Delgado-Lopez PD, Corrales-Garcia EM, Martino J et al. , . Diffuse low-grade glioma: A review on the new molecular classification, natural history and current management strategies. Clin Transl Oncol 2017;19:931–44 [DOI] [PubMed] [Google Scholar]
- 10. Hodges TR, Ott M, Xiu J et al. , . Mutational burden, immune checkpoint expression, and mismatch repair in glioma: Implications for immune checkpoint immunotherapy. Neuro Oncol 2017;19:1047–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Szopa W, Burley TA, Kramer-Marek G et al. , . Diagnostic and therapeutic biomarkers in glioblastoma: Current status and future perspectives. Biomed Res Int 2017;2017:8013575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cohen A, Sato M, Aldape K et al. , . DNA copy number analysis of Grade II–III and Grade IV gliomas reveals differences in molecular ontogeny including chromothripsis associated with IDH mutation status. Acta Neuropathol Commun 2015;3:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rao SK, Edwards J, Joshi AD et al. , . A survey of glioblastoma genomic amplifications and deletions. J Neurooncol 2010;96:169–79 [DOI] [PubMed] [Google Scholar]
- 14. Hatanpaa KJ, Hu T, Vemireddy V et al. , . High expression of the stem cell marker nestin is an adverse prognostic factor in WHO grade II–III astrocytomas and oligoastrocytomas. J Neurooncol 2014;117:183–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Richardson TE, Snuderl M, Serrano J et al. , . Rapid progression to glioblastoma in a subset of IDH-mutated astrocytomas: A genome-wide analysis. J Neurooncol 2017;133:183–92 [DOI] [PubMed] [Google Scholar]
- 16. Brat DJ, Verhaak RGW, Aldape KD et al. , . Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med 2015;372:2481–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cerami E, Gao J, Dogrusoz U et al. , . The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2:401–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gao J, Aksoy BA, Dogrusoz U et al. , . Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Colaprico A, Silva TC, Olsen C et al. , . TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res 2016;44:e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mermel CH, Schumacher SE, Hill B et al. , . GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 2011;12:R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Beroukhim R, Getz G, Nghiemphu L et al. , . Assessing the significance of chromosomal aberrations in cancer: Methodology and application to glioma. Proc Natl Acad Sci U S A 2007;104:20007–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reich M, Liefeld T, Gould J et al. , . GenePattern 2.0. Nat Genet 2006;38:500–1 [DOI] [PubMed] [Google Scholar]
- 23. Chan SH, Ngeow J.. Germline mutation contribution to chromosomal instability. Endocr Relat Cancer 2017;24:T33–46 [DOI] [PubMed] [Google Scholar]
- 24. Glover TW, Wilson TE, Arlt MF.. Fragile sites in cancer: More than meets the eye. Nat Rev Cancer 2017;17:489–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Forbes SA, Bindal N, Bamford S et al. , . COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 2011;39:D945–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sherry ST, Ward MH, Kholodov M et al. , . dbSNP: The NCBI database of genetic variation. Nucleic Acids Res 2001;29:308–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Landrum MJ, Lee JM, Benson M et al. , . ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res 2016;44:D8628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li J, Duncan DT, Zhang B.. CanProVar: A human cancer proteome variation database. Hum Mutat 2010;31:219–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Abecasis GR, Auton A, Brooks LD et al. , . An integrated map of genetic variation from 1,092 human genomes. Nature 2012;491:56–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shihab HA, Rogers MF, Gough J et al. , . An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 2015;31:1536–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sturm D, Bender S, Jones DT et al. , . Paediatric and adult glioblastoma: Multiform (epi)genomic culprits emerge. Nat Rev Cancer 2014;14:92–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maher EA, Brennan C, Wen PY et al. , . Marked genomic differences characterize primary and secondary glioblastoma subtypes and identify two distinct molecular and clinical secondary glioblastoma entities. Cancer Res 2006;66:11502–13 [DOI] [PubMed] [Google Scholar]
- 33. Vanan MI, Underhill DA, Eisenstat DD.. Targeting epigenetic pathways in the treatment of pediatric diffuse (high grade) gliomas. Neurotherapeutics 2017;14:274–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Aldape K, Zadeh G, Mansouri S et al. , . Glioblastoma: Pathology, molecular mechanisms and markers. Acta Neuropathol 2015;129:829–48 [DOI] [PubMed] [Google Scholar]
- 35. Burford A, Little SE, Jury A et al. , . Distinct phenotypic differences associated with differential amplification of receptor tyrosine kinase genes at 4q12 in glioblastoma. PLoS ONE 2013;8:e71777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Holtkamp N, Ziegenhagen N, Malzer E et al. , . Characterization of the amplicon on chromosomal segment 4q12 in glioblastoma multiforme. Neuro-Oncology 2007;9:291–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hulleman E, Helin K.. Molecular mechanisms in gliomagenesis. Adv Cancer Res 2005;94:1–27 [DOI] [PubMed] [Google Scholar]
- 38. Konopka G, Bonni A.. Signaling pathways regulating gliomagenesis. Curr Mol Med 2003;3:73–84 [DOI] [PubMed] [Google Scholar]
- 39. Lam PY, Di Tomaso E, Ng HK et al. , . Expression of p19INK4d, CDK4, CDK6 in glioblastoma multiforme. Br J Neurosurg 2000;14:28–32 [DOI] [PubMed] [Google Scholar]
- 40. Masui K, Mischel PS, Reifenberger G.. Molecular classification of gliomas. Handb Clin Neurol 2016;134:97–120 [DOI] [PubMed] [Google Scholar]
- 41. Hassler MR, Vedadinejad M, Flechl B et al. , . Response to imatinib as a function of target kinase expression in recurrent glioblastoma. SpringerPlus 2014;3:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Finn RS, Dering J, Conklin D et al. , . PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res 2009;11:R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Finn RS, Martin M, Rugo HS et al. , . Palbociclib and letrozole in advanced breast cancer. N Engl J Med 2016;375:1925–36 [DOI] [PubMed] [Google Scholar]
- 44. Patnaik A, Rosen LS, Tolaney SM et al. , . Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non-small cell lung cancer, and other solid tumors. Cancer Discov 2016;6:740–53 [DOI] [PubMed] [Google Scholar]
- 45. Alberici P, Fodde R.. The role of the APC tumor suppressor in chromosomal instability. Genome Dyn 2006;1:149–70 [DOI] [PubMed] [Google Scholar]
- 46. Caldwell CM, Kaplan KB.. The role of APC in mitosis and in chromosome instability. Adv Exp Med Biol 2009;656:51–64 [DOI] [PubMed] [Google Scholar]
- 47. Deakyne JS, Mazin AV.. Fanconi anemia: At the crossroads of DNA repair. Biochemistry Mosc 2011;76:36–48 [DOI] [PubMed] [Google Scholar]
- 48. Levitus M, Joenje H, de Winter JP.. The Fanconi anemia pathway of genomic maintenance. Cell Oncol 2006;28:3–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Moldovan GL, D'Andrea AD.. How the fanconi anemia pathway guards the genome. Ann Rev Genet 2009;43:223–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sahm F, Schrimpf D, Jones DT et al. , . Next-generation sequencing in routine brain tumor diagnostics enables an integrated diagnosis and identifies actionable targets. Acta Neuropathol 2016;131:903–10 [DOI] [PubMed] [Google Scholar]
- 51. Sturm D, Orr BA, Toprak UH et al. , . New brain tumor entities emerge from molecular classification of CNS-PNETs. Cell 2016;164:1060–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Crespo I, Tao H, Nieto AB et al. , . Amplified and homozygously deleted genes in glioblastoma: Impact on gene expression levels. PLoS ONE 2012;7:e46088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jeuken J, van den Broecke C, Gijsen S et al. , . RAS/RAF pathway activation in gliomas: The result of copy number gains rather than activating mutations. Acta Neuropathol 2007;114:121–33 [DOI] [PubMed] [Google Scholar]
- 54. de Tayrac M, Etcheverry A, Aubry M et al. , . Integrative genome-wide analysis reveals a robust genomic glioblastoma signature associated with copy number driving changes in gene expression. Genes Chromosom Cancer 2009;48:55–68 [DOI] [PubMed] [Google Scholar]
- 55. Kim YW, Koul D, Kim SH et al. , . Identification of prognostic gene signatures of glioblastoma: A study based on TCGA data analysis. Neuro Oncol 2013;15:829–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Waugh MG. Chromosomal instability and phosphoinositide pathway gene signatures in glioblastoma multiforme. Mol Neurobiol 2016;53:621–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rausch T, Jones DT, Zapatka M et al. , . Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 2012;148:59–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.