

SUMMARY
Dietary supplementation with fermentable fiber suppresses adiposity and the associated parameters of metabolic syndrome. Microbiota-generated fiber-derived short-chain fatty acids (SCFA) and free fatty acid receptors including GPR43 are thought to mediate these effects. We find that while fermentable (inulin), but not insoluble (cellulose) fiber, markedly protected mice against high-fat diet (HFD)-induced metabolic syndrome, the effect was not significantly impaired by either inhibiting SCFA production or genetic ablation of GPR43. Rather, HFD decimates gut microbiota resulting in loss of enterocyte proliferation, leading to microbiota encroachment, low-grade inflammation (LGI) and metabolic syndrome. Enriching HFD with inulin restored microbiota loads, IL-22 production, enterocyte proliferation, and anti-microbial gene expression in a microbiota dependent manner, as assessed by antibiotic and germ-free approaches. Inulin-induced IL-22 expression, which required innate lymphoid cells, prevented microbiota encroachment and protected against LGI and metabolic syndrome. Thus, fermentable fiber protects against metabolic syndrome by nourishing microbiota to restore IL-22-mediated enterocyte function.
Keywords: Metabolic syndrome, short-chain fatty acids, germ-free mice, microbiota encroachment, intestinal inflammation
In Brief
Dietary fiber supplements suppress adiposity and the associated parameters of metabolic syndrome. Zou et al. show that the fermentable fiber inulin impacts gut microbiota to increase intestinal epithelial proliferation, prevent colonic atrophy, reduce microbiota encroachment into the mucosa, and thereby protect against metabolic syndrome in a microbiota- and IL-22-dependent manner.
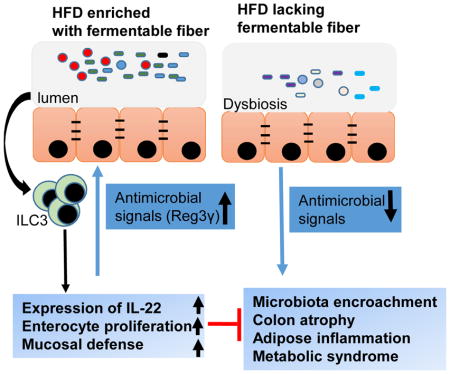
INTRODUCTION
Obesity and its associated metabolic disorders, collectively referred to as metabolic syndrome, are amongst humanity’s most pressing public health problems (Collaborators, 2017). Metabolic syndrome is increasingly viewed as a chronic inflammatory disease in which altered host-microbiota interactions in the gut contribute to disease development (Everard and Cani, 2013). Accordingly, in humans, metabolic syndrome is associated with alterations in gut microbiota composition and gut bacteria infiltrating the inner mucus layer thus encroaching upon gut epithelial cells in a manner that may promote inflammation (Chassaing et al., 2017). Societal changes in dietary habits, especially increased consumption of processed foods lacking fiber, are thought to have impacted the microbiota and contributed to increased incidence of chronic inflammatory disease, including metabolic syndrome (Sonnenburg et al., 2016; Sonnenburg and Sonnenburg, 2014). Accordingly, administering mice or humans supplements of the fermentable fiber inulin suppresses adiposity and associated parameters of metabolic syndrome (Dewulf et al., 2011; Salazar et al., 2015). However, the doses of inulin needed to provide a marked health benefit are difficult to achieve both because of logistical considerations and adverse effects, especially bloating and flatulence. Hence, it remains important to use tractable models to define mechanisms by which fermentable fiber protects against metabolic syndrome, thus providing the rationale for the central question addressed herein. Previous studies have reported a role for microbiota-generated inulin-derived short-chain fatty acids (SCFA) in promoting beneficial aspects of host metabolism and reducing inflammation, acting in large part free fatty acid receptor GPR43 (also known as Ffar2) (Brooks et al., 2017). In contrast, our results indicate that much of the ability of inulin to suppress diet-induced obesity may not require SCFA but is driven by inulin-elicited bacteria inducing IL-22, which fortifies the intestine thus reducing microbiota encroachment and ameliorates metabolic syndrome.
RESULTS
Fermentable fiber inulin prevented HFD-induced metabolic syndrome
A widely-used mouse model of diet-induced obesity (DIO) compares mice fed a grain-based, poorly defined rodent chow with mice fed a compositionally-defined obesogenic purified diet, commonly referred to as a “high-fat diet” (HFD), that is typically 35% fat by weight (60% by calories) and contains 5% cellulose as a source of fiber. This is in contrast to chows, which typically contain 5% fat by weight (10 – 15% by calories) and 15 – 25% fiber, coming from diverse sources. Relative to chow-fed mice, this obesogenic diet results in a marked increase in adiposity associated with numerous features of metabolic syndrome. Moreover, in addition to its high-fat content, its relatively low fiber content, particularly its lack of fermentable fiber, drives the metabolic syndrome phenotype induced by this diet (Chassaing et al., 2015b). Accordingly, manipulating the fiber content of this diet, particularly adding fermentable fiber, ameliorates the severity of metabolic syndrome although the extent of such amelioration is highly variable across different studies (Brooks et al., 2017; Chassaing et al., 2015b; Hamilton et al., 2017). Hence, we first sought to establish a well-controlled experimental platform whereby supplementation of an obesogenic diet with fiber would result in a strong degree of protection against indices of metabolic syndrome that would be amenable to mechanistic study. We found that relative to the “standard” HFD, a diet comprised of 20% inulin (wt/wt) reduced weight gain and markedly attenuated HFD-induced adiposity, as assessed by amount of epididymal, mesenteric, and subcutaneous fat (Figure 1A and B). This reduction in adiposity was accompanied by a reduction in adipocyte size (Figures 1C, S1A). In contrast, no changes were found in brown adipose tissue adipocyte size (Figure S1B) and expression of mRNA of UCP1 (data not shown) was similar between mice fed inulin or cellulose enriched diets, suggesting fiber content did not affect thermogenesis of brown adipose tissue. Dietary enrichment with inulin did not significantly impact serum levels of free fatty acids or triglycerides (Figure S1D and E), but markedly lowered levels of cholesterol, which were increased by HFD relative to chow (Figure S1F). Supplementation of the HFD with inulin also largely prevented dysglycemia, as assessed by measuring blood glucose after administration of glucose (Figure 1D and E) or in a fasted state and following injection of insulin (Figure 1F and G), although the statistical significance of differences following insulin administration varied depending upon metric used for the analysis (Figures 1H and S1G). Inulin also reduced hepatosteatosis, as assessed by staining liver sections with H&E (Figure S1C) or oil-red O (Figure 1I and J), which otherwise resulted from exposure to the standard HFD (HFD-50 Cell). Inulin’s ability to reduce indices of metabolic syndrome were associated with a modest but significant reduction in food consumption, based on both food weight and calorie content (Figure S1H and I). Such reduction in food consumption was not likely a simple “bulking effect” as food consumption was not significantly impacted by cellulose. Moreover, enriching HFD with cellulose, (HFD-200 Cell) which is not easily fermentable by mice, only very modestly reduced adiposity and dysglycemia (Figures 1, S1). Together, these results suggest that comparing phenotypes of mice being fed high-fat diets comprised of about 20% cellulose or inulin provides a tractable means to study how fermentable fiber is impacting DIO.
Figure 1. Inulin prevented metabolic syndrome induced by HFD.

C57BL/6 male mice were fed chow, HFD, or HFD supplemented with cellulose or inulin for 4 weeks. A) Body mass over time (n=10). B) Epididymal fat (n=10), mesenteric fat (n=5) and subcutaneous fat (n=5) were measured at the end of experiment. C) The epididymal fat was stained with H&E, adipocyte size distribution was determined. D–E) Mice (n=10) were administered glucose, 2 g/kg intraperitoneally following an overnight fast. Blood glucose levels were measured at the indicated time point (D) and area under curve (AUC) calculated (E). F–H) Mice (n=10) were fasted 5 h, the glucose was measured at 0 (F), 30, 60, 90 min after intraperitoneally injected with insulin (G) and area under curve calculated (H). I–J) Fat in the liver (n=5) was observed by oil red staining (I), and quantified by image analysis (J), Scale bars, 50 μm. Data were expressed as mean ± SEM. Statistical significance was assessed by unpaired Student t test. *p<0.05; **p<0.01. See also Figure S1.
Enrichment of HFD with inulin increased gut epithelial cell proliferation and prevented colon atrophy
The promotion of adiposity by diets lacking fermentable fiber, irrespective of fat content, correlates with a marked loss of colon mass, quantifiable by weighing the organ, that occurs within a few days of the change in diet (Chassaing et al., 2015b). Such loss of colon length and mass (Figure 2A and B) largely reflects a reduction in colon cross sectional area, which is proportional to crypt length (Figure 2D and E). We hypothesize that such HFD-induced intestinal atrophy contributes to the low-grade inflammation and metabolic syndrome-like phenotype that eventuates from HFD consumption. In accord with this notion, the reductions in colon mass and crypt length that resulted from switching from chow to HFD were fully restored by enrichment of the diet with inulin, but not cellulose, thus correlating with inulin’s protection against HFD-induced metabolic syndrome. Such promotion of colon mass by inulin enrichment correlated with increased enterocyte proliferation, as measured by the number of BrdU+ cells per crypt in the proximal colon section (Figure 2F and G). A similar pattern of results was yielded from immunostaining for PCNA (Figure S2A). We hypothesize that inulin’s ability to promote enterocyte growth may have broad impacts upon a range of cell types. Accordingly, we observed that inulin, but not cellulose, increased the level of Paneth cells in ileum as assessed by quantitating the number of lysozyme-expressing cells per crypt by immunostaining (Fig 2H and I). Inulin, but not cellulose, also increase colon expression of tight junction proteins Occludin and Claudin-2 (Fig 2C), although such changes did not correlate with a significant change in overall gut permeability to FITC-dextran (data not shown). In contrast to the colon, neither a generalized change in tissue growth (Figure S2B and C), nor changes in tight junction proteins (Figure S2D), were observed in the small intestine in response to HFD or inulin. However, enrichment of HFD with inulin but not cellulose did result in a significant increase in GLP1-expressing L-cells (Figure S2E and F), which can be envisaged to contribute to inulin’s impact upon glucose metabolism. Thus, inulin, but not cellulose, promotes enterocyte proliferation, which correlates with protection against HFD-induced metabolic syndrome.
Figure 2. Enrichment of HFD with inulin increased epithelial cell proliferation and prevented gut atrophy.

C57BL/6 male mice were fed chow, HFD, or HFD supplemented with cellulose or inulin for 4 weeks. A) Colon length (n=10). B) Colon mass (n=10). C) The mRNA was extracted from colon of mice fed with chow (n=4), HFD supplemeted with cellulose (n=8) or inulin (n=8), and expression level of ZO-1, Occludin and Claudin-2 was analyzed by RT-PCR. D) Colon histopathologic appearance by H&E staining. Scale bars, 200 μm. E) Colon crypt length (n=5), as shown by a double- headed arrow, was measured. F–G) BrdU was injected intraperitoneally 24 h before euthanizing (n=5). Visualization of BrdU positive cells in proximal colon by fluorescence microscopy following staining with FITC-anti-BrdU (F). Scale bars, 100 μm. Enumeration of BrdU positive cells (G). H–I) The paneth cells in ileum (n=5) were stained for lysozyme (H), and the number of paneth cells per crypt was counted (I), scale bars, 50 μm. Data were expressed as mean ± SEM. Statistical significance was assessed by unpaired Student t test. *p<0.05; **p<0.01. See also Figure S2.
Inulin restored HFD-induced microbiota depletion and host-microbiota separation at the mucosal surface
Obesity and metabolic syndrome are associated with alterations in intestinal microbiota composition in both mice and humans, including an increase in the ratio of Firmicutes to Bacteroidetes and increases in relative abundance of Proteobacteria (Turnbaugh et al., 2006; Verdam et al., 2013). Conversely, inulin is a well-recognized prebiotic known to promote growth of select beneficial bacteria such as Bifidobacteria species (Gibson, 1999). Hence, we next examined the extent to which inulin’s protection against HFD-induced metabolic disease would correlate with microbiota alterations. Relative to chow-fed mice, feeding of the standard HFD (i.e. HFD-50 Cell) resulted in about 10-fold reduction in total fecal bacterial loads as assessed by qPCR, suggesting that, relative to chow, this CDD does not support a normal level of bacterial growth (Figure 3A). Importantly, such reduction in fecal bacterial load was fully restored by addition of inulin but not cellulose. Moreover, analysis of microbiota composition by 16S sequencing indicated that replacing cellulose with inulin also corrected some of the HFD-induced changes in microbiota that were observed at the phylum level. Specifically, supplementation of HFD with inulin but not cellulose ameliorated HFD-induced increases in Firmicutes/Bacteroidetes ratio and lowered levels of Proteobacteria (Figure 3B–D). Such sequence-based analysis also showed an inulin-induced increase in Bifidobacteria and Akkermansia (Figure S3A and B) in accord with previous reports (Liu et al., 2016), although the latter was also observed upon supplementation of HFD with cellulose. Using 16S data to look broadly at microbiota composition by an unbiased method, namely principal component analysis of the UniFrac distance, revealed that addition of inulin but not cellulose to the HFD dramatically altered the microbiota composition (Figures 3E, S3C). Such change in microbiota composition that associated with inulin feeding was driven by enrichment in various Bacteroidetes and depletion of Ruminococcus, Clostridium, and Enterococcaceae (Figure S3D). However, it should be noted that inulin did not restore the microbiota composition of HFD-fed mice toward that of chow-fed mice. Rather, there remained a marked difference in microbiota composition between chow-fed and inulin-HFD fed mice that was driven by enrichment and depletion of a wide variety of species (Figure S3E). Such inulin-induced changes in the microbiota resulted in reduced levels of alpha diversity (Figure S3F), which we and others have generally considered a feature of dysbiosis. In contrast, enrichment of HFD with cellulose has a relatively mild impact on microbiota composition.
Figure 3. Inulin restored microbiota loads and prevented HFD-induced microbiota encroachment.

C57BL/6 male mice were fed chow, HFD, or HFD supplemented with cellulose or inulin for 4 weeks and feces collected at 28 days post diets treatment. A) Levels of fecal bacterial DNA were quantitated by qPCR (n=5). B–E) Fecal microbiota composition was analyzed by 16S RNA sequencing (n=4–5). B–D displays relative abundance of bacteria at phylum level. E Global composition as expressed by UniFrac PCoA analysis. F–G) Analysis of microbiota-mucus-epithelial localization in transverse colon via carnoy fixation, FISH, immunohistochemical staining (F). Scale bars, 20 μm. Bacterial epithelial distance was quantified (G). H) Quantitation of colonic Reg3γ by RT-PCR. Data were expressed as mean ± SEM. Statistical significance was assessed by unpaired Student t test. *p<0.05; **p<0.01. See also Figure S3.
While mechanisms by which alterations in microbiota alter metabolic phenotype of the host are not well understood, we have shown that both mice and humans with dysglycemia exhibit microbiota that infiltrate the mucus layer to attain a close proximity to host cells (Chassaing et al., 2015a; Chassaing et al., 2014; Chassaing et al., 2017). Herein, we observed that such microbiota encroachment, which was previously observed in mice fed emulsifiers and mice with a genetic deficiency in TLR5, was also observed in response to HFD (Figure 3F and G). Moreover, such encroachment was largely reversed by adding 4-fold more fiber as inulin but not cellulose (Figure 3F and G). Such a reduction of microbiota encroachment by inulin was associated with restoration of the expression of anti-microbial peptide Reg3γ in whole colon tissue (Figure 3H) or in colon epithelial cells (Figure S3G). Altogether, these results are consistent with the notion that inulin’s protection against HFD is mediated by promoting epithelial proliferation and antimicrobial gene expression that increases host-microbiota interactions in a manner that may reduce the tendency of microbiota encroachment and thus low-grade inflammation (LGI).
Microbiota ablation eliminates inulin’s ability to impact colon, suppress adiposity, and improve glycemic control
We next sought to examine the extent to which inulin’s ability to restore HFD-induced loss of colon mass required the presence of a microbiota. First, we eliminated the contribution of the microbiota by using germfree (GF) mice. These experiments utilized Swiss-Webster mice, which, unlike C57BL/6 mice, are readily maintained in a seemingly healthy state in GF conditions. While HFD is not autoclavable, irrespective of fiber content, we found that subjecting such diets to 2 rounds of γ-irradiation did not compromise the GF status of mice consuming these diets, as evidenced by similar levels of fecal bacterial DNA before and after cessation of HFD (Figure S4A). The increase in levels of fecal bacterial DNA while consuming HFD reflects levels of bacterial products in these diets, which is not removed by irradiation, with the observation of bacterial DNA mainly being from Lactococcus (Figure S4A). The source in the purified HFDs was likely the casein, which is precipitated from skim milk by lactic cultures. Comparison of chow-fed conventional and GF Swiss-Webster mice revealed that the absence of a microbiota by itself on a chow diet resulted in colon atrophy, specifically reduced mass, crypt length, reminiscent of what exhibited conventional mice fed a compositionally-defined diet with cellulose as a sole source of fiber (Figure S4B–D). Such gut atrophy that resulted from the GF status was not further enhanced by administration of HFD. Moreover, in GF conditions, inulin supplementation did not rescue gut atrophy and enterocyte proliferation as assayed by any of these measurements (Figure S4B–F). These results suggest that HFD-induced colon atrophy reflects a reduction in gut bacterial loads, and/or key species, and that inulin’s restoration of the former reflects its restoration of the latter. Because GF mice are known to have a number of developmental abnormalities, particularly in regards to the gut associated immune system (Bouskra et al., 2008), which is thought to influence development of metabolic syndrome (Winer et al., 2016), we also sought to ablate the microbiota by use of antibiotics. Specifically, we examined the extent to which supplementation of HFD with inulin vs. cellulose would promote intestinal mass in mice maintained on a cocktail of oral antibiotics that reduced fecal bacterial loads by about 3 logs (Fig S4G). We observed that, analogous to our results with GF mice, antibiotic treatment greatly reduced the ability of inulin to promote microbiota growth and was paralleled by markedly reducing its promotion of colon mass (Figure 4B–C).
Figure 4. Microbiota ablation eliminated inulin’s beneficial effects in HFD-induced metabolic syndrome.

C57BL/6 male mice (n=10) were fed HFD supplemented with cellulose (HFD-200 Cell) or inulin (HFD-200 Inul) with or without antibiotic cocktail in drink water. A) Body mass over time, B) Colon length, C) Colon weight, D) Epididymal fat and mesenteric fat were measured at the end of experiment. E–G). Glucose tolerance was measured and area under curve (AUC) calculated. H) 5 h fasting glucose. I) Mice were fasted 5 h and intraperitoneally injected with insulin to measure insulin sensitivity. J) Correlation between 5 h fasting glucose and colon weight using data from Figure 1&4. Data were expressed as mean ± SEM. Statistical significance was assessed by unpaired Student t test. *p<0.05; **p<0.01; n.s, not significance. See also Figure S4.
HFD-induced metabolic syndrome is, itself, known to be largely dependent upon the presence of a microbiota (Backhed et al., 2007; Rabot et al., 2010), particularly in GF mice, thus making it difficult to discern the extent to which amelioration of such a phenotype by a particular treatment is also dependent upon the microbiota. Such caveat notwithstanding, we sought to determine if, under conditions of microbiota ablation via antibiotics, inulin might retain any ability to improve parameters of metabolic syndrome in HFD-treated mice. We observed that, upon antibiotic cocktail-mediated suppression of the microbiota, inulin’s ability to reduce weight gain, adiposity and improve glycemic control was not merely eliminated but, rather, inulin promoted adiposity and dysglycemia (Figures 4A, D–I, S4H), perhaps reflecting that mice with minimal microbiota are more readily to digest inulin-enriched diets and harvest energy therefrom (Weitkunat et al., 2015). In any case, these results indicate that microbiota are necessary for both inulin’s promotion of colon mass and beneficial effects on parameters of metabolic syndrome. Lastly, in accord with the general notion that increased colonic mass is generally protective against metabolic syndrome, we note that, over a range of experimental conditions, colon mass negatively correlates with fasting blood glucose concentration (Figure 4J). To further investigate the role of inulin-induced changes in microbiota in impacting the gut and influencing metabolic parameters, fecal microbiota transplants were performed. Specifically, feces from mice fed chow or HFD supplemented with inulin or cellulose, were administered to 4-week old GF mice, which were then maintained on autoclaved chow for an additional 4 weeks. We observed that mice transplanted with microbiotas from inulin-fed mice exhibited a trend toward improved glycemic control, reduced adiposity, increased colon mass (Figure S4I–O). That these effects were of modest magnitude and significance might reflect that the inulin-induced alterations in microbiota are not fully maintained when mice are fed a chow diet but are nonetheless in accord with the notion that such alterations play a role in mediating inulin’s impact on gut and metabolic phenotype.
Manipulation of SCFA levels in intestine did not impact colonic health or adiposity induced by HFD enriched with cellulose or inulin
Next, we examined the mechanism by which the microbiota mediates inulin’s restoration of colon mass and amelioration of metabolic syndrome. First, we considered the role of short-chain fatty acids (SCFA), which have been reported to mediate many of the beneficial effects of fermentable fiber in general and inulin in particular (Woting and Blaut, 2016). In accord with this notion, consumption of HFD resulted in a marked loss of feces levels of acetate, propionate and butyrate that was fully restored by addition of inulin (Figure S5A–C). To investigate the role of inulin-induced SCFA in promotion of colon mass and amelioration of metabolic syndrome, we sought a means to inhibit SCFA without significantly altering bacterial loads per se. Hence, we utilized β-acids, an ingredient derived from hops which have been shown to decrease bacterial-mediated SCFA production by gastrointestinal or fecal bacteria ex vivo (Flythe and Aiken, 2010; Harlow et al., 2014). We observed that administration of β-acids to mice via drinking water potently blocked the inulin-induced increase in SCFA in vivo (Figure S5D–F) but, importantly, did not significantly reduce inulin’s ability to restore microbiota growth (Data not show). Such β-acids-mediated inhibition of SCFA production did not reduce inulin’s ability to promote intestinal mass or suppress adiposity and only mildly impaired inulin’s ability to improve glycemic control (Figure 5A–F) arguing against the notion that SCFA production is a limiting factor in inulin’s protective effects in this model.
Figure 5. Manipulation of SCFA levels in intestine did not impact colonic health or adiposity induced by HFD enriched with cellulose or inulin.

A–F) C57BL/6 male mice (n=5) were fed HFD supplemented with cellulose (HFD-200 Cell) or inulin (HFD-200 Inul) with drink water containing β acid or not. A) Colon weight. B) Epididymal fat pad. C) Mesenteric fat pad. D–E). Glucose tolerance was measured and area under curve (AUC) calculated. F) 5 h fasting glucose. G–N) C57BL/6 male mice (n=5) were fed chow, standard HFD or HFD containing inulin while being administered drinking water containing SCFA (L or H as described in material and method) for 28 days. G) Colon weight. H) Epididymal fat pad. I) fat percentage. J) lean percentage. K–L). Glucose tolerance was measured (K) and area under curve (AUC) calculated (L). M) 5 h fasting glucose. N) Mice were fasted 5 h and intraperitoneally injected with insulin to measure insulin sensitivity. Data were expressed as mean ± SEM. Statistical significance was assessed by unpaired Student t test.
*p<0.05; **p<0.01; n.s, not significance. See also Figure S5.
Next, we examined the extent to which direct administration of SCFA might impact the intestinal atrophy and metabolic syndrome induced by HFD. A mixture of SCFA was provided in drinking water to HFD fed mice using two different doses that were shown to be anti-inflammatory or health-promoting in other studies (Smith et al., 2013). In contrast to inulin enrichment in diet, direct administration of SCFA did not significantly restore colonic mass nor ameliorate metabolic syndrome induced by HFD (Figure 5G–N). Finally, we sought a means to impede the actions of SCFA, specifically, by use of mice deficient in the free fatty acid receptor GPR43 (also referred to FFar2), which is reported to mediate many of the beneficial effects of SCFA (Brooks et al., 2017). Our experimental design was not optimized to discern the role of GPR43 in mediating the response to HFD per se, on which there is conflicting literature (Ang and Ding, 2016), but rather to define if this receptor was required for inulin’s promotion of colon mass and/or its ability to improve indices of metabolic syndrome. We observed that, irrespective of whether mice expressed GPR43 or not, inulin promoted intestinal mass, suppressed HFD-induced adiposity and dysglycemia (Figure S5G–K). SCFA are reported to promote expansion of regulatory T-cells, which dampen chronic inflammatory diseases (Smith et al., 2013). Hence, in light of the notion that HFD-induced metabolic syndrome can be viewed as chronic inflammatory disease, we considered role for T-cells in mediating inulin’s protective effect in metabolic syndrome. Inulin promoted colonic mass, suppressed adiposity, and improved glycemic control in HFD-fed Rag1KO mice, which lack T- and B-lymphocytes arguing against this possibility (Figure S5L–P). Together, these results indicate that inulin’s promotion of colon mass and improvement of metabolic syndrome are not largely mediated by SCFA production.
Inulin restoration of colonic and metabolic health is dependent on IL-22
We next began to consider other potential mechanisms whereby inulin might impact the microbiota to promote epithelial cell proliferation and production of antimicrobial gene expression that, together, might reduce HFD-induced microbiota encroachment and associated parameters of metabolic syndrome. Interlukin-22 (IL-22), induced by gut bacteria, is known to both promote epithelial cell proliferation and induce antimicrobials such as Reg3γ and thereby promote intestinal restitution following acute inflammation (Kinnebrew et al., 2010; Lindemans et al., 2015). We view HFD-induced metabolic syndrome as a state of low-grade inflammation (LGI) and thus envisaged that inulin’s impact on the microbiota might result in IL-22 production that could prevent or resolve such LGI. In accord with this hypothesis, genetic ablation of IL-22 receptor potentiates HFD-induced metabolic syndrome while administration of recombinant IL-22 ameliorates it (Wang et al., 2014). Hence, we next considered the possibility that inulin’s protective effect against HFD might be mediated by IL-22. In accord with this notion, HFD supplementation with inulin increased IL-22 production (Figure 6A). Such inulin-induced IL-22 production, which was detectable in the colon but not serum, was dependent upon the presence of a microbiota in that was absent in GF and antibiotic-treated mice (Figures 6A, S6A), wherein it correlated with lack of induction of Reg3 γ expression in GF mice (Figure S6B). IL-22 has been reported to be produced by neutrophils (Zindl et al., 2013), a subset of T-helper lymphocytes (Th22 cells) (Basu et al., 2012), and type innate lymphoid cells (ILC3) (Rankin et al., 2016). We observed that inulin-induced IL-22 was not impacted by antibody-mediated neutrophil depletion (data not shown), maintained in Rag1 KO mice, which lack most populations of T- and B-lymphocytes, but was absent in Rag/IL2Rγ-DKO mice, which also lack ILC thus indicating a key role for the ILC3 in this response (Figure S7A). Next, we examined whether deficiency IL-22 impacted inulin’s ability to restore gut mass and protect against HFD-induced metabolic syndrome. We observed that inability to produce IL-22 in response to consumption of an inulin-enriched diet, due to loss of the IL-22 gene or ILC3, markedly impaired inulin’s ability to restore colon health (Figures 6B–C, S7B–G), and to suppress indices of metabolic syndrome (Figures 6D–I, S6C).
Figure 6. Inulin’s rescue of HFD-induced colon atrophy and metabolic syndrome is mediated by IL-22.

A) C57BL/6 female conventional (n=5) mice and Swiss Webster GF (n=3–4) mice fed indicated diets were euthanized and distal colon cut into small pieces and cultured overnight. The supernatant was used to measure IL-22 by ELISA. B–I) IL-22KO (n=13) male mice were fed chow, HFD supplemented with cellulose (HFD-200 Cell) or inulin (HFD-200 Inul) for 4 weeks. B) Colon length. C) Colon weight. D) Epididymal fat pad. E) Mesenteric fat. F–G) Mice (n=10) were administered glucose, 2 g/kg intraperitoneally following an overnight fast. Blood glucose levels were measured at the indicated time point (F) and area under curve (AUC) calculated (G). H–I) Mice (n=5) were fasted 5 h, the glucose was measured at 0 (H), 30, 60, 90 min after intraperitoneally injected with insulin (I). Data were expressed as mean ± SEM. Statistical significance was assessed by unpaired Student t test. *p<0.05; **p<0.01; n.s, not significance. See also Figure S6 and S7
In considering how IL-22 might protect against metabolic syndrome, we hypothesized that IL-22-induced fortification of the gut might protect against bacterial translocation that promotes LGI, which has classically been defined as mild elevations in pro-inflammatory gene expression in adipose tissue (Lee et al., 2011). In accord with this possibility, we observed that in the absence of IL-22, inulin no longer significantly induced Reg3γ (Figure 7A) and no longer prevented HFD-induced microbiota encroachment (Figure 7C). Rather, inulin-treated IL-22KO mice exhibited markedly enhanced microbial staining in the mucus layer, which appeared to have an increased tendency to breach intestinal defenses (Figure 7C and D). Specifically, supplementation of HFD with inulin resulted in a marked increase in levels of bacterial 16S RNA in the liver of IL-22KO but not WT mice (Figure 7B) suggesting increased bacterial translocation. Furthermore, inulin reversed some indices of HFD-induced LGI, specifically, adipose tissue expression of CXCL1, TNFα, and IL-6, in WT but not IL-22KO mice (Figure 7E and F). Together, these results indicate that inulin impacts microbiota to induce expression of IL-22, which fortifies the intestine, resulting in reduced microbiota encroachment, and pro-inflammatory gene expression that, together, may underlie inulin’s protection against HFD-induced metabolic syndrome.
Figure 7. Inulin’s restoration of microbiota containment and prevention of low-grade inflammation required IL-22.

IL-22 KO mice were fed chow, HFD supplemented with cellulose (HFD-200 Cell) or inulin (HFD-200 Inul) for 4 weeks. A) The mRNA was extracted from colon to analyze the expression of Reg3γ. B) Bacterial 16S rRNA in the livers were analyzed by qRT-PCR. C) Analysis of microbiota-mucus-epithelial localization in transverse colon of IL-22 KO mice via carnoy fixation, FISH, immunohistochemical staining. D) Bacterial vs epithelial distance was quantified. E–F) Quantitation of CXCL1, TNF-α and IL-6 in white adipose tissue (WAT) of wild type (E) and IL-22 KO mice (F) by qRT-PCR. Data were expressed as mean ± SEM. Statistical significance was assessed by unpaired Student t test. *p<0.05; **p<0.01; n.s, not significance.
DISCUSSION
The central goal of this study was to elucidate the mechanism whereby enrichment of an obesogenic high-fat diet (HFD) with inulin suppresses adiposity and its associated parameters of metabolic syndrome. We hypothesized that bacterial metabolism of inulin, specifically generation of short-chain fatty acids (SCFA), would promote enterocyte proliferation and thus broadly fortify innate mucosal defense, leading to reduce bacterial encroachment. Subsequently, this would reduce low-grade inflammation (LGI) that promotes numerous events that promote and define the metabolic syndrome. Our results supported some aspects of this hypothesis, particularly the notion that inulin restores the HFD-induced loss of enterocyte proliferation, reduced microbiota encroachment, and protects against metabolic syndrome in a microbiota-dependent manner. However, our data did not support a major role for SCFA in inulin’s restoration of colonic health or amelioration of metabolic syndrome. Rather, our results indicate that inulin restores gut health and protects against metabolic syndrome in a manner that correlates with and is dependent upon microbiota-dependent induction of IL-22 expression. We now hypothesize that such inulin-induced IL-22 expression promotes colon health in a manner that reduces microbiota encroachment by fortifying the epithelium via promoting crypt regeneration and increasing expression of antibacterial proteins. These results add to understanding of pathophysiologic mechanisms that underlie DIO and provide a previously unappreciated means by which fermentable fibers might promote health.
Our previous study revealed that HFD resulted in a loss of gut mass, and that such gut atrophy was not caused by dietary fat content per se but, rather, by its lack of fermentable fiber (Chassaing et al., 2015b). Herein, we observed that gut atrophy correlated with reduced enterocyte proliferation and, moreover, microbiota encroachment, all of which were reversed by enrichment of HFD with inulin but not with additional insoluble fiber, cellulose. Interestingly, such microbiota encroachment was inversely proportional to total bacterial loads in the gut. Specifically, relative to chow-fed mice, HFD administration resulted in bacterial infiltration into the mucus layer despite an approximate 10-fold reduction in fecal bacteria. Inulin supplementation of HFD restored bacterial loads, and resulted in some changes in gut microbiota composition that have been associated with health. Specifically, inulin enrichment of HFD restored the Firmicutes/Bacteroidetes ratio, which associates with and thought to contribute to leanness, reduced levels of Proteobacteria, which have been proposed to promote an array of chronic inflammatory diseases, and increased levels of Bifidobacteria, which are depleted in inflammatory diseases and have been used as probiotics to ameliorate such diseases. While there are a myriad of potential means by which these and other inulin-induced changes in microbiota might ameliorate metabolic disease, we hypothesize that reducing microbiota encroachment plays a central role in this process. We envision that such reduced microbiota encroachment should reduce inflammatory signaling, which is thought to impair a range of metabolic signaling pathways including those involved in glycemic control and satiety signaling such as insulin and leptin respectively. Thus, we propose inulin-induced fortification of the mucosa enhances both insulin and satiety signaling in an intertwined manner to ameliorate metabolic syndrome. In accord with our hypothesis that microbiota encroachment, which we recently showed is a feature of metabolic syndrome in humans (Chassaing et al., 2017), plays a role in driving this low-grade chronic inflammatory disorder. While reduced microbiota encroachment can be imagined to result in reduced expression of IL-22 and Reg3γ, we propose that, in fact, the inulin-induced microbiotas resulted in a relatively stable host-microbiota equilibrium state. We speculate that inulin-induced IL-22 dependent fortification of the mucosa likely involves increased barrier function but further studies are needed to determine the extent to which this involves exclusion of bacteria themselves and/or their products.
While our observation that soluble fiber improves parameters of metabolic syndrome via a microbiota-dependent process is generally in accord with most published work in this area, we note that Bindels et. al recently reported that such fibers improved glycemic control even in GF mice (Bindels et al., 2017). While this apparent discrepancy between their and our results could reflect differences in specific fibers, source and/or mouse strains utilized, we speculate that the absolute absence of bacteria in GF mice makes it very difficult for them to digest these fibers resulting in reduced appetite and, consequently, alterations in glycemic control by mechanisms that would not be relevant in mice with a microbiota. Such apparent inability to digest inulin-enriched diets was not seen in antibiotic treated mice, perhaps reflecting that a minimal microbiota is sufficient to at least partially digest this fiber (Weitkunat et al., 2015) and, in any case, arguing that the inability of inulin to suppress adiposity or improve glycemic control in antibiotic-treated mice reflects an essential role for the microbiota in mediating inulin’s amelioration of HFD-induced metabolic syndrome.
That inulin’s restoration of gut bacterial loads correlated with a microbiota-dependent increase in fecal SCFA levels supported our presumption that SCFA would be pivotal in mediating inulin’s beneficial effects. Yet, suppressing SCFA production, to levels approaching or below baseline levels in the cecum, which is the most sensitive locale to measure changes in SCFA resulted in only a modest loss of glucose tolerance and did not ameliorate inulin’s ability to suppress adiposity and fasting glucose levels. Nor did loss of the free fatty acid receptor GPR43 impact upon inulin’s ability to ameliorate HFD-induced dysglycemia or adiposity. This latter result is an apparent contradiction with a recent publication from Brooks et. al that reported inulin suppressed HFD-induced weight gain and adiposity in WT but not GPR43-KO mice (Brooks et al., 2017). The reason for this discrepancy is unclear but given that our study used a higher level of inulin, which appears to have a stronger suppression of adiposity, we speculate that some portion of inulin’s beneficial metabolic effects might be mediated by SCFA acting via GPR43 but that SCFA-independent effects may mask the need for this receptor. We also note that our approaches to manipulate SCFA levels have significant limitations. Indeed, the specificity of β acid is not well defined, and administration of SCFA in drinking water may not reach the colon in adequate levels. Thus, further work is needed in this area to better define the role of SCFA in protecting against metabolic syndrome. Specific needs include determining what metabolites are produced when SCFA production is blocked and discern how they impact the host. The need for such studies notwithstanding, our results indicate that SCFA may not be a limiting factor in the microbiota-dependent ability of inulin to protect against HFD-induced metabolic syndrome.
It should be noted that, although inulin restored total gut bacterial loads, it did not restore microbiota composition toward that of chow fed mice but, rather, resulted in a microbiota composition seemingly equally distinct from chow and HFD-fed mice, at least as assessed by unbiased metrics such as UniFrac analysis. While such altered microbiota composition did not result in an obvious phenotypic difference relative to chow-fed mice, we recently reported that exposure of mice fed inulin-enriched diets, irrespective of fat content, exhibited very severe colitis upon challenge with dextran sodium sulfate (DSS), a chemical colitogen (Miles et al., 2017b). This observation highlights potential risk in promoting bacterial growth in the gut, particularly in inducing a microbiota composition whose functional attributes are not well defined. Moreover, it underscores the need to define specific mechanisms whereby inulin impacts microbiota to protect against metabolic disease. That inulin’s protection against HFD-induce metabolic syndrome correlated with IL-22 induction and is IL-22 dependent, combined with the observation that recombinant IL-22 ameliorates HFD-induced obesity indicates that IL-22 is one such potential mechanism (Wang et al., 2014). This notion is in accord with recent work that direct IL-22 administration protects against HFD-induced metabolic syndrome and the observation that IL-22R KO mice potentiated disease in this model. Yet, interestingly, IL-22KO mice were not found to share this latter phenotype (Wang et al., 2014). Our results were in accord with this observation in that we did not observe a difference in the extent of standard HFD-induced metabolic disease between IL-22 and WT mice but it should be noted that, although maintained in the same facility in similar conditions, knockout mice had not been bred with the WT control mice so we don’t view our comparison to be a rigorous confirmation of this seemingly enigmatic result.
In contrast to inulin supplementation, which can exacerbate severity of DSS colitis(Miles et al., 2017a), IL-22 administration seems broadly protective against a range of intestinal infectious and inflammatory challenges including DSS, C. rodentium, and rotavirus (Sugimoto et al., 2008; Zhang et al., 2014; Zheng et al., 2008). We hypothesize that a common mechanism by which IL-22 protects against these inflammatory challenges, including HFD, which we view as a low-grade inflammatory challenge, is by increasing the metabolic rate of the epithelium, resulting in greater expression of antimicrobials, more rapid proliferation, which correlates with longer crypts thus, together, creating a more protective shield against the gut microbiota. IL-22’s recently recognized ability to strengthen the intestinal barrier by reducing ER stress might contribute to this process (Gulhane et al., 2016). We speculate that such effects on the epithelium result in an enhanced shield from the microbiota protect against an array of inflammatory challenges and, in particular, underlie inulin’s and recombinant IL-22’s protection against HFD-induced metabolic syndrome.
STAR METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Ki67 | Abcam | Cat.# Ab15580 |
| Anti-PCNA | Cell Signaling Technology | Cat.# 2586S |
| Anti-Lysozyme | Abcam | Cat.# ab108508 |
| FITC labeled anti-BrdU | Abcam | Cat.# Ab74545 |
| Anti-Mucin 2 | Santa Cruz Biotechnology | Cat.# H-300 |
| Anti-GLP1 | Abcam | Cat.# Ab22625 |
| Alexa Fluor 488 Goat Anti-Rabbit IgG | Invitrogen | Cat.# A11008 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| D-(+)-GLUCOSE | Sigma-Aldrich | Cat.# G8270-1KG |
| Insulin | Sigma-Aldrich | Cat.# 12643-25MG |
| Oil red O | Sigma-Aldrich | Cat.# O0625-100G |
| Sodium acetate | Sigma-Aldrich | Cat.# S2889-250G |
| Sodium butyrate | Sigma-Aldrich | Cat.# 303410-100G |
| Sodium propionate | Sigma-Aldrich | Cat.# P1880-500G |
| Ampicillin | Sigma-Aldrich | Cat.# A9518-100G |
| Vancomycin | Sigma-Aldrich | Cat.# V2002-250MG |
| Neomycin | Sigma-Aldrich | Cat.# N1876-100G |
| Metronidazole | Sigma-Aldrich | Cat.# M1547-25G |
| Critical Commercial Assays | ||
| Free Fatty Acid Assay Kit | Abcam | Cat.# Ab65341 |
| Triglycerides Reagent | Thermo Scientific | Lot. # 723341 |
| Cholesterol Reagent | Thermo Scientific | Lot. # 848958 |
| IL-22 ELISA Kit | R&D Systems | Cat.# DY582 |
| One-Step RT–PCR Kit with SYBR Green | Bio-Rad | Cat. # 172-5151 |
| QIAamp DNA Stool Mini Kit | Qiagen | Cat.# 51504 |
| QuantiFast SYBR Green PCR kit | Bio-Rad | Cat.# 204054 |
| Experimental Models: Organisms/Strains | ||
| Mice: C57BL/6 | The Jackson Laboratory | Cat. # 000664 |
| Mice: Rag1 knockout | The Jackson Laboratory | Cat. # 002216 |
| Mice: GPR43 knockout | The Jackson Laboratory | Cat. # 027241 |
| Mice: Rag2−/−IL2rg−/− | The Jackson Laboratory | Cat. # 014593 |
| Mice: IL-22 knockout | Genentech, Inc | NA |
| Mice: Swiss-Webster mice | Charles River Laboratories | Cat. # 024 |
| Oligonucleotides | ||
| See Figure S2 | This study | |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad Software | NA |
| Image J | NIH | NA |
| Other | ||
| halloidin–Tetramethylrhodamine B isothiocyanate | Sigma-Aldrich | Cat.# P1951-.1MG |
| β-acid | S.S. Steiner, Inc | NA |
| DAPI Fluoromount-G® Mounting Medium | SouthernBiotech | Cat.# 0100-20 |
| Rodent chow | LabDiet | Cat. # 5001 |
| Standard high-fat diet | Research Diets, Inc. | Cat. # D12492 |
| Inulin-enriched high-fat diet | Research Diets, Inc. | Cat. # D13081106 |
| Cellulose-enriched high-fat diet | Research Diets, Inc. | Cat. # D13081107 |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andrew T. Gewirtz (agewirtz@gsu.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice and diets
C57BL/6 WT, Rag1 knockout (Rag1KO) and GPR43knockout (GPR43KO) mice, Rag2−/−IL2rg−/− were purchased from Jackson Laboratories, Bar Harbor, ME. IL-22 KO mice were provided by Genentech, Inc. South San Francisco, CA. Swiss-Webster mice were purchased from Charles River Laboratories. All mice were bred and housed at Georgia State University, Atlanta, Georgia, USA under institutionally-approved protocols (IACUC # A14033). Mice, 6–10 weeks of age, were fed either a grain-based rodent chow (cat# 5001) from LabDiet (hereafter referred as ‘chow’), St. Louis, MO, or indicated compositionally defined diets (CDD) made with purified ingredients, which were produced by Research Diets, Inc, New Brunswick, NJ. The composition of all purified-ingredient CDDs used in this study are listed in Table S1, and wherein levels of fiber in supplemented HFD (about 20% fiber) is approximately similar that of grain-based chows (15–25% fiber). As shown, there were 3 different compositionally defined high fat diets with 60 kcal% fat (mainly lard) and different levels and types of fiber: A high fat purified diet with 50 g cellulose per 4,057 kcals (D12492, HFD-50 Cell) and the remaining diets with 200 g cellulose (HFD-200 Cell) or 200 g inulin (HFD-200 Inul) per 4,057 kcals. Three other diets (not shown) contained 50% more vitamins to compensate for potential losses during double irradiation for germ-free mice. Prior to the experimentation period, all mice were maintained on chow, which we view as a reference diet. During the experimental period, male mice were fed chow or specified CDD for a period of 28 days except where indicated. At the end of the experimental period, feces and blood were collected before euthanasia, organs including colon and liver were collected for measuring colon length, weight, fat pad weight and other downstream analysis. For antibiotic treatment, two days prior to HFD initiation, mice were administered drinking water containing ampicillin (1 g/L), vancomycin (0.25 g/L), neomycin (1 g/L), and metronidazole (1 g/L), water supply was renewed every 3 days. To inhibit short chain fatty acid production by inhibiting bacterial fermentation, mice were treated with vehicle control (Propylene Glycol) or 20 ppm β-acid (extracted from the hops plant; S.S. Steiner, Inc.) (Flythe and Aiken, 2010; Harlow et al., 2014).
METHOD DETAILS
Food consumption measurements
A known amount of food was placed in a cage of five mice, the amount of remaining food was measured twenty-four hours later. The amount of food consumed was calculated by the difference divided by 5 and expressed as food intake per mouse per 24 h. Food intake was monitored every day for two weeks.
SCFA treatment
Mice consuming standard HFD (D12492) were treated with the following high (H) and low (L) dose cocktail of short chain fatty acid (SCFA) mixture in drink water for 28 days: H= 67.5 mM sodium acetate, 40 mM sodium butyrate, and 25.9 mM sodium propionate; L= 33.75 mM sodium acetate, 20 mM sodium butyrate, and 12.95 mM sodium propionate. Measurement of % fat mass and lean mass was performed via MRI (Bruker MiniSpec) at day 27 post diets treatment.
Glucose measurement
Glucose tolerance was measured 26 days following initiation of indicated diet. Mice were placed in a clean cage and provided with water but without food overnight (15h). Baseline blood glucose was then measured using a Nova Max plus Glucose meter. Mice were administered glucose, 2 mg of glucose/gm body weight, and blood glucose levels measured 30, 60, and 90 minutes later. Data are expressed as mg glucose/dL blood. 2 days later, (i.e. day 28), to measure “fasting glucose levels”, mice were placed in a clean cage and provided with water but without food for 5h, at which point, fasting glucose levels were determined and insulin tolerance test initiated. Specifically, 5h-fasted mice were injected with 0.5 U insulin/kg body weight and blood glucose levels were measured at 30, 60, 90 min after injection.
Assays for cholesterol, triglyceride and FFA
Cholesterol and triglyceride levels in serum were measured using kits from Thermo Scientific. Serum total free fatty Acids (FFA) was measured by using a FFA quantification assay kit from Abcam, Cambridge MA.
Germ-free (GF) mice experiments
Germ-free Swiss Webster mice were bred in a Park Bioservices isolator and then transferred to “Isocages” (Techniplast USA, West Chester, PA), which maintain a germ-free state. GF male mice were fed autoclaved chow or CDD, which was sterilized by 2 rounds of γ-irradiation (10 – 20 kGy, 2 times). Such diets were supplemented with extra vitamins to guard against depletion of key micronutrients by radiation. After 4 weeks of treatment, mice were removed from isocages and immediately euthanized. To verify that the irradiated diets are free of bacteria, GF mice were treated with irradiated HFD diet for 14 days, the feces were collected before and after irradiated HFD diet treatment. Then these mice were switched to autoclaved chow diet for 14 days before the feces were collected to measure bacteria load (Figure S4 A).
Microbiota transplantation
Feces from mice fed chow or HFD supplemented with cellulose or inulin were suspended in PBS. Germ-free Swiss Webster mice (4 weeks old) were removed from the isolator and were orally administered 200 μl of faecal suspension. Transplanted mice were fed with chow for 4 weeks.
Bacteria localization by FISH staining
The staining of bacteria localization at the surface of the intestinal mucosa was conducted as previously described (Chassaing et al., 2014). Briefly, transverse colonic tissues full of fecal material were placed in methanol-Carnoy’s fixative solution (60% methanol, 30% chloroform, 10% glacial acetic acid) for a minimum of 3 h at room temperature. Tissues were then washed in methanol 2x 30 min, ethanol 2x 20 min, and xylene 2x 20 min and embedded in paraffin for 5 μm sections on glass slides. The tissue sections were dewaxed by preheating at 60°C for 10 min, followed by xylene 60°C for 10 min, xylene for 10 min and 99.5% ethanol for 10 minutes. Deparaffinized sections were incubated at 50°C overnight with EUB338 probe (5′-GCTGCCTCCCGTAGGAGT-3′, with a 5′ labeling using Alexa 647) diluted to 10 μg/mL in hybridization buffer (20 mM Tris–HCl, pH 7.4, 0.9 M NaCl, 0.1% SDS, 20% formamide). After incubating with wash buffer (20 mM Tris–HCl, pH 7.4, 0.9 M NaCl) for 10 min and 3x 10 min in PBS sequentially, the slides were blocked with 5% fetal bovine serum for 1h at 4°C. Samples were then incubated with Mucin-2 primary antibody (Santa Cruz Biotechnology, 1:500) overnight at 4°C. After washing 3x 10 min in PBS, the slides were incubated with anti-rabbit Alexa 488 secondary antibody diluted 1:500, Phalloidin Tetramethyl rhodamine B isothiocyanate (Sigma, St. Louis, MO) at 1μg/mL. After washing 3x in PBS, allowed to dry, the slides were mounted in DAPI containing mounting medium.
RNA isolation and qRT-PCR
Total RNA was extracted from transverse colonic tissues or epithelial cells using TRIzol (Invitrogen, Carlsbad, California) according to the manufacturer’s protocol. Quantitative RT–PCR was performed using the iScriptTM One-Step RT–PCR Kit with SYBR Green (Bio-Rad, Hercules, Californai) in a CFX96 apparatus (Bio-Rad, Hercules, California) with the primers listed in Table S2. Difference in transcript levels were quantified by normalization of each amplicon to housekeeping gene 36B4.
Hematoxylin and eosin staining
Mouse colons, small intestine including duodenum and ileum, adipose tissue, and liver were fixed in 10% buffered formalin at room temperature before embedding in paraffin. Tissues were sectioned at 4 μm thickness and stained with hematoxylin and eosin (H&E). The adipose cells size and colonic cypt length and villus length in small intestine was determined using Image J software from H&E.
Immunohistochemistry
The tissue sections were immersed in a citrate buffer solution (0.01M, pH 6.0) and heated at 120°C in an autoclave sterilizer for 10 min for antigen retrieval. After cooling to room temperature the following steps were performed in a moist chamber. The sections were washed in PBS, blocked with protein block (BioGenex, CA, USA), and the proximal colon section was incubated with rabbit anti-Ki67, or anti-GLP1 (Abcam, Cambridge, MA) diluted 1:1000 for 1 h, the ileum section was incubated with rabbit anti-lysozyme for 1 h. After washing in PBS, these sections for Ki67, GLP1 and lysozyme staining were stained with secondary fluorescent antibody, and the tissues were counterstained with mounting medium containing DAPI. The section for PCNA staining was detected by DAB. The expression level of Ki67 in chow-fed Swiss Webster conventional and GF mice were set as “1” respectively. The relative level of ki67 in conventional mice or GF mice with different HFD diets treatment were expressed fold change relative to conventional mice or GF mice which fed chow diet respectively. The number of GLP1 positive cells per crypt in proximal colon, and lysozyme positive Paneth cells per crypt in ileum was counted.
Oil red O staining
Mouse livers were embedded in OCT (Optimal cutting temperature) compound after euthanasia. Tissues were sectioned at 4 μm thickness and stained with Oil red O. Briefly, 1.9g of Oil red O (Sigma) was dissolved in 300mL of isopropyl alcohol. After homogenization, 200 mL of distilled water was added. The reagent was filtered after incubating at 4°C for 30 minutes, then used for staining liver sections. After staining for 6 min, the sections were washed using running tap water, and mounted using a sterile glycerol 40% solution. Image J was used to quantify the red staining.
BrdU staining
Mice were injected with Brdu (50 μg of BrdU/g) intraperitoneally. After 24 h, proximal colon tissue was harvested and embedded in OCT. Tissues sections (4 μm thickness) were cut and, fixed with 4% formaldehyde for 30 min at room temperature. After washing with PBS, the sections were incubated with prewarmed (37°C) 1N HCL for 45 min in 37°C incubator, blocked with protein block (BioGenex, CA, USA) before incubating with FITC labeled anti-BrdU antibody (Abcam) overnight at 4°C. Brdu-labeled nuclei were counted in each crypt of colon under fluorescent microscope. While Brdu positive cells in the lamina propria, which might be immune cells, were not excluded.
ELISA
The distal colons were washed in HBSS supplemented with penicillin and streptomycin, and cut into 1 cm segments. These segments were cultured in 24-well flat-bottom culture plates in RPMI 1640 medium supplemented with 10% FBS, penicillin and streptomycin, l-glutamine. After 24 h, supernatant was collected and centrifuged at 5,000 g for 10 minutes at 4°C and the concentration of IL-22 was measured according to the ELISA kit provided by R&D Systems.
Bacterial quantification in feces and liver
For quantification of total faecal bacterial load, total DNA was isolated from known amounts of feces using QIAamp DNA Stool Mini Kit (Qiagen). DNA was then subjected to quantitative PCR using QuantiFast SYBR Green PCR kit (Biorad) with universal 16S rRNA primers listed in Table S2 to measure total bacteria number. Results are expressed as bacteria number per mg of stool, using a standard curve. The quantification of bacteria in the liver was measured as previously described by Etienne-Mesmin et al (Etienne-Mesmin et al., 2016).
Isolation of intestinal epithelial cells
The colons were washed extensively in PBS, cut longitudinally and generated 1.0 cm pieces and incubated with HBSS containing 2 mM EDTA for 30 min with shanking at 200 rpm. After vortexing, the supernatant was passed through 100 μm cell strainer. The filtrate containing intestinal epithelial cells (IEC) were centrifuged, washed with cold PBS and centrifuged and pellet was stored at −80°C until analyzed.
Short-chain fatty acids analysis
Levels of acetate, propionate, and butyrate in feces and cecal content, from non-fasted mice, were measured by gas chromatography as previously described (Zheng et al., 2013).
16S rRNA gene sequence analysis
16S rRNA gene amplification and sequencing were conducted as described (Chassaing et al., 2015a). Briefly, DNA was extracted from feces of C57BL/6, which was collected at 28 day post diet initiation, using a PowerSoil-htp kit from MO BIO Laboratories (Carlsbad, California). The region V4 of 16S rRNA genes were amplified by the primers listed in Table S2; The PCR product of each samples were combined and purified with Ampure magnetic purification beads (Agencourt). Products were then quantified (BIOTEK Fluorescence Spectrophotometer) using a Quant-iT PicoGreen dsDNA assay. The pooled products were quantified and sequenced by using an Illumina MiSeq sequencer (paired-end reads, 2 × 250 base pairs) at Cornell University, Ithaca. The sequences were demultiplexed, quality filtered using the Quantitative Insights into Microbial Ecology (QIIME, version 1.8.0) software package before analysis as previously described (Chassaing et al., 2015a).
QUANTIFICATION AND STATISTICAL ANALYSIS
Results were expressed as mean ± SEM. Statistical significance was analyzed by unpaired Student t test (GraphPad Prism 5). Differences between experimental groups were considered significant at *P ≤ 0.05 or **P ≤ 0.01.
Supplementary Material
Highlights.
The fermentable fiber inulin prevented high fat diet (HFD)-induced metabolic syndrome
HFD enriched with inulin increased gut epithelial proliferation, prevented colon atrophy
Inulin restored HFD-induced microbiota depletion and microbiota-mucosa separation
Inulin effects are microbiota and IL-22, but not short-chain fatty acid, dependent
Acknowledgments
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK099071 and DK083890. B. Chassaing is supported by a Career Development Award from the Crohn’s and Colitis Foundation of America. We thank Dr. Edward Ulman from Research Diets, Inc. for his insightful comments regarding the differences in fiber level/type between grain-based and purified diets and how this might impact the animals’s phenotype.
Abbreviations
- DIO
diet-induced obesity
- CDD
compositionally-defined diet
- HFD
high-fat diet
- SCFA
short-chain fatty acid
- IL-22
Interleukin 22
Footnotes
AUTHOR CONTRIBUTIONS
J.Z, B.C. and A.T.G. conceived the project and designed experiments. J.Z and A.T.G. prepared the manuscript with input from all other authors. J.Z, B.C. and V.S performed the experiments and data analysis. M.P, M.R, M.F and M.V.K provided key reagents and guided experimental design and data interpretation.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ang Z, Ding JL. GPR41 and GPR43 in Obesity and Inflammation - Protective or Causative? Front Immunol. 2016;7:28. doi: 10.3389/fimmu.2016.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci U S A. 2007;104:979–984. doi: 10.1073/pnas.0605374104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu R, O’Quinn DB, Silberger DJ, Schoeb TR, Fouser L, Ouyang W, Hatton RD, Weaver CT. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity. 2012;37:1061–1075. doi: 10.1016/j.immuni.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindels LB, Segura Munoz RR, Gomes-Neto JC, Mutemberezi V, Martinez I, Salazar N, Cody EA, Quintero-Villegas MI, Kittana H, de Los Reyes-Gavilan CG, et al. Resistant starch can improve insulin sensitivity independently of the gut microbiota. Microbiome. 2017;5:12. doi: 10.1186/s40168-017-0230-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouskra D, Brezillon C, Berard M, Werts C, Varona R, Boneca IG, Eberl G. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature. 2008;456:507–510. doi: 10.1038/nature07450. [DOI] [PubMed] [Google Scholar]
- Brooks L, Viardot A, Tsakmaki A, Stolarczyk E, Howard JK, Cani PD, Everard A, Sleeth ML, Psichas A, Anastasovskaj J, et al. Fermentable carbohydrate stimulates FFAR2-dependent colonic PYY cell expansion to increase satiety. Mol Metab. 2017;6:48–60. doi: 10.1016/j.molmet.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassaing B, Koren O, Goodrich JK, Poole AC, Srinivasan S, Ley RE, Gewirtz AT. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature. 2015a;519:92–96. doi: 10.1038/nature14232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassaing B, Ley RE, Gewirtz AT. Intestinal epithelial cell toll-like receptor 5 regulates the intestinal microbiota to prevent low-grade inflammation and metabolic syndrome in mice. Gastroenterology. 2014;147:1363–1377. e1317. doi: 10.1053/j.gastro.2014.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassaing B, Miles-Brown J, Pellizzon M, Ulman E, Ricci M, Zhang L, Patterson AD, Vijay-Kumar M, Gewirtz AT. Lack of soluble fiber drives diet-induced adiposity in mice. American journal of physiology Gastrointestinal and liver physiology. 2015b;309:G528–541. doi: 10.1152/ajpgi.00172.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassaing B, Shreya MR, Lewis JD, Srinivasan S, Gewirtz AT. Colonic Microbiota Encroachment Correlates With Dysglycemia in Humans. Cell Mol Gastroenterol Hepatol. 2017 doi: 10.1016/j.jcmgh.2017.04.001. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborators, G.B.D.O. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N Engl J Med. 2017 doi: 10.1056/NEJMoa1614362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewulf EM, Cani PD, Neyrinck AM, Possemiers S, Van Holle A, Muccioli GG, Deldicque L, Bindels LB, Pachikian BD, Sohet FM, et al. Inulin-type fructans with prebiotic properties counteract GPR43 overexpression and PPARgamma-related adipogenesis in the white adipose tissue of high-fat diet-fed mice. J Nutr Biochem. 2011;22:712–722. doi: 10.1016/j.jnutbio.2010.05.009. [DOI] [PubMed] [Google Scholar]
- Etienne-Mesmin L, Vijay-Kumar M, Gewirtz AT, Chassaing B. Hepatocyte Toll-Like Receptor 5 Promotes Bacterial Clearance and Protects Mice Against High-Fat Diet-Induced Liver Disease. Cell Mol Gastroenterol Hepatol. 2016;2:584–604. doi: 10.1016/j.jcmgh.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everard A, Cani PD. Diabetes, obesity and gut microbiota. Best Pract Res Clin Gastroenterol. 2013;27:73–83. doi: 10.1016/j.bpg.2013.03.007. [DOI] [PubMed] [Google Scholar]
- Flythe MD, Aiken GE. Effects of hops (Humulus lupulus L.) extract on volatile fatty acid production by rumen bacteria. J Appl Microbiol. 2010;109:1169–1176. doi: 10.1111/j.1365-2672.2010.04739.x. [DOI] [PubMed] [Google Scholar]
- Gibson GR. Dietary modulation of the human gut microflora using the prebiotics oligofructose and inulin. J Nutr. 1999;129:1438S–1441S. doi: 10.1093/jn/129.7.1438S. [DOI] [PubMed] [Google Scholar]
- Gulhane M, Murray L, Lourie R, Tong H, Sheng YH, Wang R, Kang A, Schreiber V, Wong KY, Magor G, et al. High Fat Diets Induce Colonic Epithelial Cell Stress and Inflammation that is Reversed by IL-22. Scientific reports. 2016;6:28990. doi: 10.1038/srep28990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton MK, Ronveaux CC, Rust BM, Newman JW, Hawley M, Barile D, Mills DA, Raybould HE. Prebiotic milk oligosaccharides prevent development of obese phenotype, impairment of gut permeability, and microbial dysbiosis in high fat-fed mice. Am J Physiol Gastrointest Liver Physiol. 2017;312:G474–G487. doi: 10.1152/ajpgi.00427.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow BE, Lawrence LM, Kagan IA, Flythe MD. Inhibition of fructan-fermenting equine faecal bacteria and Streptococcus bovis by hops (Humulus lupulus L.) beta-acid. J Appl Microbiol. 2014;117:329–339. doi: 10.1111/jam.12532. [DOI] [PubMed] [Google Scholar]
- Kinnebrew MA, Ubeda C, Zenewicz LA, Smith N, Flavell RA, Pamer EG. Bacterial flagellin stimulates Toll-like receptor 5-dependent defense against vancomycin-resistant Enterococcus infection. J Infect Dis. 2010;201:534–543. doi: 10.1086/650203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Li P, Huh JY, Hwang IJ, Lu M, Kim JI, Ham M, Talukdar S, Chen A, Lu WJ, et al. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes. 2011;60:2474–2483. doi: 10.2337/db11-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindemans CA, Calafiore M, Mertelsmann AM, O’Connor MH, Dudakov JA, Jenq RR, Velardi E, Young LF, Smith OM, Lawrence G, et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature. 2015;528:560–564. doi: 10.1038/nature16460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TW, Cephas KD, Holscher HD, Kerr KR, Mangian HF, Tappenden KA, Swanson KS. Nondigestible Fructans Alter Gastrointestinal Barrier Function, Gene Expression, Histomorphology, and the Microbiota Profiles of Diet-Induced Obese C57BL/6J Mice. J Nutr. 2016;146:949–956. doi: 10.3945/jn.115.227504. [DOI] [PubMed] [Google Scholar]
- Miles JP, Zou J, Kumar MV, Pellizzon M, Ulman E, Ricci M, Gewirtz AT, Chassaing B. Supplementation of Low- and High-fat Diets with Fermentable Fiber Exacerbates Severity of DSS-induced Acute Colitis. Inflammatory bowel diseases. 2017a;23:1133–1143. doi: 10.1097/MIB.0000000000001155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabot S, Membrez M, Bruneau A, Gerard P, Harach T, Moser M, Raymond F, Mansourian R, Chou CJ. Germ-free C57BL/6J mice are resistant to high-fat-diet-induced insulin resistance and have altered cholesterol metabolism. FASEB J. 2010;24:4948–4959. doi: 10.1096/fj.10-164921. [DOI] [PubMed] [Google Scholar]
- Rankin LC, Girard-Madoux MJ, Seillet C, Mielke LA, Kerdiles Y, Fenis A, Wieduwild E, Putoczki T, Mondot S, Lantz O, et al. Complementarity and redundancy of IL-22-producing innate lymphoid cells. Nature immunology. 2016;17:179–186. doi: 10.1038/ni.3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar N, Dewulf EM, Neyrinck AM, Bindels LB, Cani PD, Mahillon J, de Vos WM, Thissen JP, Gueimonde M, de Los Reyes-Gavilan CG, et al. Inulin-type fructans modulate intestinal Bifidobacterium species populations and decrease fecal short-chain fatty acids in obese women. Clin Nutr. 2015;34:501–507. doi: 10.1016/j.clnu.2014.06.001. [DOI] [PubMed] [Google Scholar]
- Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, Glickman JN, Garrett WS. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg ED, Smits SA, Tikhonov M, Higginbottom SK, Wingreen NS, Sonnenburg JL. Diet-induced extinctions in the gut microbiota compound over generations. Nature. 2016;529:212–215. doi: 10.1038/nature16504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg ED, Sonnenburg JL. Starving our microbial self: the deleterious consequences of a diet deficient in microbiota-accessible carbohydrates. Cell Metab. 2014;20:779–786. doi: 10.1016/j.cmet.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, Blumberg RS, Xavier RJ, Mizoguchi A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest. 2008;118:534–544. doi: 10.1172/JCI33194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Verdam FJ, Fuentes S, de Jonge C, Zoetendal EG, Erbil R, Greve JW, Buurman WA, de Vos WM, Rensen SS. Human intestinal microbiota composition is associated with local and systemic inflammation in obesity. Obesity (Silver Spring) 2013;21:E607–615. doi: 10.1002/oby.20466. [DOI] [PubMed] [Google Scholar]
- Wang X, Ota N, Manzanillo P, Kates L, Zavala-Solorio J, Eidenschenk C, Zhang J, Lesch J, Lee WP, Ross J, et al. Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature. 2014;514:237–241. doi: 10.1038/nature13564. [DOI] [PubMed] [Google Scholar]
- Weitkunat K, Schumann S, Petzke KJ, Blaut M, Loh G, Klaus S. Effects of dietary inulin on bacterial growth, short-chain fatty acid production and hepatic lipid metabolism in gnotobiotic mice. J Nutr Biochem. 2015;26:929–937. doi: 10.1016/j.jnutbio.2015.03.010. [DOI] [PubMed] [Google Scholar]
- Winer DA, Luck H, Tsai S, Winer S. The Intestinal Immune System in Obesity and Insulin Resistance. Cell metabolism. 2016;23:413–426. doi: 10.1016/j.cmet.2016.01.003. [DOI] [PubMed] [Google Scholar]
- Woting A, Blaut M. The Intestinal Microbiota in Metabolic Disease. Nutrients. 2016;8:202. doi: 10.3390/nu8040202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Chassaing B, Shi Z, Uchiyama R, Zhang Z, Denning TL, Crawford SE, Pruijssers AJ, Iskarpatyoti JA, Estes MK, et al. Viral infection. Prevention and cure of rotavirus infection via TLR5/NLRC4-mediated production of IL-22 and IL-18. Science. 2014;346:861–865. doi: 10.1126/science.1256999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Qiu Y, Zhong W, Baxter S, Su M, Li Q, Xie G, Ore BM, Qiao S, Spencer MD, et al. A targeted metabolomic protocol for short-chain fatty acids and branched-chain amino acids. Metabolomics. 2013;9:818–827. doi: 10.1007/s11306-013-0500-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- Zindl CL, Lai JF, Lee YK, Maynard CL, Harbour SN, Ouyang W, Chaplin DD, Weaver CT. IL-22-producing neutrophils contribute to antimicrobial defense and restitution of colonic epithelial integrity during colitis. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:12768–12773. doi: 10.1073/pnas.1300318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.