

Phage therapy is the use of bacteriophages—viruses that infect and replicate within a bacterium—to treat pathogenic bacteria. This approach had a short history in the premolecular era of Western medicine, but it died out before the mid-20th century mainly as a result of justly critical reports from the American Medical Association and the development of chemical antibiotics (1). Now, the global antibiotic resistance crisis and a new appreciation for the importance of the human microbiota have led to a resurgence of interest in phage therapy, not only in the classic sense of treating bacterial infections (2) but also for its potential role in modulating microbiota (3). A landmark 2015 meeting (4) on phage-based therapeutics hosted by the U.S. National Institute of Allergy and Infectious Diseases (NIAID) included not only phage biologists but also participants from private companies, public and governmental research organizations, clinicians, and the federal regulatory community. Opinions on the practicality of phage applications replacing traditional antibiotic regimens ranged from full-speed ahead, mostly from the biotechnology industry, to overt skepticism on the part of some physicians. In any case, participants left the meeting convinced that a “Phage Therapy 2.0” is on its way.
“…‘Phage Therapy 2.0′ is on its way.”
The primary focus in this new impetus is on the tailed phages, or Caudovirales, the most numerous and diverse biological entities in the biosphere (5). These have a head, containing a linear double-stranded DNA molecule of 15 to 500 kilo–base pairs; a tail; and, in most cases, tail fibers (see the figure). The phage makes specific contacts with surface features (receptors) on its bacterial prey, using the tail itself or the tail fibers, or both. Once the DNA is injected into the cell through the tail, the phage pursues a replicative cycle that culminates in host lysis, liberating typically hundreds of progeny virions. Some Caudovirales phages, designated as temperate or lysogenic, have the additional ability to become stable components of the host genome by forming dormant prophages.
Figure. Phage therapy reboot.
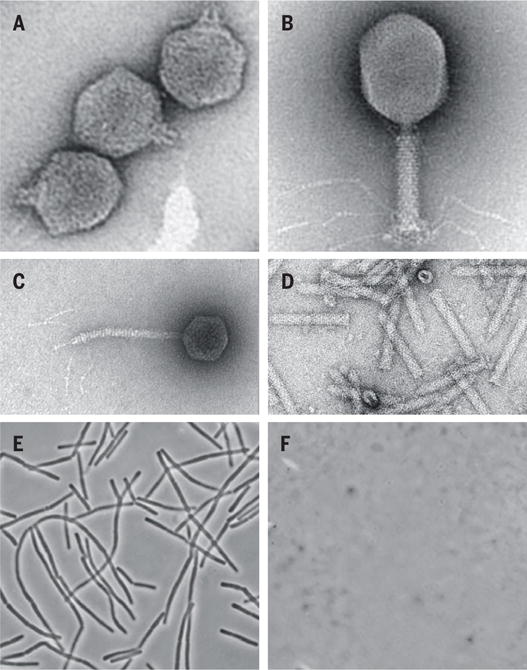
(A to C) Three paradigm Caudovirales. (A) podophage T7, stubby tail; (B) myophage T4, contractile tail; (C) siphophage lambda, flexible tail; (D) tailocin (contractile type) (14); (E and F) a field of Bacillus anthracis before and 3 s after addition of 1 μg/ml lysin.
Classic phage therapy relied on the isolation and use of naturally occurring phages found in the environment, and this trend dominates the recent literature. Phages are isolated, screened for their host ranges among pathogenic bacterial strains, and then evaluated in vitro or in vivo in animal models, sometimes formulated as mixtures of individual phages to cover the broadest host range. Besides phages themselves, two other phage-related therapeutics are emerging: lysins (6) and tailocins (7). Muralytic enzymes, or lysins, are used by phages both in the penetration of the DNA through the cell envelope and in the host lysis process that releases the progeny virions (6). These enzymes kill Gram-positive bacteria very rapidly, exhibit genus-level specificity, and, to date, do not generate resistant organisms. Tailocins, or phage tail-like bacteriocins, are essentially “headless” phages. Upon adsorption to a receptor, a tailocin particle effects lethal damage to the host cell envelope and thus exhibits single-hit lethality (7, 8). Engineered tailocins can be retargeted to heterologous hosts and have several inherent advantages, in that they can be deployed without the prospect of the environmental release of recombinant DNA and can be administered as a defined dose (8).
In the clinical and commercial sphere, recent consolidation of several phage therapy companies has brought more focused pipelines for clinical product development. As would be expected, bacterial pathogens that are already difficult to treat with conventional antibiotic therapy are receiving most of this early attention, including Pseudomonas aeruginosa, Staphylococcus aureus, and Clostridium difficile. All three of the phage-based strategies described above (phage therapy, tailocins, and lysins) are being pursued commercially, with products now in varying stages of preclinical development. A handful of phase 1 and 2 clinical trials have been conducted or are in progress. Perhaps the largest of these is the ambitious Phagoburn study, evaluating phages as a treatment for wounds (9). Several companies have already developed and marketed phage products for controlling food-borne pathogens such as Escherichia coli O157:H7 and Listeria monocytogenes (10). These developments were attractive early applications for phages, owing to the genetic homogeneity of the target bacteria and the lower regulatory barriers for registration of phages as food-processing aids.
Among the next steps that will be required for continued momentum is a prudent reassessment of the regulatory environment. The positive engagement of members of the regulatory community at the NIAID meeting was reassuring in this regard. Phages are inherently harmless to eukaryotic cells, so the health risks associated with phage therapeutics stem primarily from the debris in the source bacterial lysates and the immunological sequelae attendant to the introduction of virions into human tissues. Nevertheless, some phage-specific rules should be established (2). For example, the use of lysogenic phages should be prohibited, mainly because they usually carry genes that alter the pathogenic potential of their hosts. In addition, the identity of the host receptor should be established for any phage proposed for therapeutic use, in part because this would seem a minimal hurdle for any therapeutic but also so that combinations of phages can be assembled that are less likely to generate resistant hosts that are defective for a single receptor.
Some effort toward standardization of phage collections and consistent use of phages across studies would also help the development of the field. To date, almost all of the published preclinical phage therapy studies have used their own phages for each study. Major hospitals should establish validated phage collections for in extremis, compassionate use applications, where, in many instances, the bacterial pathogen has been identified and could be tested for sensitivity to a library of phages (2). As with many other narrow-spectrum antibacterials, the specificity of phages requires the availability of rapid methods for identifying bacterial pathogens as a necessary development in any scenario involving phage-based therapeutics.
The early abandonment of phage therapy by the 1940s was largely due to the poor understanding of what phages were or how they functioned (1). Several newer phage therapy initiatives are attempting to leverage modern synthetic biology techniques such as in vitro genome assembly or yeast-based engineering platforms, based on the idea that virulence and specificity features of effective phages can be identified and reassorted combinatorially to produce “ideal” killer phages optimized for various pathogens (11, 12). On this point, phage biology is still a “mile deep and an inch wide,” much of it derived from studies of classic phages like T4, T7, and lambda, chosen for their facile genetics in domesticated laboratory strains. Despite its depth and rigor, this record is too narrow and elderly to be a stable underpinning for the coming tsunami of phage genomics, or for the application or engineering of phages as antibacterials in systems far removed from E. coli K-12.
Aside from receptor recognition, many phages require intimate interactions with other host components, such as chaperones and transcription factors, to complete their life cycles, and these interactions must be understood in greater detail if routine engineering of phages is to become a reality. Moreover, plaque formation on agar plates or lysis of bacteria in shaker flasks is not likely to be a good predictor for efficacy in a therapeutic scenario. How phages are transported into the niches inhabited by their bacterial targets in vivo, and how they interact with bacteria in their normal physiological state in the body, need serious attention. In addition, there are new reports that phages have evolved features to exploit the surface architecture of human tissues for enhancing bacterial predation (13). This discovery points to enhanced prospects for the application of phage engineering but also is indicative of how many surprises may be in store for us now that phage biology is moving beyond its historic role as a model system. Unlike chemical antibiotics, phages are biological entities of incredible diversity and adaptability. That’s the good news and the bad news, especially since the majority of phage genes are of unknown function. In sum, caution, diligence, rigor, and patience are necessary if phage biology is to realize its full potential in support of human health. ■
References
- 1.Summers WC. Annu Rev Microbiol. 2001;55:437. doi: 10.1146/annurev.micro.55.1.437. [DOI] [PubMed] [Google Scholar]
- 2.Gill JJ, Young R. In: Emerging Trends in Antibacterial Discovery: Answering the Call to Arms. Miller AA, Miller PF, editors. Horizon Scientific; Norwich, UK: 2011. pp. 367–410. [Google Scholar]
- 3.Reyes A, Wu M, McNulty NP, Rohwer FL, Gordon JI. Proc Natl Acad Sci USA. 2013;110:20236. doi: 10.1073/pnas.1319470110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.NIAID Conference: Bacteriophage Therapy: An Alternative Strategy to Combat Drug Resistance. https://respond.niaid.nih.gov/conferences/bacteriophage/
- 5.Brussow H, Hendrix RW. Cell. 2002;108:13. doi: 10.1016/s0092-8674(01)00637-7. [DOI] [PubMed] [Google Scholar]
- 6.Fischetti VA. Curr Opin Microbiol. 2008;11:393. doi: 10.1016/j.mib.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghequire MG, De Mot R. Trends Microbiol. 2015;23:587. doi: 10.1016/j.tim.2015.07.011. [DOI] [PubMed] [Google Scholar]
- 8.Williams SR, Gebhart D, Martin DW, Scholl D. Appl Environ Microbiol. 2008;74:3868. doi: 10.1128/AEM.00141-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.www.phagoburn.eu/about-phagoburn.html
- 10.Nannapaneni R, Soni KA. In: Biofilms in the Food Environment. Pometto AL III, Demirci A, editors. Wiley Blackwell; Chichester, UK: 2015. pp. 131–144. [Google Scholar]
- 11.Nobrega FL, Costa AR, Kluskens LD, Azeredo J. Trends Microbiol. 2015;23:185. doi: 10.1016/j.tim.2015.01.006. [DOI] [PubMed] [Google Scholar]
- 12.Ando H, et al. Cell Systems. 2015;1:187. doi: 10.1016/j.cels.2015.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barr JJ, et al. Proc Natl Acad Sci USA. 2015;112:13675. doi: 10.1073/pnas.1508355112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ge P, et al. Nat Struct Mol Biol. 2015;22:377. doi: 10.1038/nsmb.2995. [DOI] [PMC free article] [PubMed] [Google Scholar]