

 We report the first synthesis of taxadiene-4(5)-epoxide, which rearranges upon acid treatment to produce products of relevance to taxol biosynthesis.
We report the first synthesis of taxadiene-4(5)-epoxide, which rearranges upon acid treatment to produce products of relevance to taxol biosynthesis.
Abstract
We have shown for the first time that taxadiene (3) can be epoxidised in a regio- and diastereoselective manner to provide taxadiene-4(5)-epoxide (12) as a single diastereoisomer, and that this epoxide can be rearranged to give taxa-4(20),11(12)-dien-5α-ol (4). Furthermore, the epoxide 12 rearranges under acidic conditions to give taxa-4(20),11(12)-dien-5α-ol (4), the known bridged ether OCT (5) and the new oxacyclotaxane (OCT2) 15. Contrary to previous speculation, taxadiene-4(5)-epoxide (12) is susceptible to rearrangement when exposed to an ironIII porphyrin, and these observations justify consideration of epoxide 12 as a chemically competent intermediate on the taxol biosynthetic pathway.
Introduction
Since its isolation from the pacific yew (Taxus brevifolia), and subsequent FDA approval in 1992, taxol and its close derivatives continue to be used as frontline drugs for the treatment of cancer.1 Its effectiveness in the clinic, coupled with an intriguing tricyclic structure, has ensured that taxol has endured as a molecule of interest to scientists for nearly 50 years.2 In this paper we show that a combination of metabolic engineering and synthetic chemistry can be used to give ready access to low oxidation state taxanes, giving new insight into the early stages of the ‘oxidase-phase’ of the taxol biosynthetic pathway.3
The first committed step in the taxol biosynthetic pathway (Scheme 1) is the taxadiene synthase-catalysed cyclisation of geranylgeranyl pyrophosphate 1 to produce taxa-4(5),11(12)-diene (3).4 The remaining biosynthetic steps involve a series of oxidation, and functional group interconversion processes, the first of which is the taxadiene-5α-hydroxylase-mediated oxidation of 3 into taxa-4(20),11(12)-dien-5α-ol (4).5
Scheme 1. Biosynthesis of Taxol® from geranylgeranyl-pyrophosphate, via taxadiene.

A number of research groups have reported the over-production of taxa-4(5),11(12)-diene (3) in a variety of chassis organisms (yeast,6 tobacco,7E. coli,8 tomato9), and the incorporation of both taxadiene synthase and its 5α-hydroxylase (tobacco,7E. coli8a) has also been described. In 2008 Rontein showed that overexpression of both taxadiene synthase and taxa-4(5),11(12)-diene 5-hydroxylase (CYP725A4) in tobacco (Nicotiana sylvestris) did not produce taxa-4(20),11(12)-dien-5α-ol (4) as expected, but instead led to the production of 5(12)-oxa-3(11)-cyclotaxane (OCT) 5 (Scheme 2).7
Scheme 2. Production of oxidised taxanes in metabolically engineered tobacco and E. coli containing both taxadiene synthase and taxadiene hydroxylase.

In 2010 Stephanopoulos reported a significant improvement in this area using E. coli as the chassis organism.8a Under their optimised conditions, taxa-4(20),11(12)-dien-5α-ol (4) could be produced, but unfortunately the desired product 4 was obtained as a 1 : 1 mixture with OCT (5), thus severely limiting the amount of 4 being produced. These two studies clearly demonstrate that the presence of both taxadiene synthase and taxadiene-5α-hydroxylase in a metabolically engineered chassis organism does not guarantee satisfactory production of taxadien-5-ol 4, and the catalytic promiscuity and multispecificity of taxadiene-5α-hydroxylase has attracted recent attention.10
Our current understanding of the taxadiene-5α-hydroxylase oxidation mechanism is derived from experiments performed by Williams and Croteau (Scheme 3).5 The observation that taxadiene-containing microsomes could convert both the 4(5)-3 (Scheme 1) and the 4(20)-6 alkene isomers of taxadiene to taxadien-5-ol 4 with equal efficiency (Scheme 3, eqn (1)), lead Williams and Croteau to suggest an H-atom abstraction/oxygen rebound mechanism, via the allylic radical 10, as being the most likely (path A, Scheme 4).
Scheme 3. Elucidating the taxadiene hydroxylase mechanism (Williams and Croteau).

Scheme 4. Taxadiene hydroxylase mediated oxidation of taxa-4(5),11(12)-diene (3) to taxa-4(20),11(12)-dien-5α-ol (4).

An alternative pathway involving epoxidation of 3 to give 12, followed by rearrangement to give 4 (path B, Scheme 4) was also considered, but was eventually discounted by the fact that the 4(20)-alkene isomer 6 is also converted to 4 by taxadiene hydroxylase (via a process unlikely to involve 12).5 This conclusion was further supported by the fact that the epoxide 12 has not been observed as an oxidation product of 3 in any studies reported thus far. In order to provide further evidence for the H-atom abstraction/oxygen rebound mechanism (path A, Scheme 4), Williams et al. prepared deuterium-labelled [C20–2H3]-taxadiene (7) and subjected this to taxadiene hydroxylase. However, under these conditions, the expected kinetic isotope effect was not observed for the transformation of 7 to 8 (Scheme 3, eqn (2)),5 which is at odds with the proposed H-atom abstraction process. Furthermore, Williams et al. report that their experiment ‘unexpectedly revealed that the deuterated substrate yielded slightly more taxa-4(20),11(12)-dien-5α-ol than did the unlabeled substrate’,5b thus indicating a small inverse isotope effect. This experimental observation actually supports the epoxide/rearrangement route for the conversion of 3 to 4, as small inverse secondary isotope effects are observed in epoxidation reactions,11 but no further experiments have been reported to examine this possibility.
The production of OCT 5, along with additional oxidation products, in engineered taxadiene synthase/taxadiene hydroxylase-containing organisms7,8a lead us to question whether epoxide 12 could be an intermediate in the taxadiene hydroxylase mechanism as we could envisage viable pathways for the production of both 4 and 5 from epoxide 12. Therefore, we decided to synthesise 12 and study it's chemistry in the context of the early stages of the taxol biosynthetic pathway.
Results and discussion
Epoxidation of taxadiene
Our studies began by isolating taxadiene from our previously described taxadiene synthase-containing tomatoes,9 using a slightly modified protocol that allows extraction directly from fresh fruit (see ESI† for details). This procedure afforded taxa-4(5),11(12)-diene (3) and taxa-4(20),11(12)-diene (6) as an inseparable 17 : 1 (3 : 6) mixture. With ready access to taxadiene we next turned our attention to epoxidation of 3, with DMDO being selected as the oxidant due to its ease of use.12 As we were concerned with the potential over-epoxidation of taxadiene, we performed the reactions with substoichiometric quantities of oxidant. Pleasingly, when taxa-4(5),11(12)-diene (3) was treated with 0.7 equivalents of DMDO, the desired epoxide 12 was obtained as the major new product (95% purity as judged by 1H NMR; see ESI†) and unreacted taxadiene was recovered (Scheme 5).
Scheme 5. DMDO epoxidation of taxa-4(5),11(12)-diene.

Whilst the epoxide derived from 6 was not observed, the recovered taxadiene (20%) was significantly enriched in 6 (1 : 2; 3 : 6) compared to the starting material (17 : 1; 3 : 6), thus indicating that 3 is much more reactive towards epoxidation than 6. Care had to be taken during chromatography on silica gel as the epoxide 12 was acid sensitive (vide infra). Treatment of taxadiene (3) with excess DMDO (2 equivalents), produced the bis-epoxide 13 in 75% yield, and this epoxide was found to be much more stable than 12 to chromatography on silica gel (Scheme 5).
Synthesis of taxa-4(20),11(12)-dien-5α-ol (4)
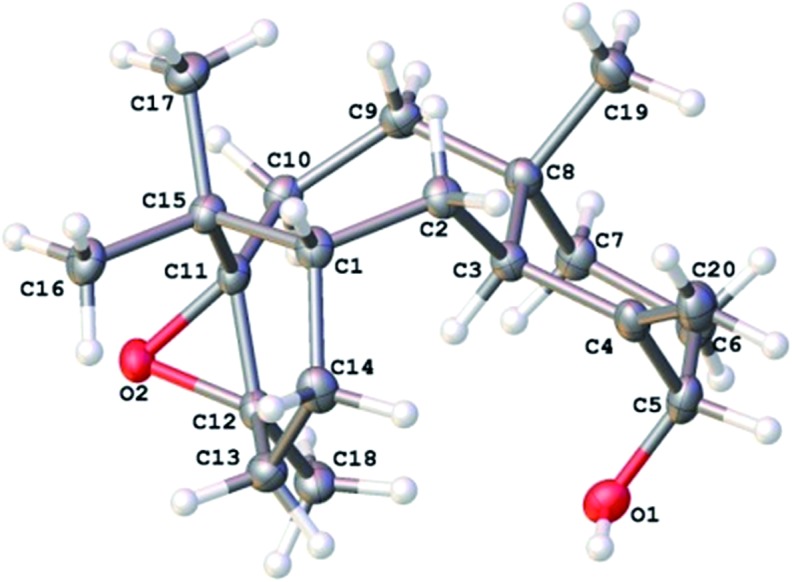
With a reliable route to the key epoxide 12 secured, we next wanted to assess its ability to act as a precursor to taxa-4(20),11(12)-dien-5α-ol (4). Before examining conditions of relevance to the biosynthesis, we first reacted 12 with Yamamoto's aluminium amide reagent (TMPAlEt2) to produce 4 as a reference sample (Scheme 6).13 As the epoxide 12 was prone to decomposition during column chromatography (vide infra), we used the epoxide in crude form directly from the DMDO oxidation. Thus, treatment of unpurified 12 with freshly prepared Yamamoto's reagent (BuLi, TMP, ClAlEt2, 0 °C, PhMe) gave taxa-4(20),11(12)-dien-5α-ol (4) in 60% isolated yield over the two steps from taxadiene (3). The spectroscopic data for 4 matched that reported by Williams for the 5α-stereoisomer,5 and this enabled us to confirm that epoxidation (3 → 12) must have occurred on the α-face of taxadiene. Having prepared bis-epoxide 13, we next examined its behaviour under the same rearrangement conditions. Thus, treatment of 13 with Yamamoto's reagent provided epoxy-alcohol 14 as the major isolable product (50%). It is interesting to note that the 11(12)-epoxide moiety is also observed in natural taxanes such as taxinine A 11(12)-epoxide.14 Fortunately, 14 was obtained as a crystalline solid and we were able to determine an X-ray crystal structure (Fig. 1) to confirm the stereochemistry of the 11(12)-epoxide, and also show that the C5-hydroxyl was on the α-face.
Scheme 6. Synthesis of taxa-4(20),11(12)-dien-5α-ol (4) via rearrangement of epoxide 12.

Fig. 1. X-ray crystal structure of the taxadiene derived epoxyalcohol 14.† .
Rearrangements of taxadine-4(5)-epoxide 12
Encouraged by the successful conversion of epoxide 12 to taxa-4(20),11(12)-dien-5α-ol (4) using Yamamoto's reagent, we next explored the behaviour of 12 under conditions of more relevance to the biosynthesis. We speculated that if taxadiene hydroxylase acts as a monooxygenase and epoxidises taxadiene 3 to produce 12, then this would initially leave a mild Lewis acidic iron centre in close proximity to the epoxide, which could catalyse subsequent rearrangement reactions. Therefore, we decided to examine the behaviour of 12 under a range of acidic conditions.
In order to simulate the acid-mediated decomposition encountered during silica gel chromatography, the epoxide 12 was treated with silica gel in C6D6 at 70 °C. Reaction progress was monitored by 1H NMR (see ESI†), and we determined that 12 converts into OCT (5), the molecule that had previously been produced in metabolically engineered tobacco by Rontein (Scheme 7),7 and the new isomeric oxacyclotaxane 15 (OCT2). Complete conversion of epoxide 12 was observed, as judged by the loss of the C19 methyl 1H NMR signal at 0.58 ppm, and the isomeric bridged ethers 5 and 15 were produced in an approximately 3 : 2 ratio (1H NMR). Chromatographic separation gave isolated samples of 5 (19%) and 15 (19%), which were then fully characterised.
Scheme 7. Rearrangement of taxadiene-4(5)-epoxide (12) under acidic conditions.

Treatment of epoxide 12 with a stronger acid (pTSA, C6D6) gave taxa-4(20),11(12)-dien-5α-ol (4) as the major new product, with OCT (5) and OCT2 (15) being produced as minor products (isolated yields: 4 (17%); 5 (7%); 15 (7%)). The formation of 4(20),11(12)-dien-5α-ol (4) from the epoxide 12 under these strongly acidic conditions is readily explained by invoking protonation of the epoxide 12 to produce 16 (Scheme 8). Ring-opening then affords the cation 17, and loss of a proton from the C20 methyl group installs the exo-methylene group in 4 (Scheme 8). The formation of OCT (5) also implicates the cation 17 as an intermediate. A 1,2-hydride shift first produces the new tertiary cation 18, which next undergoes transannulation with the C11(12)-alkene leading to the cation 19. Etherification, involving trapping the cation 19 with the secondary hydroxyl, then gives OCT (5). Similarly, the formation of OCT2 (15) can be rationalised by invoking a 1,2-alkyl shift of the tertiary cation 19, leading to the new tertiary cation 20, which is then trapped as the ether 15 by reacting with the C5-hydroxyl (Scheme 8).
Scheme 8. Proposed mechanisms for the formation of taxa-4(20),11(12)-dien-5α-ol (4), OCT (5) and OCT2 (15) from taxadiene-4(5)-epoxide (12).

As the biological oxidant (taxadiene hydroxylase5) acting upon taxadiene is a cytochrome P450, it is tempting to speculate that the reduced ironIII porphyrin (11, Scheme 4) is capable of facilitating a Lewis acid-catalysed rearrangement of the epoxide in vivo. Rontein, however, discounted this proposal17 on the basis that previous work on very different chemical systems has shown that ironIII porphyrins are poor catalysts for the rearrangement of epoxides.15 As we had access to the epoxide 12, we could test this hypothesis experimentally, and we decided to treat 12 with an ironIII porphyrin.
Contrary to the literature hypothesis, we were pleased to find that treatment of 12 with FeIII(TPP)Cl (2 equiv.) in C6D6 at 25 °C for 72 hours, lead to epoxide rearrangement, with the production of OCT (5) and OCT2 (15) as the main new products in a 1 : 1 ratio (1H NMR). Formation of taxa-4(20),11(12)-dien-5α-ol (4) was not observed under these Lewis acidic conditions (Scheme 9). As a control experiment, we exposed the similarly-substituted cyclogeraniol-derived epoxide 2216 to the same Fe(TPP)Cl rearrangement conditions,17 and as expected from previous reports,15 no rearrangement was observed, thus highlighting the propensity of 12 to rearrange.
Scheme 9. IronIII porphyrin mediated rearrangement of taxadiene-4(5)-epoxide (12).

Having shown that the two step epoxidation/FeIII induced rearrangement mimics that seen in vivo (tobacco) mediated by taxa-4(5),11(12)-diene 5-hydroxylase (CYP725A4), we wondered if the initial oxidation of taxadiene could also be achieved using the FeIII(TPP)Cl catalyst and a suitable stoichiometric oxidant (Scheme 10). Thus, treatment of taxadiene (3) with FeIII(TPP)Cl (10 mol%) and hydrogen peroxide (1 equiv.)18 lead to complete consumption of starting material (as judged by t.l.c. and 1H NMR), and the subsequent production of oxidation products. Although the isolated yields were low, 1H NMR of the crude reaction mixture showed that the two major products were OCT (5) and the OCT2 (15). The production of taxa-4(20),11(12)-dien-5α-ol (4) was not observed under these conditions (Scheme 10).
Scheme 10. IronIII porphyrin mediated oxidation of taxa-4(5),11(12)-diene (3).

Implications for the taxol biosynthetic pathway
As discussed in the introduction (Scheme 2), the current proposal for the biosynthesis of 4 from taxadiene 3 is that taxadiene hydroxylase performs an H-atom abstraction from the C20 methyl group of the 4(5)-alkene isomer of 3 to form the allyl radical 10, and involvement of the epoxide 12 was rejected. Further support for the involvement of a common allyl radical 10 came from the fact that the 4(20)-alkene isomer 6 was also converted to taxa-4(20),11(12)-dien-5α-ol (4) by taxadiene hydroxylase. However, our experiments, coupled with the previously published kinetic isotope effect data,5 demonstrate that the epoxide 12 cannot be discounted as an intermediate on the taxol biosynthetic pathway. We have shown that the major, naturally occurring, 4(5)-alkene isomer of taxadiene 3 can be converted to taxa-4(20),11(12)-dien-5α-ol (4) via the epoxide 12, and this suggests that the 4(5)-3 and 4(20)-6 alkene isomers of taxadiene are processed differently by taxadiene hydroxylase (Scheme 11).19
Scheme 11. Proposal for the role of epoxide 12 in the biosynthesis of taxa-4(20),11(12)-dien-5α-ol (4).

It is possible that 4(5)-alkene isomer 3 is epoxidised to produce 12, which is then rearranged to 4, by the action of the reduced form of the hydroxylase 11. In contrast, the 4(20)-alkene isomer 6 could be converted directly to 4via the accepted H-atom abstraction mechanism. The involvement of epoxide 12 in the pathway provides an explanation for the lack of a significant primary kinetic isotope effect and the presence of an inverse secondary isotope effect when deuterium labelled [C20–2H3]-taxadiene (7) was oxidized by taxadiene hydroxylase. The labelled C20 methyl likely plays only a small role in the epoxidation process (i.e. leads to small inverse isotope effect), and loss of a proton from C20 in an intermediate such as 19 (Scheme 8) is unlikely to be rate-limiting.
Conclusions
In this study, we have shown that taxa-4(5),11(12)-diene (3) can be isolated from the fruit of metabolically engineered tomatoes using our new optimised procedure. Furthermore, we have shown that taxadiene (3) can be epoxidised in a regio- and diastereoselective manner to provide taxadiene-4(5)-epoxide (12), and that this epoxide can be rearranged to give taxa-4(20),11(12)-dien-5α-ol (4) in 60% over the two chemical steps. We have shown that the epoxide 12 is sensitive to acids, and that both taxa-4(20),11(12)-dien-5α-ol (4), the known bridged ether OCT (5) and the new oxacyclotaxane (OCT2) 15 can be obtained from this material. We have shown that contrary to previous speculation, taxadiene-4(5)-epoxide (12) is susceptible to rearrangement when exposed to an ironIII porphyrin, and these observations combine to warrant reconsideration of the epoxide 12 as a chemically competent intermediate on the taxol biosynthetic pathway.
Supplementary Material
Acknowledgments
We thank the EPSRC for providing DTG studentships for NAB and BJM, and the University of Nottingham for additional financial support of this work.
Footnotes
†Electronic supplementary information (ESI) available: Full experimental procedures and copies of 1H and 13C NMR spectra. CCDC 1030909. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc03463a
References
- (a) Wani M. C., Taylor H. L., Wall M. E., Coggon P., McPhail A. T. J. Am. Chem. Soc. 1971;93:2325. doi: 10.1021/ja00738a045. [DOI] [PubMed] [Google Scholar]; (b) Cragg G. M. Med. Res. Rev. 1998;18:315. doi: 10.1002/(sici)1098-1128(199809)18:5<315::aid-med3>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]; (c) Kingston D. G. I. Chem. Commun. 2001:867. [Google Scholar]
- (a) Holton R. A., Somoza C., Kim H. B., Liang F., Biediger R. J., Boatman P. D., Shindo M., Smith C. C., Kim S. J. Am. Chem. Soc. 1994;116:1597. [Google Scholar]; (b) Holton R. A., Kim H. B., Somoza C., Liang F., Biediger R. J., Boatman P. D., Shindo M., Smith C. C., Kim S. J. Am. Chem. Soc. 1994;116:1599. [Google Scholar]; (c) Nicolaou K. C., Yang Z., Liu J. J., Ueno H., Nantermet P. G., Guy R. K., Claiborne C. F., Renaud J., Couladouros E. A., Paulvannan K., Sorensen E. J. Nature. 1994;367:630. doi: 10.1038/367630a0. [DOI] [PubMed] [Google Scholar]; (d) Danishefsky S. J., Masters J. J., Young W. B., Link J. T., Snyder L. B., Magee T. V., Jung D. K., Isaacs R. C. A., Bornmann W. G., Alaimo C. A., Coburn C. A., Di Grandi M. J. J. Am. Chem. Soc. 1996;118:2843. [Google Scholar]; (e) Wender P. A., Badham N. F., Conway S. P., Floreancig P. E., Glass T. E., Houze J. B., Krauss N. E., Lee D., Marquess D. G., McGrane P. L., Meng W., Natchus M. G., Shuker A. J., Sutton J. C., Taylor R. E. J. Am. Chem. Soc. 1997;119:2757. [Google Scholar]; (f) Morihira K., Hara R., Kawahara S., Nishimori T., Nakamura N., Kusama H., Kuwajima I. J. Am. Chem. Soc. 1998;120:12980. [Google Scholar]; (g) Mukaiyama T., Shiina I., Iwadare H., Saitoh M., Nishimura T., Ohkawa N., Sakoh H., Nishimura K., Tani Y., Hasegawa M., Yamada K., Saitoh K. Chem.–Eur. J. 1999;5:121. [Google Scholar]; (h) Doi T., Fuse S., Miyamoto S., Nakai K., Sasuga D., Takahashi T. Chem.–Asian J. 2006;1:370. doi: 10.1002/asia.200600156. [DOI] [PubMed] [Google Scholar]
- (a) Holton R. A., Juo R. R., Kim H. B., Williams A. D., Harusawa S., Lowenthal R. E., Yogai S. J. Am. Chem. Soc. 1988;110:6558. [Google Scholar]; (b) Rubenstein S. M., Williams R. M. J. Org. Chem. 1995;60:7215. [Google Scholar]; (c) Huang Q., Pennington J. D., Williams H. J., Scott A. I. Synth. Commun. 2006;36:2577. [Google Scholar]; (d) Mendoza A., Ishihara Y., Baran P. S. Nat. Chem. 2012;4:21. doi: 10.1038/nchem.1196. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ishihara Y., Mendoza A., Baran P. S. Tetrahedron. 2013;69:5685. doi: 10.1016/j.tet.2013.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Wilde N. C., Isomura M., Mendoza A., Baran P. S. J. Am. Chem. Soc. 2014;136:4909. doi: 10.1021/ja501782r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Koepp A. E., Hezari M., Zajicek J., Vogel B. S., LaFever R. E., Lewis N. G., Croteau R. J. Biol. Chem. 1995;270:8686. doi: 10.1074/jbc.270.15.8686. [DOI] [PubMed] [Google Scholar]; (b) Köksal M., Jin Y., Coates R. M., Croteau R., Christianson D. W. Nature. 2011;469:116. doi: 10.1038/nature09628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Jennewein S., Long R. M., Williams R. M., Croteau R. Chem. Biol. 2004;11:379. doi: 10.1016/j.chembiol.2004.02.022. [DOI] [PubMed] [Google Scholar]; (b) Hefner J., Rubenstein S. M., Ketchum R. E. B., Gibson D. M., Williams R. M., Croteau R. Chem. Biol. 1996;3:479. doi: 10.1016/s1074-5521(96)90096-4. [DOI] [PubMed] [Google Scholar]
- (a) DeJong J., Liu Y., Bollon A. P., Long R. M., Jennewein S., Williams D., Croteau R. B. Biotechnol. Bioeng. 2006;93:212. doi: 10.1002/bit.20694. [DOI] [PubMed] [Google Scholar]; (b) Engels B., Dahm P., Jennewein S. Metab. Eng. 2008;10:201. doi: 10.1016/j.ymben.2008.03.001. [DOI] [PubMed] [Google Scholar]
- Rontein D., Onillon S., Herbette G., Lesot A., Werck-Reichhart D., Sallaud C., Tissier A. J. Biol. Chem. 2008;283:6067. doi: 10.1074/jbc.M708950200. [DOI] [PubMed] [Google Scholar]
- (a) Ajikumar P. K., Xiao W.-H., Tyo K. E. J., Wang Y., Simeon F., Leonard E., Mucha O., Phon T. H., Pfeifer B., Stephanopoulos G. Science. 2010;330:70. doi: 10.1126/science.1191652. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Huang Q., Roessner C. A., Croteau R., Scott A. I. Bioorg. Med. Chem. 2001;9:2237. doi: 10.1016/s0968-0896(01)00072-4. [DOI] [PubMed] [Google Scholar]; (c) Huang K., Huang Q., Wildung M. R., Croteau R., Scott A. I. Protein Expression Purif. 1998;13:90. doi: 10.1006/prep.1998.0870. [DOI] [PubMed] [Google Scholar]
- Kovacs K., Zhang L., Linforth R. S. T., Whittaker B., Hayes C. J., Fray R. G. Transgenic Res. 2007;16:121. doi: 10.1007/s11248-006-9039-x. [DOI] [PubMed] [Google Scholar]
- Yadav V. G. J. Mol. Catal. B: Enzym. 2014;110:154. [Google Scholar]
- (a) Angelis Y. S., Orfanopoulos M. J. Org. Chem. 1997;62:6083. [Google Scholar]; (b) Hanzlik R. P., Shearer G. O. Biochem. Pharmacol. 1978;27:1441. doi: 10.1016/0006-2952(78)90099-0. [DOI] [PubMed] [Google Scholar]
- Adam W., Bialas J., Hadjiarapoglou L. Chem. Ber. 1991;124:2377. [Google Scholar]
- Yasuda A., Yamamoto H., Nozaki H. Bull. Chem. Soc. Jpn. 1979;52:1705. [Google Scholar]
- (a) Murakami R., Shi Q., Oritani T. Phytochemistry. 1999;52:1577. [Google Scholar]; (b) Wang Y.-F., Shi Q.-W., Dong M., Kiyota H., Gu Y.-C., Cong B. Chem. Rev. 2011;111:7652. doi: 10.1021/cr100147u. [DOI] [PubMed] [Google Scholar]
- Liebler D. C., Guengerich F. P. Biochemistry. 1983;22:5482. doi: 10.1021/bi00293a005. [DOI] [PubMed] [Google Scholar]
- For the synthesis of epoxide 22 see: Uroos M., Hayes C. J., Org. Lett., 2010, 12 , 5294 . [DOI] [PubMed] [Google Scholar]
- For Lewis acid-mediated rearrangements of cyclohexene oxides see:; (a) Braude E. A., Webb A. A., Sultanbawa M. U. S. J. Chem. Soc. 1958:3328. [Google Scholar]; (b) Parker R. E., Isaacs N. S. Chem. Rev. 1959;59:737. [Google Scholar]; (c) Maruoka K., Ooi T., Yamamoto H. J. Am. Chem. Soc. 1989;111:6431. [Google Scholar]
- (a) Groves J. T., Nemo T. E., Myers R. S. J. Am. Chem. Soc. 1979;101:1032. [Google Scholar]; (b) Traylor T. G., Tsuchiya S., Byun Y.-S., Kim C. J. Am. Chem. Soc. 1993;115:2775. [Google Scholar]; (c) Barbosa Sousa D. P., Fricks A. T., Alvarez H. M., Salomao G. C., Neves Olsen M. H., Cardozo Filho L., Fernandes C., Antunes O. A. C. Catal. Commun. 2007;8:1041. [Google Scholar]
- Whilst this manuscript was under review, a complementary study by Stephanopoulos et al. has been reported that also proposes taxadiene epoxidation by taxadiene-5α-hydroxylase as being a step on the taxol biosynthetic pathway. Please see: Edgar S., Zhou K., Qiao K., King J. R., Simpson J. H., Stephanopoulos G., ACS Chem. Biol., 2016. 10.1021/acschembio.5b00767 . [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.