

Abstract
Autosomal dominant polycystic kidney disease (ADPKD) is a debilitating disease that is characterized by the accumulation of numerous fluid-filled cysts in the kidney. ADPKD is primarily caused by mutations in two genes, PKD1 and PKD2. Long noncoding RNAs (lncRNA), defined by a length >200 nucleotides and absence of a long ORF, have recently emerged as epigenetic regulators of development and disease; however, their involvement in PKD has not been explored previously. Here, we performed deep RNA-Seq to identify lncRNAs that are dysregulated in two orthologous mouse models of ADPKD (kidney-specific Pkd1 and Pkd2 mutant mice). We identified a kidney-specific, evolutionarily conserved lncRNA called Hoxb3os that was down-regulated in cystic kidneys from Pkd1 and Pkd2 mutant mice. The human ortholog HOXB3-AS1 was down-regulated in cystic kidneys from ADPKD patients. Hoxb3os was highly expressed in renal tubules in adult WT mice, whereas its expression was lost in the cyst epithelium of mutant mice. To investigate the function of Hoxb3os, we utilized CRISPR/Cas9 to knock out its expression in mIMCD3 cells. Deletion of Hoxb3os resulted in increased phosphorylation of mTOR and its downstream targets, including p70 S6 kinase, ribosomal protein S6, and the translation repressor 4E-BP1. Consistent with activation of mTORC1 signaling, Hoxb3os mutant cells displayed increased mitochondrial respiration. The Hoxb3os mutant phenotype was partially rescued upon re-expression of Hoxb3os in knockout cells. These findings identify Hoxb3os as a novel lncRNA that is down-regulated in ADPKD and regulates mTOR signaling and mitochondrial respiration.
Keywords: kidney; long noncoding RNA (long ncRNA, lncRNA); mTOR complex (mTORC); CRISPR/Cas; epithelial cell
Introduction
Autosomal dominant polycystic kidney disease (ADPKD)2 is one of the most common monogenic diseases, affecting 1 in 800–1000 people worldwide, and is the fourth-leading cause of end-stage renal failure (1, 2). ADPKD is caused primarily by mutations in PKD1 or PKD2 (3). The typical renal phenotype of ADPKD is the insidious development of hundreds of renal cysts. Cysts begin forming during early stages of development and grow progressively throughout the life of the individual (4, 5). About half of those affected with ADPKD will progress to end-stage renal failure necessitating dialysis or kidney transplantation (6).
The functions of the PKD1 and PKD2 gene products are incompletely understood. PKD1 encodes polycystin-1, a large glycoprotein with an N-terminal extracellular region containing protein-protein interaction motifs, a transmembrane domain, and a C-terminal cytoplasmic tail (7, 8). PKD2 encodes polycystin-2, a Ca2+-permeable cation channel that interacts with, and is regulated by, polycystin-1 (9–11). A common consequence of malfunction of the polycystin-1/2 complex is reduced intracellular Ca2+ levels and increased levels of cAMP (12, 13). PKD1 and PKD2 regulate multiple signaling pathways that maintain normal tubular structure and function (14). For example, some mutant mice display defects in growth factor/receptor tyrosine kinase pathways (15), whereas others have abnormalities in Wnt/β-catenin signaling (16), and in the mTOR pathway (17). It is still unclear which pathways are primarily involved in pathogenesis and which ones are affected secondarily.
Long noncoding RNAs (lncRNAs) are a class of nonprotein-coding RNAs that are >200 nucleotides in length and are characterized by the absence of a long ORF (18). LncRNAs function as activators, guides, or scaffolds for transcription factors and histone modifiers. They also play important roles in posttranscriptional regulation by affecting mRNA activity and stability, as well as translational efficiency (19). LncRNAs have been implicated in a range of diseases, including cancer (20). However, their involvement in ADPKD has not been reported previously. LncRNAs are attractive therapeutic targets because they can regulate multiple genes and pathways that are affected in a disease, and their activity can be modulated in vivo (21–23). Recent studies reveal that lncRNAs are dysregulated in human diseases. For example, lnc-PCA3 promotes aggressive prostate cancer and is up-regulated 60-fold in prostate tumors compared with normal tissue (24). Another lncRNA, MALAT1, is overexpressed in lung cancer (25), bladder cancer (26), liver cancer (27), and colorectal cancer (28). MALAT1 is regarded as a decisive regulator of metastasis in lung cancer cells (29). Recent evidence suggests that lncRNAs play a significant role in kidney diseases. For example, Tug1 is an lncRNA that improves diabetic nephropathy in the mouse kidney by regulating mitochondrial bioenergetics (30). These findings make lncRNAs attractive targets for efforts aimed at improving ADPKD treatment.
In this study, we have identified a kidney-specific, evolutionarily conserved lncRNA called Hoxb3os, which is down-regulated in two orthologous mouse models of ADPKD (kidney-specific Pkd1 and Pkd2 mutant mice). Knockout of Hoxb3os with CRISPR/Cas9 in mIMCD3 kidney epithelial cells results in hyperactivation of mTOR signaling, and increased rates of oxygen consumption. Conversely, re-expression of Hoxb3os in knockout cells partially rescues the mutant phenotype. These findings identify Hoxb3os as an lncRNA that is down-regulated in ADPKD and negatively regulates mTOR signaling.
Results
Identification of lncRNAs that are dysregulated in Pkd1 and Pkd2 mutant kidneys
Our laboratory has previously generated two mouse models of ADPKD: Ksp/Cre;Pkd1f/f mice (Pkd1-KO), in which Pkd1 is deleted in renal tubules, and Pkhd1/Cre;Pkd2f/f mice (Pkd2-KO), in which Pkd2 is deleted in collecting ducts (31, 32). Compared with WT mice, extensive renal cyst formation was present in Pkd1-KO kidney at postnatal day 10 (P10) and in Pkd2-KO kidney at P21 (Fig. 1A). To identify lncRNAs that are dysregulated in both KO mouse models, we conducted whole-transcriptome analysis (RNA-Seq) of RNA from WT, Pkd1-KO, and Pkd2-KO kidneys. We discovered 120 lncRNAs that were dysregulated in Pkd1-KO kidneys and 135 that were dysregulated in Pkd2-KO kidneys (p < 0.05, log cpm >−1). Sixty-six lncRNAs were similarly dysregulated in both models, and 41 showed >2-fold change in expression (Fig. 1B). Next, we sought to identify novel lncRNAs that are dysregulated in ADPKD by performing de novo transcriptome assembly. We identified 358 previously unannotated transcripts from mouse adult kidney. These putative transcripts were then filtered to distinguish transcripts that might encode proteins from those that are noncoding. We used the phylogenetic codon substitution frequency software (PhyloCSF) (33) to assess the protein-coding potential of each transcript. Twenty-four transcripts had protein-coding potential and were excluded from further analysis. Of the remaining transcripts, 139 were dysregulated in Pkd1-KO kidneys and 106 were dysregulated in Pkd2-KO kidneys (p < 0.05, log cpm >−1). Sixty unique transcripts were dysregulated in both animal models, and 50 displayed more than 2-fold change in expression (Fig. 1C). The majority of the novel transcripts were down-regulated, whereas six were up-regulated. Mapping of the lncRNAs to the mouse genome revealed that most annotated lncRNAs were classified as antisense and intergenic, whereas the majority of the novel transcripts were classified as sense and intergenic (Fig. 1D).
Figure 1.
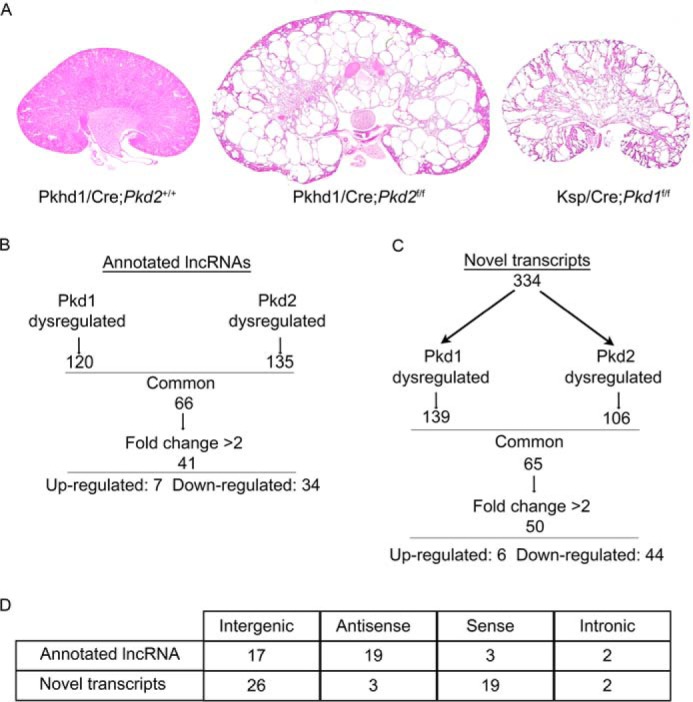
Identification of lncRNAs that are dysregulated in mouse models of ADPKD. A, kidney-specific inactivation of Pkd1 (Ksp/Cre;Pkd1f/f) at postnatal day 10 (P10) and Pkd2 (Pkhd1/Cre;Pkd2f/f) at P20 produces kidney cysts compared with control (Pkhd1/Cre;Pkd2+/+). Kidneys were stained with H&E. B and C, identification by pair-end RNA-Seq of annotated (B) and novel (C) lncRNAs that are dysregulated in both Pkd1 and Pkd2 mutant kidneys (p < 0.05, log cpm >−1). D, mapping of dysregulated lncRNAs to genomic regions relative to protein-coding genes.
Characterization of lncRNAs in the mouse kidney
We characterized the expression of the 41 known lncRNAs that were highly dysregulated in both Pkd1-KO and Pkd2-KO kidneys. We first determined the developmental pattern of expression by performing quantitative RT-PCR (qRT-PCR) on RNA from embryonic (E16.5), newborn (P1), and adult (P30) mouse kidney. Our results show that the majority of lncRNAs in the mouse kidney were developmentally regulated. Twenty-seven lncRNAs were up-regulated, 10 were down-regulated, and 4 were unchanged between E16.5 and P30 (Fig. S1). Next, we compared expression of the dysregulated lncRNAs in the kidney relative to five other organs in the mouse. The majority of lncRNAs were expressed in all six organs, whereas five were kidney-specific (Fig. S2). We then examined the subcellular localization of lncRNAs by performing RNA fractionation in WT adult mouse kidney tissue. Fractionation experiments revealed that 32 lncRNAs were predominantly nuclear and 9 were predominantly cytoplasmic (Fig. S3).
Identification of Hoxb3os as a candidate lncRNA in the pathogenesis of ADPKD
Hoxb3os was among the most dysregulated lncRNAs that were identified in our screen. Hoxb3os is an antisense transcript that is located on chromosome 11 and spans a genomic region of about 7 kb. Hoxb3os is located 5′ to Hoxb2 and partially overlaps with the 3′-end of Hoxb3 (Fig. 2A). Rapid amplification of cDNA ends (RACE) in the adult mouse kidney showed that Hoxb3os exists as two major isoforms that are transcribed from two distinct promoter regions. Hoxb3os isoform 1, which constitutes the longest transcript (557 nucleotides), comprises four exons and represents the major fraction of the total transcript as determined by qRT-PCR. Expression analysis revealed that Hoxb3os was highly abundant in the normal kidney but was almost undetectable in five other organs (Fig. 2B). In addition, expression of Hoxb3os was developmentally up-regulated between E16.5 and adult kidney (Fig. 2C). In contrast, the nearby protein-coding genes, Hoxb2 and Hoxb3, were developmentally down-regulated (Fig. S4A). RNA fractionation experiment revealed that 80% of spliced Hoxb3os transcripts were localized to the cytoplasm and 20% were nuclear (Fig. 2D).
Figure 2.

Expression of Hoxb3os in the mouse kidney. A, RACE identifies two major Hoxb3os isoforms in the mouse kidney. Upper panel shows RNA-Seq reads from a WT kidney visualized with Integrative Genomics Viewer software (blue bars indicate reads from positive strand and red bars indicate negative strand). The two Hoxb3os isoforms (lower panel) are located 5′ to Hoxb2 and partially overlap with Hoxb3 (arrowheads indicate direction of transcription and solid rectangles indicate exons). B, quantitative RT-PCR showing expression of Hoxb3os in different mouse organs relative to the kidney. C, developmental expression of Hoxb3os in the embryonic (E16.5), newborn (P1), and adult (P30) mouse kidney measured by qRT-PCR. D, percentage of Hoxb3os expression in cytoplasmic and nuclear fractions from an adult mouse kidney measured by qRT-PCR. Error bars indicate S.E.
Hoxb3os is down-regulated in cystic kidney
RNA-Seq studies revealed that Hoxb3os was down-regulated in both Pkd1-KO and Pkd2-KO kidneys. We performed qRT-PCR and confirmed down-regulation of Hoxb3os by 90% in Pkd1-KO kidneys and by 75% in Pkd2-KO kidneys (Fig. 3A). We also measured Hoxb3os expression before the onset of cyst formation in Pkd2-KO kidney at P10. Relative to WT kidney, expression of Hoxb3os was reduced by 35% in the pre-cystic kidney (Fig. 3B). Hoxb3os was also down-regulated in a Pkd2-null kidney cell line (Fig. 3C). We determined the localization of Hoxb3os in the normal and cystic kidney by in situ hybridization (ISH) utilizing a probe that recognizes both Hoxb3os isoforms. ISH revealed that Hoxb3os was expressed in epithelial tubules of the normal kidney (Fig. 3, D and E). In cystic kidneys, expression of Hoxb3os was significantly reduced throughout the kidney and was almost undetectable within the epithelial lining of the cysts (Fig. 3, F and G). In contrast to the decreased expression of Hoxb3os in Pkd1 and Pkd2 mutant kidneys, the expression of Hoxb2 was up-regulated and Hoxb3 was unchanged (Fig. S4C).
Figure 3.
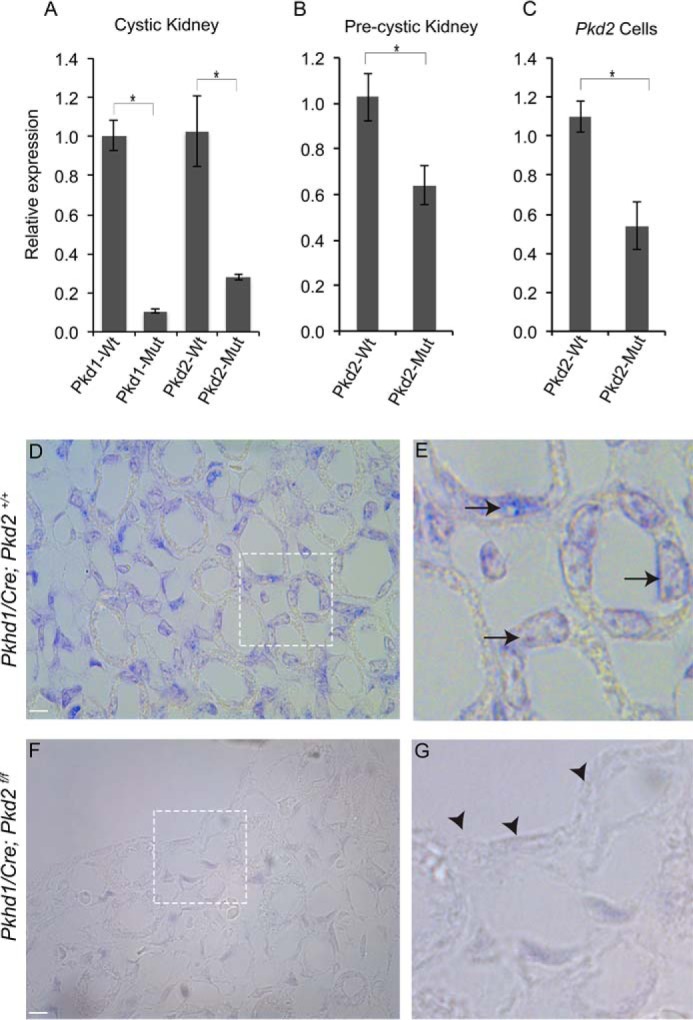
Down-regulation of Hoxb3os in cystic kidney. A–C, quantitative RT-PCR showing decreased expression of Hoxb3os in cystic kidneys from Pkd1 (P10) and Pkd2 (P20) mutant mice (A), pre-cystic Pkd2 mutant kidney at P10 (B), and Pkd2 mutant cells (C). D–G, in situ hybridization showing down-regulation of Hoxb3os in cystic kidneys (Pkhd1/Cre;Pkd2f/f) (F and G) compared with control kidneys (Pkhd1/Cre;Pkd2+/+) (D and E) at P15. Insets show higher magnification images. Arrows show expression in kidney tubules, and arrowheads indicate loss of expression within the epithelial lining of the cyst. Scale bar is 5 μm. Error bars indicate S.E. *, p < 0.05.
Because Hoxb3os is highly evolutionarily conserved, we measured the expression of its human ortholog, HOXB-AS1 (34), in human ADPKD. HOXB-AS1 comprises four transcripts that partially overlap with HOXB2 and HOXB3 (Fig. S5A). Expression analysis utilizing primer sets that detected multiple HOXB-AS1 isoforms revealed that HOXB-AS1 was down-regulated by 60% in cystic kidneys from humans with ADPKD (Fig. S5B). Similar to results in the mouse, the expression of the protein-coding gene HOXB2 was up-regulated and the expression of HOXB3 was unchanged in cystic human kidneys (Fig. S4D).
Hoxb3os regulates a network of genes and signaling pathways that are commonly altered in ADPKD
To determine the function of Hoxb3os, we inhibited its expression in mIMCD3 renal epithelial cells using GapmeRs, which are potent antisense oligonucleotides (Exiqon). Transfection with GapmeRs against Hoxb3os reduced levels of the mature transcript by 65% (Fig. 4A). Next, we performed RNA-Seq to identify genes that are differentially expressed in response to Hoxb3os knockdown. A total of 1964 genes were differentially expressed between knockdown and control cells (false discovery rate <0.05). The expression of 77 genes changed by more than 2-fold between control and knockdown cells (Fig. 4B). Importantly, expression of 40 of these genes was also altered in vivo in cystic kidneys from Pkd1-KO and Pkd2-KO mice (Fig. 4C). We then performed pathway analysis on the complete set of genes that were differentially expressed in knockdown cells (n = 1964). This analysis identified EIF2, mTOR, EIF4, and P70S6K signaling as the most dysregulated pathways in Hoxb3os mutant cells (Fig. 4D). These pathways are involved in enhanced anabolic processes such as mRNA translation, and share a common gene-signature profile. To determine whether the predicted dysregulations in pathways translated to molecular alterations, we measured the levels of mTOR and phosphorylated mTOR (p-mTOR). Western blot analysis revealed that p-mTOR was increased in cell lysates from Hoxb3os knockdown (Fig. 4E). These data suggested that Hoxb3os regulates mTOR signaling.
Figure 4.

Knockdown of Hoxb3os in kidney cells results in abnormal mTOR signaling. A, treatment of mIMCD3 cells with GapmeRs decreases Hoxb3os expression levels by more than 60% as measured by qRT-PCR. B, RNA-Seq reveals differential expression of genes between Hoxb3os knockdown and control mIMCD3 cells (n = 3). C, heat map showing most deregulated genes in response to Hoxb3os knockdown that were also altered in both Pkd1 and Pkd2 cystic kidneys (n = 40). D, pathway analysis using Ingenuity Pathway Analysis (IPA) of differentially regulated genes indicating the most affected signaling pathways in Hoxb3os knockdown cells. E, Western blot analysis demonstrates an increase in the phosphorylation of mTOR (p-mTOR) in response to knockdown of Hoxb3os. Right panel shows densitometric analysis of the Western blots. Error bars indicate S.E. *, p < 0.05.
Ablation of Hoxb3os results in hyperactivation of mTORC1 and enhanced mitochondrial respiration
To confirm that Hoxb3os regulates mTOR signaling, we used CRISPR/Cas9 gene editing to ablate Hoxb3os in mIMCD3 cells. We designed single guide RNAs (sgRNAs) to remove the endogenous promoter and first exon of isoform 1 of Hoxb3os (Fig. S6A). Because expression of Hoxb3os isoform 2 was not detected in mIMCD3 cells (data not shown), only isoform 1 was targeted. Successful deletion of the Hoxb3os locus (Hoxb3os-KO) was confirmed by PCR analysis on genomic DNA (Fig. S6B). To determine the effect of genomic deletion on the Hoxb3os transcript, we performed qRT-PCR using primer sets designed to amplify different exons. Elimination of the full-length Hoxb3os transcript that harbors the first exon was confirmed by qPCR. However, low levels of a truncated transcript that harbors downstream exons were detected, indicating that expression was not completely abolished (Fig. S6C). Quantitative RT-PCR showed that similar to Hoxb3os-KD cells, the expression of genes in the mTOR pathway was dysregulated in Hoxb3os-KO cells (Fig. S6E). Importantly, the expression of nearby protein-coding genes, Hoxb2 and Hoxb3, was unaltered in Hoxb3os-KO cells (Fig. S4B), which indicates that Hoxb3os is unlikely to regulate the transcription of Hoxb2 and Hoxb3.
mTOR exists in two distinct multiprotein complexes (mTORC1 and mTORC2) (35). Phosphorylation of p70S6 kinase (p-S6K), ribosomal S6 protein (p-RS6), and eukaryotic translation initiation factor 4E–binding protein 1 (p-4E–BP1) are known downstream targets of mTORC1 (36), whereas phosphorylation of AKT at Ser-473 is targeted by mTORC2 (37). Western blot analysis of lysates from Hoxb3os-KO and WT cells revealed that ablation of Hoxb3os increased the levels of p-mTOR, p-S6K, p-RS6, and p-4E–BP1 at Ser-65 (Fig. 5A). The levels of the nonphosphorylated forms of these proteins did not change nor did the phosphorylation of AKT at Ser-473 (data not shown). These results suggest that Hoxb3os regulates the activity of mTORC1 but not mTORC2. A key physiological readout of mTORC1 activation is enhanced cell metabolism (38). To uncover metabolic differences between WT and Hoxb3os-KO cells, we measured extracellular acidification (ECAR) and oxygen consumption (OCR) rates. Compared with WT cells, Hoxb3os-KO cells displayed increased mitochondrial respiration as evidenced by higher OCR at baseline and upon addition of FCCP, an uncoupling agent that increases proton permeability and dissociates the electron transport chain from ATP production (Fig. 5B). The enhanced cellular respiration is consistent with activation of mTORC1 signaling in Hoxb3os-KO cells. Hoxb3os-KD cells also showed increased OCR, although the change was not statistically significant, presumably because of incomplete inhibition in KD cells (data not shown).
Figure 5.

Ablation of Hoxb3os in kidney cells results in up-regulation of mTOR signaling and enhanced mitochondrial respiration. A, Western blot analysis showing the levels of mTOR and p-mTOR, RS6, and p-RS6, eukaryotic translation initiation factor 4E-BP1 and p-4E-BP1, p-S6K, and GAPDH in WT and Hoxb3os knockout (KO) cells. Right panel shows densitometric analysis of the Western blots. B, measurement of OCR in WT (blue) and Hoxb3os KO (red) cells under basal conditions and in response to the indicated mitochondrial inhibitors. Addition of the protonophore FCCP reveals the maximal respiratory capacity of the cells. The rate of respiration at baseline and in response to FCCP is quantified in the right panel. Error bars indicate S.E. *, p < 0.05.
Re-expression of Hoxb3os in knockout cells partially rescues mTOR signaling and mitochondrial respiration
Next, we tested whether restoring the expression of Hoxb3os in knockout cells rescues the abnormalities in mTOR signaling and mitochondrial respiration. We cloned full-length Hoxb3os into a lentiviral vector containing a modified backbone that is suitable for lncRNA expression (39). The resulting vector (LeGO-Hoxb3os) featured a spleen focus-forming virus promoter (SFFV) that drives the expression of Hoxb3os at levels similar to its endogenous expression. In addition, the LeGO-Hoxb3os vector included a poly(A) signal immediately downstream of the Hoxb3os insert that prevented transcription of adjacent proviral DNA and maintained the secondary structure of Hoxb3os. We established three independent cell lines with stable expression of Hoxb3os. qRT-PCR confirmed re-expression of Hoxb3os transcript in the original Hoxb3os-KO cells at a level similar to WT mIMCD3 cells (Fig. S4D). To determine whether restoring Hoxb3os expression in Hoxb3os-KO cells rescued the increase in mTOR signaling, we performed Western blot analysis on lysates from Hoxb3os-KO cells transduced with either LeGO-Hoxb3os (Hoxb3os-rescued) or LeGO-empty (nonrescued) vector. Compared with nonrescued cells, Hoxb3os-rescued cells displayed lower levels of p-mTOR, p-S6K, and p-RS6. No difference was observed in p-4E–BP1 at Ser-65 levels (Fig. 6A). Next, we measured ECAR and OCR in nonrescued and Hoxb3os-rescued cells. Compared with nonrescued cells, Hoxb3os-rescued cells displayed significantly lower OCR levels, both at baseline and after FCCP treatment (Fig. 6B). Furthermore, OCR levels in rescued cells were similar to WT cells at baseline, but remained higher after addition of FCCP (Fig. 6B). These data further confirm that Hoxb3os is a negative regulator of mTORC1 signaling and mitochondrial respiration.
Figure 6.

Re-expression of Hoxb3os in knockout cells partially rescues mTOR signaling and mitochondrial respiration. A, Western blot analysis showing lower levels of p-mTOR, p-RS6, and p-S6K in knockout cells transfected with Hoxb3os-expressing lentiviral construct (Rescue) compared with cells transfected with the empty lentiviral construct (KO). Right panel shows densitometric analysis of the Western blots. B, measurement of OCR in nonrescued (red), rescued (green), and WT (blue) cells demonstrate partial restoration of OCR to WT levels in rescued compared with nonrescued cells. The rate of respiration at baseline and in response to FCCP is quantified in the right panel. Error bars indicate S.E. *, p < 0.05. NS, not significant.
Discussion
In this study, we comprehensively identified lncRNAs that are dysregulated in mouse models of ADPKD. Utilizing two independent and orthologous mouse models is a powerful approach to discover lncRNAs that are implicated in the pathogenesis of ADPKD. A total of 120 and 135 lncRNA were dysregulated in Pkd1-KO and Pkd2-KO mice, respectively. Among the differentially expressed lncRNAs, 77 were commonly dysregulated, and 41 displayed more than 2-fold change in expression. Endogenous lncRNAs are expressed at low levels, and studies suggest that the contribution of lncRNAs in a disease process is modest in many cases. Our aim was to discover lncRNAs that could potentially be pathogenic in ADPKD; as a result, we focused on identifying targets that are highly expressed and significantly dysregulated in ADPKD. The stringency of our selection criteria was reflected by the relatively small number of annotated transcripts that were identified in our screen.
We specifically focused on investigating Hoxb3os, a lncRNA of unknown function, which is highly down-regulated in both Pkd1-KO and Pkd2-KO kidneys. Hoxb3os was a compelling lncRNA in our study for the following reasons: 1) evolutionarily conserved and abundantly expressed within the kidney epithelium, 2) dysregulated in both cystic and pre-cystic kidneys, 3) loss of expression in the cyst lining, and 4) decreased expression of its human ortholog, HOXB-AS1, in kidneys from ADPKD patients. We uncovered a novel role for Hoxb3os in regulating mTOR pathway and metabolism in mouse kidney cells. Our results reveal that Hoxb3os is a negative regulator of mTORC1 activity and mitochondrial respiration. The evidence for our findings is demonstrated using two experimental approaches: 1) knockdown and genetic deletion of Hoxb3os in kidney cells resulted in enhanced mTORC1 activity and mitochondrial respiration, and 2) re-expression of Hoxb3os transcript in the knockout cells using lentiviral vector partially rescued the Hoxb3os-mutant phenotype.
Hoxb3os is located on chromosome 11 within the HoxB cluster of homeotic genes. Recent studies reveal that lncRNAs embedded within the HOX cluster of homeotic genes possess critical gene-regulatory functions through both cis and trans mechanisms on HOX and non-HOX genes. For example, Hotair is located in the HoxC cluster and is expressed in distal limb buds and posterior trunk of the developing mouse. Deletion of Hotair in the mouse resulted in vertebral transformations and abnormal development of metacarpals and carpals (40). A similar mutant phenotype was reported for the HoxA-associated lncRNA Hottip (41). In our study, knockout of Hoxb3os in kidney cells did not affect the expression of the overlapping Hoxb3 gene. Expression of Hoxb3 was developmentally regulated, but its expression was unchanged in Pkd1 and Pkd2 mutant kidneys. These results suggest that dysregulation of Hoxb3 is unlikely to explain the pathological effects of Hoxb3os dysregulation. However, 77 genes displayed more than 2-fold change in expression in response to Hoxb3os knockdown. These results suggest that Hoxb3os may exert transcriptional effects in trans toward distant promoters. Moreover, 40 genes that represented more than 50% of the highly dysregulated targets (n = 77) were also changed in both Pkd1 and Pkd2 mutant kidneys. These findings indicate a similarity in the transcriptional alterations between Hoxb3os knockdown cells and ADPKD and suggest that a subset of the dysregulated genes in Pkd1 and Pkd2 mutant kidneys may result from altered Hoxb3os expression.
Pathway analysis of RNA-Seq data from Hoxb3os knockdown cells identified defects in signaling networks that are involved in translational control, including the mTOR pathway. The predicted changes in mTOR signaling translated into alteration at the molecular level, whereby phospho-mTOR levels were significantly induced in response to Hoxb3os knockdown. We used CRISPR/Cas9 gene editing to delete Hoxb3os in mIMCD3 cells and confirmed regulation of the mTOR pathway. Similar to knockdown cells, ablation of the first exon and promoter region of Hoxb3os isoform 1 resulted in increased p-mTOR levels. We next focused on mapping additional proteomic and phosphoproteomic changes within the mTOR pathway as a result of Hoxb3os deficiency. We found an increase in phosphorylation of downstream effectors of mTORC1, including p-S6K, p-RS6, and p-4E–BP1. No changes were observed in proteins that are upstream regulators of mTOR (ERK1/2 and RHEB) as well as those that are known to function in mTORC2 (IRS2 and p-AKT at Ser-473) (data not shown). Taken together, our data suggest that Hoxb3os specifically regulates the activity of mTORC1 but not mTORC2. mTOR hyperactivation has been reported in many mouse models of ADPKD, including Pkd1-KO and Pkd2-KO mouse models (42). Although clinical trials with mTOR inhibitors have produced mixed results, a recent study showed that low-dose rapamycin (mTOR inhibitor) improved kidney function (43). We are currently investigating whether down-regulation of Hoxb3os contributes to mTOR hyperactivation and cyst progression in ADPKD. Other studies have reported regulation of mTOR signaling by lncRNAs. Qian et al. (44) showed that the mammalian-imprinted Dlk1-Gtl2 noncoding RNA inhibited PI3K-mTOR signaling, which in turn maintained mitochondrial biogenesis and metabolic activity in check and protected cells from excessive reactive oxygen species (ROS). Another lncRNA, NEAT1, is up-regulated in nonalcoholic fatty liver disease and has been shown to influence the activation of mTOR. Overexpression of NEAT1 in cells increased the levels of p-mTOR and p-S6K, and inhibition of NEAT1 down-regulated mTOR signaling (45).
Using metabolic profiling, we investigated whether the increase in mTORC1 activity affected cellular metabolism. Compared with WT cells, Hoxb3os-KO displayed higher OCR at baseline and in response to FCCP without changes in ECAR. These data suggest that the defects in Hoxb3os knockout cells primarily result from alterations in mitochondrial respiration and not from glycolysis. This finding contrasts with previous studies that showed lower mitochondrial respiration and higher glycolysis in PKD (46). The molecular mechanism underlying the Hoxb3os-dependent metabolic abnormality is not yet clear. Mitochondria play a critical role in normal development and disease through various processes including metabolism and generation of ROS (47). Increased oxidative phosphorylation has been observed in many cancers, including lymphoma (48) and leukemia (49). It is possible that the enhanced mitochondrial respiration in Hoxb3os knockout cells is pathogenic because of an increase in ROS. Alternatively, deficiency of Hoxb3os may promote survival and prevent apoptosis of mutant cells. Future studies are required to examine these possibilities.
Finally, re-expression of Hoxb3os in mutant cells restored p-mTOR as well as p-RS6 and p-S6K to normal levels and partially rescued the metabolic abnormality. The Hoxb3os re-expression results provide further evidence for Hoxb3os regulation of mTORC1 and mitochondrial respiration. Whether the regulation of mTOR signaling by Hoxb3os is because of direct association of Hoxb3os with mTOR complex or is indirect remains to be determined.
Collectively, our data suggest that Hoxb3os regulates mTORC1 signaling and mitochondrial respiration. Because mTOR signaling is known to be dysregulated in ADPKD, our results provide important proof of concept for future studies on re-expressing Hoxb3os as a therapeutic strategy in ADPKD.
Experimental procedures
Animals and human studies
Ksp/Cre (50), Pkhd1/Cre (51), Pkd1f/f (52), and Pkd2f/f (52) mice were used in this study. The genetic background of the mice was C57BL/6, and animals of both sexes were used. All experiments involving animals were performed under the auspices of the Institutional Animal Care and Research Advisory Committees at the University of Minnesota. Human tissue from normal and ADPKD kidneys was provided by the Biological Materials Procurement Network (Bionet) at the University of Minnesota. Kidneys were immediately sealed in sterile bags, immersed in ice, and delivered to the laboratory. The protocol for the use of surgically discarded kidney tissues complied with federal regulations for excess surgical specimens obtained at the University of Minnesota Medical Center-Fairview Campus. All studies were approved by the Institutional Review Board at the University of Minnesota and complied with the Declaration of Helsinki principles.
Quantitative real-time PCR
Total RNA was extracted from mouse inner medullary collecting duct (mIMCD3) cell line or mouse kidneys using RNeasy Mini Kit (Qiagen, Germantown, MD) according to the manufacturer's protocol. mIMCD3 cells were purchased from American Type Culture Collection (ATCC) and were genotyped upon receipt using short tandem repeat (STR) DNA profiling. cDNA was synthesized using the iScript cDNA Synthesis Kit (Bio-Rad), and quantitative real-time PCR was performed with the iTAG Universal SYBR Green Supermix (Bio-Rad). Reactions were performed on the CFX Connect Real-Time System (Bio-Rad). Gene expression levels were normalized to U6 small nuclear RNA or 18S rRNA. Primers for detecting Hoxb3os expression were agagctggaactagaaact (forward) and aacctagcttattactgcttc (reverse).
RNA-Seq
RNA extracted from mouse kidney was checked for quality and sequenced on Illumina instruments. Paired-end reads were checked for quality using FastQC (v0.11.2) and FastQ Screen (v0.4.4) and were quality trimmed using fastq-mcf (ea-utils, v1.1.2-806). Trimmed reads were aligned to the mouse reference genome (UCSC mm10 version from iGenomes) using STAR (v2.4.2a) and known transcripts from GENCODE (vM9). Cufflinks (v2.2.1) was then used for reference annotation–based transcript assembly (RABT) of the transcriptome for each of the individual samples. The individual assemblies were then merged and annotated using GENCODE (vM9) into a single collection of transcripts using Cuffmerge. Expression quantification and differential expression analysis were performed on the transcripts using RSEM (v1.2.22) and edgeR, respectively. Raw and processed data have been deposited in NCBI GEO under accession number GSE108864. For the GapmeR knockdown experiment, poly(A)+ mRNA was used to create an Illumina TruSeq library that was sequenced (50 cycles paired-end) on an Illumina HiSeq 2500 System to a depth of greater than 30 million reads per sample. Reads were quality checked (FastQC v0.11.5), adapter trimmed (Trimmomatic v0.32), and mapped to the GRCm38 reference genome with STAR (2.4.2a_modified). Mapped reads were indexed and sorted with SAMtools (v1.0). The number of reads mapping to each transcript in ENSEMBL annotation set (v84) was estimated with summarizeOverlaps (v1.8.3). Moderated log2-fold change values and Benjamini-Hochberg–corrected p values were calculated using DESeq2 (1.12.3). bigWig files were created using rtracklayer (1.32.2) and scaled based on the normalization in DESeq2.
lncRNA identification pipeline
Transcripts with log cpm >−1 and false discovery rate <0.05 were considered statistically significant and were included in the analysis. PhyloCSF was used to assess the protein-coding potential of de novo transcripts by examining the overrepresentation of evolutionary signatures characteristic of alignments of conserved coding regions such as the high frequencies of synonymous codon substitutions and conservative amino acid substitution. For each locus, the ORF with maximum PhyloCSF score was taken.
RNA fractionation
Freshly harvested adult mouse kidney was cut into small pieces, re-suspended in 800 μl cytoplasmic buffer (10 mm Tris-HCl, pH 7.4, 10 mm NaCl, 3 mm MgCl2, 10% glycerol, 0.5% NP40, and 0.5 mm DTT). Samples were homogenized with a Dounce homogenizer into a single cell suspension and centrifuged at 13000 × g for 2 min to obtain the cytoplasmic fraction. The process was repeated twice, and the three fractions containing cytosolic RNA were combined. The remaining pellet contained nuclear RNA. RNA was extracted from cytosolic and nuclear fractions using the miRNeasy kit (Qiagen). Success of the fractionation procedure was confirmed by quantifying the levels of U6 small nuclear RNA and 18S RNA in each fraction.
In situ hybridization
Exiqon miRCURY LNA ISH FFPE Protocol (Exiqon miRCURY LNA ISH Optimization Kit 2, no. 90002) was utilized for in situ hybridization. Briefly, kidneys were perfused with 4% paraformaldehyde, fixed in paraformaldehyde for 48 h, transferred to 70% ethanol and embedded in paraffin blocks. Blocks were sectioned at 4 μm then incubated in an oven at 65 °C for 30 min before proceeding to deparaffinization. Sections were then washed with PBS three times then treated with proteinase K for 10 min at 37 °C. Double DIG-Hoxb3os–labeled detection probe (AGCAGAAGGTTGTGGTGGT) was hybridized to sections at 53 °C for 1 h. Anti–digoxigenin-AP Fab fragment antibody was used at 1:1000 dilution (Roche, 11093274910), and AP substrate was reapplied after 1 h. Slides were mounted with Eukitt mounting medium. Samples were examined using light microscopy.
Knockdown experiments
GapmeRs were purchased from Exiqon. mIMCD3 cells (2 × 105 cells/well) were seeded in a 6-well plate and grown for 24 h. After reaching 60–70% confluency, cells were transfected with 10 nm GapmeR against Hoxb3os (ATTACTGCTTCTTCCA) or negative control using Lipofectamine 3000. The cells were harvested 48 h after transfection and lysed in TRIzol (Invitrogen) for RNA extraction or in RIPA buffer (Sigma) for protein extraction.
CRISPR/Cas9 gene editing
Generation of the Hoxb3os mutant cell line was performed as described (53). Briefly, the following sgRNAs were used to generate three Hoxb3os mutant cell lines: sgRNA-1, caccgaaggtgattctgggagatct; sgRNA-2, caccgactgctcttaactaggccag. Each sgRNA was annealed and cloned into the pSpCas9(BB)-2A-puromycin construct (Addgene, no. 48139). Proper insertion of each sgRNA was confirmed by sequencing. One microgram of each sgRNA construct or empty vector was transfected into mIMCD3 cells using Lipofectamine 3000. Cells were trypsinized 72 h post transfection, and serial dilution was performed to obtain single-cell colonies in 96-well plates. Hoxb3os deleted clones were identified by PCR analysis of genomic DNA using the following detection primers: 5′-tgaggcgatcaatcttagcc-3′ (forward) and 5′-agctaaggtacacaacaccc-3′ (reverse).
RACE
RACE was performed using SMARTer® RACE 5′/3′ Kit from Clontech Laboratories, as described in the kit manual. The sequence of Hoxb3os primers used in RACE experiments were 5′-tcacgtgacagcctcattg-3′ (3′RACE) and 5′-aagagattccagcagatgca-3′ (5′RACE).
Western blotting
Cells were cultured in 6-well tissue culture plates. After reaching confluency, protein was extracted using RIPA lysis buffer (Sigma). Five micrograms of protein lysate was run on 4–12% Bis-tris or 2–8% Tris-acetate protein gels (Invitrogen) and transferred to nitrocellulose membranes using the wet transfer method. Blocking was performed using 5% milk in TBS with Tween for 1 h at room temperature. Membranes were incubated with antibodies at 1:1000 dilution in TBS with Tween with 5% BSA at 4 °C overnight. Secondary rabbit or mouse antibody was added at 1:20000 dilution in TBS with Tween for 1 h at room temperature. The following antibodies were purchased from Cell Signaling Technology: mTOR (2972), p-mTOR (2971), RS6 (2217), p-RS6 (2211), p-S6K (9234), 4E-BP1 (9451), p-4E–BP1 (9451). GAPDH was purchased from Santa Cruz (sc-25778) and used as loading control.
Extracellular flux measurements
Cells were seeded at 10,000 cells per well on Matrigel-coated Seahorse microplates for flux measurements on the XFe96 Extracellular Flux Analyzer (Agilent Seahorse Technologies). Glycolysis and oxidative phosphorylation were measured using the XF Cell Mito Stress Test Kit (Agilent). The stress tests require sequential addition of specific pathway inhibitors to query various aspects of these two metabolic pathways. The XF sensor cartridges were hydrated overnight at 37 °C without carbon dioxide in the Seahorse XF Calibrant Solution, as recommended in the manufacturer's protocol. The assay medium was Seahorse XF base medium (DMEM) supplemented with 5 mm pyruvate, 5 mm d-glucose, and 2 mm l-glutamine (pH 7.4). Oligomycin (1 mm), FCCP (1 mm), rotenone (0.5 mm), and antimycin (0.5 mm) were added to the ports. Both ECAR and OCR measurements were performed at 8-min intervals (2 min mixing, 2 min recovery, and 4 min measuring) for 3 h using the Seahorse XFe96 analyzer. Measurements were recorded from three biological replicates with eight technical replicates per experiment. All data were normalized to cell number.
Statistical analysis
Student's two-tailed unpaired t test was used for pairwise comparisons. Analysis of variance (ANOVA) followed by Tukey's post hoc test were used for multiple comparisons. p < 0.05 was considered to be significant.
Author contributions
K. A. conceptualization; K. A., S. F., M. K., M. D. G., and C. X. formal analysis; K. A. and S. F. investigation; K. A., S. C. C., S. A., A. M., and D. L. methodology; K. A. writing-original draft; S. F. validation; M. K., M. D. G., and C. X. resources; M. K. and C. X. data curation; V. P., C. X., and P. I. supervision; P. I. funding acquisition.
Supplementary Material
Acknowledgments
We thank Jeremy Herrera for his help in performing ISH experiments. We also thank Stefan Somlo for providing the Pkd1f/f and Pkd2f/f mice. We thank Dr. Ameeta Kelekar and the MN Partnership Infrastructure Award MNP IF 16.09 for access to the Seahorse XFe96 Analyzer.
This work was supported by the National Institutes of Health Grants R37DK042921 (to P. I.) and UL1TR001105 (to C. X.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The RNA sequences for this project can be accessed through the NCBI under Gene Expression Omnibus accession number GSE108864.
This article contains Figs. S1–S6.
- ADPKD
- autosomal dominant polycystic kidney disease
- lncRNAs
- long noncoding RNAs
- PhyloCSF
- phylogenetic codon substitution frequency software
- qRT-PCR
- quantitative RT-PCR
- RACE
- rapid amplification of cDNA ends
- P
- postnatal
- E
- embryonic
- ISH
- in situ hybridization
- ECAR
- extracellular acidification rate
- OCR
- oxygen consumption rates
- p-
- phospho-
- FCCP
- carbonyl cyanide p-trifluoromethoxyphenylhydrazone
- ROS
- reactive oxygen species
- sgRNAs
- single guide RNAs.
References
- 1. Grantham, J. J., Gardner, K. D., and PKR Foundation (1985) Problems in Diagnosis and Management of Polycystic Kidney Disease: Proceedings of the First International Workshop on Polycystic Kidney Disease. PKR Foundation, Kansas City, MO. [Google Scholar]
- 2. Iglesias, C. G., Torres, V. E., Offord, K. P., Holley, K. E., Beard, C. M., and Kurland, L. T. (1983) Epidemiology of adult polycystic kidney disease, Olmsted County, Minnesota: 1935–1980. Am. J. Kidney. Dis. 2, 630–639 10.1016/S0272-6386(83)80044-4 [DOI] [PubMed] [Google Scholar]
- 3. Harris, P. C., and Torres, V. E. (2002) Polycystic kidney disease, autosomal dominant. In GeneReviews® (Adam, M. P., Ardinger, H. H., Pagon, R. A., Wallace, S. E., Bean, L. J. H., Mefford, H. C., Stephens, K., Amemiya, A., and Ledbetter, N., eds.), University of Washington, Seattle, WA. [PubMed] [Google Scholar]
- 4. Audrézet, M. P., Corbiere, C., Lebbah, S., Morinière, V., Broux, F., Louillet, F., Fischbach, M., Zaloszyc, A., Cloarec, S., Merieau, E., Baudouin, V., Deschênes, G., Roussey, G., Maestri, S., Visconti, C., et al. (2016) Comprehensive PKD1 and PKD2 mutation analysis in prenatal autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 27, 722–729 10.1681/ASN.2014101051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cornec-Le Gall, E., Torres, V. E., and Harris, P. C. (2017) Genetic complexity of autosomal dominant polycystic kidney and liver diseases. J. Am. Soc. Nephrol. 29, 13–23 10.1681/ASN.2017050483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hateboer, N., v Dijk, M. A., Bogdanova, N., Coto, E., Saggar-Malik, A. K., San Millan, J. L., Torra, R., Breuning, M., and Ravine, D. (1999) Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 Study Group. Lancet 353, 103–107 10.1016/S0140-6736(98)03495-3 [DOI] [PubMed] [Google Scholar]
- 7. Hughes, J., Ward, C. J., Peral, B., Aspinwall, R., Clark, K., San Millán, J. L., Gamble, V., and Harris, P. C. (1995) The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat. Genet. 10, 151–160 10.1038/ng0695-151 [DOI] [PubMed] [Google Scholar]
- 8. Kurbegovic, A., Kim, H., Xu, H., Yu, S., Cruanès, J., Maser, R. L., Boletta, A., Trudel, M., and Qian, F. (2014) Novel functional complexity of polycystin-1 by GPS cleavage in vivo: Role in polycystic kidney disease. Mol. Cell Biol. 34, 3341–3353 10.1128/MCB.00687-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mochizuki, T., Wu, G. Q., Hayashi, T., Xenophontos, S. L., Veldhuisen, B., Saris, J. J., Reynolds, D. M., Cai, Y. Q., Gabow, P. A., Pierides, A., Kimberling, W. J., Breuning, M. H., Deltas, C. C., Peters, D. J. M., and Somlo, S. (1996) PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 272, 1339–1342 10.1126/science.272.5266.1339 [DOI] [PubMed] [Google Scholar]
- 10. Qian, F., Germino, F. J., Cai, Y., Zhang, X., Somlo, S., and Germino, G. G. (1997) PKD1 interacts with PKD2 through a probable coiled-coil domain. Nat. Genet. 16, 179–183 10.1038/ng0697-179 [DOI] [PubMed] [Google Scholar]
- 11. González-Perrett, S., Kim, K., Ibarra, C., Damiano, A. E., Zotta, E., Batelli, M., Harris, P. C., Reisin, I. L., Arnaout, M. A., and Cantiello, H. F. (2001) Polycystin-2, the protein mutated in autosomal dominant polycystic kidney disease (ADPKD), is a Ca2+-permeable nonselective cation channel. Proc. Natl. Acad. Sci. U.S.A. 98, 1182–1187 10.1073/pnas.98.3.1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aguiari, G., Banzi, M., Gessi, S., Cai, Y. Q., Zeggio, E., Manzati, E., Piva, R., Lambertini, E., Ferrari, L., Peters, D. J., Lanza, F., Harris, P. C., Borea, P. A., Somlo, S., and Del Senno, L. (2004) Deficiency of polycystin-2 reduces Ca2+ channel activity and cell proliferation in ADPKD lymphoblastoid cells. FASEB J. 18, 884–886 10.1096/fj.03-0687fje [DOI] [PubMed] [Google Scholar]
- 13. Hanaoka, K., and Guggino, W. B. (2000) cAMP regulates cell proliferation and cyst formation in autosomal polycystic kidney disease cells. J. Am. Soc. Nephrol. 11, 1179–1187 [DOI] [PubMed] [Google Scholar]
- 14. Song, X., Di Giovanni, V., He, N., Wang, K., Ingram, A., Rosenblum, N. D., and Pei, Y. (2009) Systems biology of autosomal dominant polycystic kidney disease (ADPKD): Computational identification of gene expression pathways and integrated regulatory networks. Hum. Mol. Genet. 18, 2328–2343 10.1093/hmg/ddp165 [DOI] [PubMed] [Google Scholar]
- 15. Boca, M., Distefano, G., Qian, F., Bhunia, A. K., Germino, G. G., and Boletta, A. (2006) Polycystin-1 induces resistance to apoptosis through the phosphatidylinositol 3-kinase/Akt signaling pathway. J. Am. Soc. Nephrol. 17, 637–647 10.1681/ASN.2005050534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lin, F., Hiesberger, T., Cordes, K., Sinclair, A. M., Goldstein, L. S., Somlo, S., and Igarashi, P. (2003) Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc. Natl. Acad. Sci. U.S.A. 100, 5286–5291 10.1073/pnas.0836980100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Baba, M., Furihata, M., Hong, S. B., Tessarollo, L., Haines, D. C., Southon, E., Patel, V., Igarashi, P., Alvord, W. G., Leighty, R., Yao, M., Bernardo, M., Ileva, L., Choyke, P., Warren, M. B., Zbar, B., Linehan, W. M., and Schmidt, L. S. (2008) Kidney-targeted Birt-Hogg-Dube gene inactivation in a mouse model: Erk1/2 and Akt-mTOR activation, cell hyperproliferation, and polycystic kidneys. J. Natl. Cancer Inst. 100, 140–154 10.1093/jnci/djm288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ma, L., Bajic, V. B., and Zhang, Z. (2013) On the classification of long non-coding RNAs. RNA Biol. 10, 925–933 10.4161/rna.24604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang, G. D., Lu, X. Z., and Yuan, L. J. (2014) LncRNA: A link between RNA and cancer. Biochim. Biophys. Acta 1839, 1097–1109 10.1016/j.bbagrm.2014.08.012 [DOI] [PubMed] [Google Scholar]
- 20. Parasramka, M. A., Maji, S., Matsuda, A., Yan, I. K., and Patel, T. (2016) Long non-coding RNAs as novel targets for therapy in hepatocellular carcinoma. Pharmacol. Ther. 161, 67–78 10.1016/j.pharmthera.2016.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fatima, R., Akhade, V. S., Pal, D., and Rao, S. M. (2015) Long noncoding RNAs in development and cancer: Potential biomarkers and therapeutic targets. Mol. Cell Ther. 3, 5 10.1186/s40591-015-0042-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schmitt, A. M., and Chang, H. Y. (2016) Long noncoding RNAs in cancer pathways. Cancer Cell 29, 452–463 10.1016/j.ccell.2016.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang, F., Ying, H. Q., He, B. S., Pan, Y. Q., Deng, Q. W., Sun, H. L., Chen, J., Liu, X., and Wang, S. K. (2015) Upregulated IncRNA-UCA1 contributes to progression of hepatocellular carcinoma through inhibition of miR-216b and activation of FGFR1/ERK signaling pathway. Oncotarget 6, 7899–7917 10.18632/oncotarget.3219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hessels, D., and Schalken, J. A. (2009) The use of PCA3 in the diagnosis of prostate cancer. Nat. Rev. Urol. 6, 255–261 10.1038/nrurol.2009.40 [DOI] [PubMed] [Google Scholar]
- 25. Ji, P., Diederichs, S., Wang, W., Böing, S., Metzger, R., Schneider, P. M., Tidow, N., Brandt, B., Buerger, H., Bulk, E., Thomas, M., Berdel, W. E., Serve, H., and Müller-Tidow, C. (2003) MALAT-1, a novel noncoding RNA, and thymosin β4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 22, 8031–8041 10.1038/sj.onc.1206928 [DOI] [PubMed] [Google Scholar]
- 26. Ying, L., Chen, Q., Wang, Y. W., Zhou, Z. H., Huang, Y. R., and Qiu, F. (2012) Upregulated MALAT-1 contributes to bladder cancer cell migration by inducing epithelial-to-mesenchymal transition. Mol. Biosyst. 8, 2289–2294 10.1039/c2mb25070e [DOI] [PubMed] [Google Scholar]
- 27. Lin, R., Maeda, S., Liu, C., Karin, M., and Edgington, T. S. (2007) A large noncoding RNA is a marker for murine hepatocellular carcinomas and a spectrum of human carcinomas. Oncogene 26, 851–858 10.1038/sj.onc.1209846 [DOI] [PubMed] [Google Scholar]
- 28. Ji, Q., Zhang, L., Liu, X., Zhou, L., Wang, W., Han, Z., Sui, H., Tang, Y., Wang, Y., Liu, N., Ren, J., Hou, F., and Li, Q. (2014) Long non-coding RNA MALAT1 promotes tumour growth and metastasis in colorectal cancer through binding to SFPQ and releasing oncogene PTBP2 from SFPQ/PTBP2 complex. Br. J. Cancer 111, 736–748 10.1038/bjc.2014.383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tano, K., Mizuno, R., Okada, T., Rakwal, R., Shibato, J., Masuo, Y., Ijiri, K., and Akimitsu, N. (2010) MALAT-1 enhances cell motility of lung adenocarcinoma cells by influencing the expression of motility-related genes. FEBS Lett. 584, 4575–4580 10.1016/j.febslet.2010.10.008 [DOI] [PubMed] [Google Scholar]
- 30. Long, J., Badal, S. S., Ye, Z., Wang, Y., Ayanga, B. A., Galvan, D. L., Green, N. H., Chang, B. H., Overbeek, P. A., and Danesh, F. R. (2016) Long noncoding RNA Tug1 regulates mitochondrial bioenergetics in diabetic nephropathy. J. Clin. Invest. 126, 4205–4218 10.1172/JCI87927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Igarashi, P., and Somlo, S. (2007) Polycystic kidney disease. J. Am. Soc. Nephrol. 18, 1371–1373 10.1681/ASN.2007030299 [DOI] [PubMed] [Google Scholar]
- 32. Patel, V., Chowdhury, R., and Igarashi, P. (2009) Advances in the pathogenesis and treatment of polycystic kidney disease. Curr. Opin. Nephrol. Hypertens. 18, 99–106 10.1097/MNH.0b013e3283262ab0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lin, M. F., Jungreis, I., and Kellis, M. (2011) PhyloCSF: A comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics 27, i275–i282 10.1093/bioinformatics/btr209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. De Kumar, B., and Krumlauf, R. (2016) HOXs and lincRNAs: Two sides of the same coin. Sci. Adv. 2, e1501402 10.1126/sciadv.1501402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hay, N., and Sonenberg, N. (2004) Upstream and downstream of mTOR. Genes Dev. 18, 1926–1945 10.1101/gad.1212704 [DOI] [PubMed] [Google Scholar]
- 36. Foster, K. G., and Fingar, D. C. (2010) Mammalian target of rapamycin (mTOR): Conducting the cellular signaling symphony. J. Biol. Chem. 285, 14071–14077 10.1074/jbc.R109.094003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sarbassov, D. D., Guertin, D. A., Ali, S. M., and Sabatini, D. M. (2005) Phosphorylation and regulation of Akt/PKB by the Rictor-mTOR complex. Science 307, 1098–1101 10.1126/science.1106148 [DOI] [PubMed] [Google Scholar]
- 38. Schieke, S. M., Phillips, D., Mccoy, J. P., Jr., Aponte, A. M., Shen, R. F., Balaban, R. S., and Finkel, T. (2006) The mammalian target of rapamycin (mTOR) pathway regulates mitochondrial oxygen consumption and oxidative capacity. J. Biol. Chem. 281, 27643–27652 10.1074/jbc.M603536200 [DOI] [PubMed] [Google Scholar]
- 39. Emmrich, S., Streltsov, A., Schmidt, F., Thangapandi, V. R., Reinhardt, D., and Klusmann, J. H. (2014) LincRNAs MONC and MIR100HG act as oncogenes in acute megakaryoblastic leukemia. Mol. Cancer 13, 171 10.1186/1476-4598-13-171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li, L., Liu, B., Wapinski, O. L., Tsai, M. C., Qu, K., Zhang, J., Carlson, J. C., Lin, M., Fang, F., Gupta, R. A., Helms, J. A., and Chang, H. Y. (2013) Targeted disruption of Hotair leads to homeotic transformation and gene derepression. Cell Rep. 5, 3–12 10.1016/j.celrep.2013.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang, K. C., Yang, Y. W., Liu, B., Sanyal, A., Corces-Zimmerman, R., Chen, Y., Lajoie, B. R., Protacio, A., Flynn, R. A., Gupta, R. A., Wysocka, J., Lei, M., Dekker, J., Helms, J. A., and Chang, H. Y. (2011) A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 472, 120–124 10.1038/nature09819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Warner, G., Hein, K. Z., Nin, V., Edwards, M., Chini, C. C. S., Hopp, K., Harris, P. C., Torres, V. E., and Chini, E. N. (2016) Food restriction ameliorates the development of polycystic kidney disease. J. Am. Soc. Nephrol. 27, 1437–1447 10.1681/ASN.2015020132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Braun, W. E., Schold, J. D., Stephany, B. R., Spirko, R. A., and Herts, B. R. (2014) Low-dose rapamycin (sirolimus) effects in autosomal dominant polycystic kidney disease: An open-label randomized controlled pilot study. Clin. J. Am. Soc. Nephrol. 9, 881–888 10.2215/CJN.02650313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Qian, P. X., He, X. C., Paulson, A., Li, Z. R., Tao, F., Perry, J. M., Guo, F. L., Zhao, M., Zhi, L., Venkatraman, A., Haug, J. S., Parmely, T., Li, H., Dobrowsky, R. T., Ding, W. X., Kono, T., Ferguson-Smith, A. C., and Li, L. H. (2016) The Dlk1-Gtl2 locus preserves LT-HSC function by inhibiting the PI3K-mTOR pathway to restrict mitochondrial metabolism. Cell Stem Cell 18, 214–228 10.1016/j.stem.2015.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang, X. (2017) Down-regulation of lncRNA-NEAT1 alleviated the non-alcoholic fatty liver disease via mTOR/S6K1 signaling pathway. J. Cell Biochem. 119, 1567–1574 [DOI] [PubMed] [Google Scholar]
- 46. Rowe, I., Chiaravalli, M., Mannella, V., Ulisse, V., Quilici, G., Pema, M., Song, X. W. W., Xu, H. X., Mari, S., Qian, F., Pei, Y., Musco, G., and Boletta, A. (2013) Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat. Med. 19, 488–493 10.1038/nm.3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pelicano, H., Xu, R. H., Du, M., Feng, L., Sasaki, R., Carew, J. S., Hu, Y. M., Ramdas, L., Hu, L. M., Keating, M. J., Zhang, W., Plunkett, W., and Huang, P. (2006) Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanism. J. Cell Biol. 175, 913–923 10.1083/jcb.200512100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Birkenmeier, K., Dröse, S., Wittig, I., Winkelmann, R., Käfer, V., Döring, C., Hartmann, S., Wenz, T., Reichert, A. S., Brandt, U., and Hansmann, M. L. (2016) Hodgkin and Reed-Sternberg cells of classical Hodgkin lymphoma are highly dependent on oxidative phosphorylation. Int. J. Cancer 138, 2231–2246 10.1002/ijc.29934 [DOI] [PubMed] [Google Scholar]
- 49. Kuntz, E. M., Baquero, P., Michie, A. M., Dunn, K., Tardito, S., Holyoake, T. L., Helgason, G. V., and Gottlieb, E. (2017) Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat. Med. 23, 1234–1240 10.1038/nm.4399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shao, X. L., Somlo, S., and Igarashi, P. (2002) Epithelial-specific Cre/lox recombination in the developing kidney and genitourinary tract. J. Am. Soc. Nephrol. 13, 1837–1846 10.1097/01.ASN.0000016444.90348.50 [DOI] [PubMed] [Google Scholar]
- 51. Patel, V., Li, L., Cobo-Stark, P., Shao, X. L., Somlo, S., Lin, F. M., and Igarashi, P. (2008) Acute kidney injury and aberrant planar cell polarity induce cyst formation in mice lacking renal cilia. Hum. Mol. Genet. 17, 1578–1590 10.1093/hmg/ddn045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shibazaki, S., Yu, Z. H., Nishio, S., Tian, X., Thomson, R. B., Mitobe, M., Louvi, A., Velazquez, H., Ishibe, S., Cantley, L. G., Igarashi, P., and Somlo, S. (2008) Cyst formation and activation of the extracellular regulated kinase pathway after kidney specific inactivation of Pkd1. Hum. Mol. Genet. 17, 1505–1516 10.1093/hmg/ddn039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A., and Zhang, F. (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.