

Abstract
Chondrocyte hypertrophy is the terminal step in chondrocyte differentiation and is crucial for endochondral bone formation. How signaling pathways regulate chondrocyte hypertrophic differentiation remains incompletely understood. In this study, using a Tbx18:Cre (Tbx18Cre/+) gene-deletion approach, we selectively deleted the gene for the signaling protein SMAD family member 4 (Smad4f/f) in the limbs of mice. We found that the Smad4-deficient mice develop a prominent shortened limb, with decreased expression of chondrocyte differentiation markers, including Col2a1 and Acan, in the humerus at mid-to-late gestation. The most striking defects in these mice were the absence of stylopod elements and failure of chondrocyte hypertrophy in the humerus. Moreover, expression levels of the chondrocyte hypertrophy–related markers Col10a1 and Panx3 were significantly decreased. Of note, we also observed that the expression of runt-related transcription factor 2 (Runx2), a critical mediator of chondrocyte hypertrophy, was also down-regulated in Smad4-deficient limbs. To determine how the skeletal defects arose in the mouse mutants, we performed RNA-Seq with ChIP-Seq analyses and found that Smad4 directly binds to regulatory elements in the Runx2 promoter. Our results suggest a new mechanism whereby Smad4 controls chondrocyte hypertrophy by up-regulating Runx2 expression during skeletal development. The regulatory mechanism involving Smad4-mediated Runx2 activation uncovered here provides critical insights into bone development and pathogenesis of chondrodysplasia.
Keywords: chondrocyte, chondrogenesis, development, SMAD transcription factor, signaling, bone, mouse, Chondrocyte differentiation, Chondrocyte hypertrophy, Endochondral bone formation, Runx2, Smad4
Introduction
Human chondrodysplasia is a heritable disorder affecting skeletal development, characterized by skeletal abnormalities with reduced length of the long bone and short stature. Several genetic mutations causing chondrodysplasia have been identified including type X collagen (Col10a1), runt-related transcription factor 2 (Runx2),3 and cartilage-derived morphogenetic protein 1 (CDMP1) (1–4). These genetic mutations affect chondrogenesis crucial for endochondral bone development. Understanding the molecular pathways underlying chondrogenesis during skeletal development may provide crucial insights into the therapeutic treatments for chondrodysplasia in humans.
Endochondral ossification is an imperative developmental process for skeletal formation in vertebrates. Endochondral bone development is initiated by aggregation and condensation of mesenchymal cells. The mesenchymal cells further differentiate into primary chondrocytes with secreting type II collagen (Col2a1) and aggrecan (Acan) to form cartilage elements. Primary chondrocytes then proliferate and develop into hypertrophic chondrocytes that secrete Col10a1. Subsequently, hypertrophic chondrocytes are invaded by blood vessels and are replaced by bone and bone marrow (5–8). In this process, chondrocyte hypertrophy is a terminal step of chondrocyte differentiation, and is essential for endochondral bone formation. A few essential molecules that control chondrocyte hypertrophic differentiation have been found (9). Indian hedgehog (Ihh), a member of the hedgehog family, is expressed in prehypertrophic chondrocytes. Ihh is required for chondrocyte proliferation and chondrocyte hypertrophy (10, 11). Bone morphogenetic protein (BMP) family members are expressed throughout limb development and play vital roles in regulating chondrogenesis (12, 13). Deletion of Bmp2 results in defective chondrocyte hypertrophy and endochondral bone formation, indicating Bmp2 is required for chondrocyte hypertrophic differentiation and endochondral bone formation (14). In addition, Runx transcription factors Runx2 and Runx3 also regulate and coordinate chondrocyte hypertrophic differentiation and proliferation (15, 16).
Smad4 is a central mediator of canonical transforming growth factor (TGF)/BMP signaling (17) and it binds to receptor-activated Smads (R-Smads) and forms a complex with R-Smads. The Smad complex translocates into the nucleus and serves as a transcription factor to regulate the expression of targeting genes involved in a wide variety of development and homeostasis processes. During mouse embryogenesis, Smad4 is expressed in the developing limbs and is required for early mesenchymal cell aggregation, chondrocyte differentiation, osteoblast differentiation, and maturation (18–21). The regulatory mechanism of the Smad4 underlying chondrocyte hypetrophic differentiation during long bone development is unknown, although studies on TGF/BMP pathway genes showed that Bmp2 regulates Runx2 expression through Smad in the C2C12 cell line (22), and Runx2 physically interacts with Smads in COS7 cells in vitro (23). In this study, we performed a genetic study in mice and found that loss of Smad4 within the limb resulted in the absence of chondrocyte hypertrophy and ossification in humerus of mice. In addition, Smad4 promotes expression of hypertrophy differentiation genes, including Runx2, Runx3, and Ihh. Further RNA-seq and ChIP-seq analyses revealed that Smad4 regulates a wide spectrum of genes associated with limb development, including direct activation of Runx2. These findings reveal an important mechanism by which Smad4 regulates chondrocyte hypertrophy during mouse skeletal development.
Results
Deletion of Smad4 disrupts skeletal development
To investigate the role of Smad4 in limb formation, we utilized new Tbx18Cre/+ knock-in mice (24), with which to remove Smad4 in the proximal domain of forelimbs and hindlimbs in mouse embryos (Fig. S1, A and B). To ascertain and characterize Tbx18-Cre-mediated Cre-loxP recombination during limb development, Tbx18Cre/+ knock-in mice were crossed to Rosa26nlacZ/+ and Rosa26GFP/+ reporter mice (25, 26). Lineage tracing showed that Tbx18 descendants (Tbx18Cre/+;Rosa26nlacZ/+ and Tbx18Cre/+;Rosa26GFP/+) include the proximal domain of the limbs at E9.5–13.5 (Fig. S1, C and D). Further immunostaining revealed that Tbx18 lineage cells are mainly co-localized with Sox9 in chondrocytes at E12.5–13.5 (Fig. S1, E and F), suggesting Tbx18Cre/+ lineages mark chondrocytes in the proximal domain of the limbs.
To delete Smad4 in the chondrocytes, Tbx18Cre/+ mice were crossed to Smad4-flox (Smad4f/f) (27) to obtain Tbx18Cre/+;Smad4f/+ compound heterozygous animals. They were further crossed to Smad4f/f mice to generate Tbx18Cre/+;Smad4f/f mutants (denoted as Smad4 CKO). We found mice with the Tbx18Cre/+;Smad4f/+ genotype are normal and therefore utilized as controls in this study. Immunostaining revealed that Smad4 is ubiquitously expressed in the WT and Tbx18Cre/+;Smad4f/+ control limb at E11.5–13.5 (Fig. 1A, data not shown for WT). In Smad4 CKO embryos, almost no Smad4 expression was detected in the chondrocytes and adjacent perichondrial cells (Fig. 1B), suggesting that Smad4 is removed in these regions. The mutant mice died shortly after birth with severe ureteral development defects (24). At E14.5, they displayed a prominently shortened limb (arrows in Fig. 1, C and D), and the defect became severe at E16.5 and E18.5 (Fig. 1, E–J). Further skeletal analysis by Alizarin red and Alcian blue staining showed that the limb shortening was mainly due to the absence of stylopod elements (humerus and femur, arrows in Fig. 1, K–R) that failed in ossification at E15.5–18.5, indicating the defect was associated with abnormal chondrocyte differentiation. Additionally, zeugopod elements of the mutant limb (radius/ulna and tibia/fibula, arrowheads in Fig. 1, K–R, Fig. S2) were shorter than the control littermates. Because the stylopod elements were the most severely affected in both forelimbs and hindlimbs, we focused on the stylopod elements (humerus) in the forelimbs.
Figure 1.

Impaired skeletal development in Smad4 mutant mice. A and B, immunostaining of Smad4 on the forelimb on the control and mutant mice at E12.5. Arrows indicate chondrocytes and arrowheads indicate perichondrial cells. A3 and B3 are overlay images for A1/2 and B1/2, respectively. A4 and B4 are high magnification images in the areas outlined in A3 and B3. C–J, whole mount view of Smad4 CKO mice at E14.5–18.5. Arrows indicate control forelimb (C, E, and G) and hindlimb (I), or mutant forelimb (D, F, and H) and hindlimb (J). K–R, skeletons of E15.5–18.5 embryos stained with Alizarin red (calcified tissue) and Alcian blue (cartilage). K, M, O, and Q are controls and L, N, P, and R are mutants. In K–P, arrows, arrowheads, and unnotched arrows indicate humerus, radius, and ulna, respectively. In Q and R, arrows, arrowheads, and unnotched arrows indicate femur, fibula, and tibia, respectively.
Loss of Smad4 causes chondrodysplasia in limb development
To investigate the cause of the shortened limbs in Smad4 CKO mice, we examined younger stage embryos from E10.5 to determine when the defects first appear. No apparent abnormalities were observed at E12.5 or earlier stages in the mutant (data not shown), except that the cartilage element was slightly shorter and smaller at E12.5 (Fig. 2, A and B). The difference became significant at E13.5 (Fig. 2, C and D), and was severe at E14.5 (Fig. 2, E and F). We analyzed the expression of chondrocyte differentiation markers including Lect1, Col2a1, and Acan (5, 28, 29) to determine whether chondrocyte differentiation was affected in the limbs at E12.5–13.5. Compared with control humerus, lower expression levels of Lect1 and Col2a1 were detected in the mutant humerus by RNA in situ hybridization (Fig. 2, G–L). Quantitative RT-PCR (qRT-PCR) confirmed that Lect1, Col2a1, and Acan mRNA expression levels were significantly lower by 50, 55, and 58% at E12.5, and by 63, 60, and 65% in the mutant forelimbs at E13.5, respectively (Fig. 2, M–O). The down-regulated Lect1, Col2a1, and Acan expression suggests that Smad4 is essential for chondrocyte differentiation.
Figure 2.

Chondrodysplasia in the limbs of Smad4 mutant mice. A–F, Alcian blue staining on the control (A, C, and E) and mutant forelimbs (B, D, and F) at E12.5–14.5. Arrows in A–F indicate humerus. G–L, RNA in situ hybridization for Lect1 and Col2a1 on the forelimb sections at E12.5 and E13.5. Arrows indicate RNA expression in humerus. M–O, Lect1, Col2a1, and Acan expression level was evaluated by qRT-PCR in the forelimbs at E12.5 and E13.5. n = 3, *, p < 0.05.
Smad4 is essential for chondrocyte hypertrophy
Failure of endochondral ossification in stylopod elements is the most striking defect in Smad4 CKO limbs at E15.5 (arrows in Fig. 1, K and L). Chondrocyte hypertrophy is a crucial developmental step for endochondral ossification. Lack of hypertrophic chondrocytes could cause failure of endochondral ossification (5). We then try to determine whether chondrocyte hypertrophy is perturbed in Smad4 CKO limb. For Tbx18Cre/+;Smad4f/+ and WT limb at E13.5. H&E staining showed that chondrocytes were present in the center of humerus with hypertrophic differentiation (Fig. 3A). These hypertrophic characteristics were unable to be detected in the Smad4 CKO humerus (Fig. 3B). At E14.5, typical hypertrophic cells were present in the control humerus (Fig. 3C), and they were absent in the mutants (Fig. 3D). Alcian blue staining further confirmed the absence of hypertrophic chondrocytes in the mutant humerus (Fig. 3, E–H). We next performed RNA in situ hybridization to detect Col10a1, a specific marker for hypertrophic chondrocytes (30, 31), and Panx3, a marker for prehypertrophic and hypertrophic chondrocytes (32, 33). Both Col10a1 and Panx3 were found in the center of humerus as early as E12.5 in the control (Fig. 3, I and O), but were undetectable in the mutant (Fig. 3, J and P). At E13.5 and E14.5, robust Col10a1 and Panx3 expression was detected in the hypertrophic or prehypertrophic region on the control mice (Fig. 3, K, M, Q, and S), and they were not seen in the mutant humerus (Fig. 3, L, N, R, and T). Further qRT-PCR analysis showed that Col10a1 and Panx3 mRNA expression was decreased by 70 and 90% at E12.5, and by 76 and 87% at E13.5 in the mutant forelimbs, respectively (Fig. 3, U and V). Taken together, these observations indicate that the primary chondrocytes of Smad4 CKO humerus failed to undergo hypertrophic differentiation, and Smad4 is critical for chondrocyte hypertrophy development during limb formation.
Figure 3.

Lack of chondrocyte hypertrophy in Smad4 CKO humerus. A–H, H&E and Alcian blue staining on the control and mutant forelimbs at E13.5 and 14.5. A2–H2 are magnified images for A1–H1 showing chondrocytes (arrows) in the humerus. Hypertrophic chondrocytes were observed in the control humerus (arrows in C2 and G2), but not in the mutant (arrows in D2 and H2) at E14.5. I–T, section RNA in situ hybridization for Col10a1 and Panx3 on the forelimb at E12.5–14.5. Arrows indicate RNA expression in humerus. U and V, Col10a1 and Panx3 expression in the forelimbs was determined by qRT-PCR at E12.5 and E13.5. n = 3, *, p < 0.01.
Smad4 deficiency results in decreased chondrocyte proliferation
Chondrocyte proliferation is important for cartilage growth prior to hypertrophic differentiation. Given the small humerus in the mutant limb at E12.5 and onward, we hypothesized that Smad4 signals may be essential for chondrocyte growth. To confirm this, EdU was injected into pregnant mice intraperitoneally at E12.5 and E13.5, and forelimbs were collected after 4 h of injection for analysis. The proliferating cells in the cartilage were analyzed by immunofluorescence to determine whether chondrocyte proliferation was affected in the mutants. The proliferation rate was calculated by the ratio of EdU/Sox9 double positive cells divided by the total of chondrocyte cells (Sox9 positive) in humerus. It showed that the mutant chondrocyte proliferation was significantly reduced more than that of the control, with 18.4% in the control versus 14.2% in the mutant at E12.5, and 17.8% in the control versus 13.1% in the mutant at E13.5 (Fig. 4). These results uncover that Smad4 signaling is required for chondrocyte proliferation.
Figure 4.
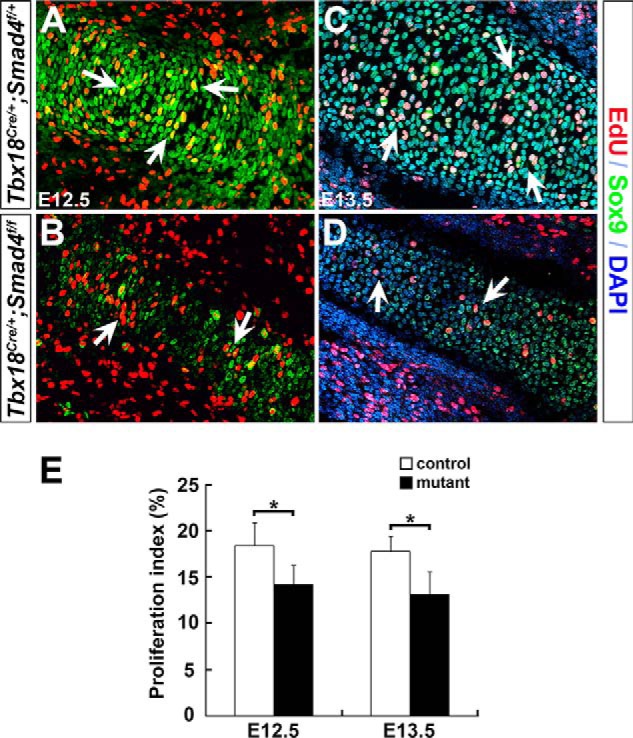
Decreased chondrocyte proliferation in Smad4 mutant mice. A–D, chondrocyte proliferation was analyzed by EdU labeling (red). Sox9 (green) was used to mark chondrocytes. Arrows indicate EdU-positive chondrocytes. E, EdU-positive cells were quantified. The proliferation rate of chondrocytes was represented by the ratio of EdU/Sox9 double positive cells normalized to Sox9 positive cells in humerus. n = 3, *, p < 0.05.
Smad4 is required for Runx2 expression in the limb
To further elucidate the molecular mechanism by which Smad4 deficiency impairs chondrocyte hypertrophy, we performed high-throughput RNA-seq to examine genes with altered expression when Smad4 is removed. Because Smad4 CKO limb defects were first detected at E12.5, and chondrocytes start the hypertrophic differentiation process at E13.5 and are largely accomplished at E14.5 (Fig. 3, A, C, E, and G), we isolated RNA at E12.5, E13.5, and E14.5 from forelimbs of mutants and controls for large-scale sequencing. Furthermore, to examine whether any molecular changes occur before E12.5, we also performed RNA-seq with E11.5 forelimb samples although no morphological abnormalities were detected.
Approximately 30 million raw reads per sample were obtained and the reads with good quality were aligned to mouse reference sequence (RefSeq) transcripts using BWA (34). In Smad4 CKO mice, 551 genes at E12.5 (Table S3) and 622 genes at E13.5 (Table S4) were down-regulated (p < 0.0001) using the DEGseq program (35), and the number increased to 2581 at E14.5 (Table S5) (Fig. 5A). As parallel, the number of up-regulated genes (p < 0.0001) was increased from 651, 775 to 3249 at E12.5 (Table S3), E13.5 (Table S4), and E14.5 (Table S5), respectively (Fig. 5B). At E11.5, very few genes (less than 10) were changed (Tables S7 and S8) and they seemed not associated with chondrocyte/limb development according to Gene Ontology (GO) analysis using DAVID 6.7 (36). At E12.5–14.5, the number of significantly changed genes appeared to reflect the degree of abnormalities between the mutant and control at corresponding stages (Fig. 2, A–F). A larger number of the overlapped genes were found between E13.5 and E14.5 (Tables S11 and S12) than E12.5 and E14.5 (Tables S13 and S14) or E12.5 and E13.5 (Tables S9 and S10) (Fig. 5, A and B). In addition, 123 genes were down-regulated (Table S15, Fig. 5A) and 30 genes were up-regulated (Table S16, Fig. 5B) at all three stages. At E12.5, the down-regulated genes are highly associated with skeletal, cartilage, and bone development (asterisks in Fig. 5C, and Tables S17 and S18). At E13.5, skeletal, cartilage, and limb/bone development are still on the top lists of term according to GO (asterisks in Fig. 5D, Tables S19 and S20). It is of importance to note that although many genes were changed at E14.5, the limb/bone, skeletal, and cartilage development are not the primary terms (Tables S21 and S22). This may suggest that the genes changed at E14.5 are secondary or may not be directly regulated by Smad4.
Figure 5.

Ablation of Smad4 in the forelimb causes severe impaired chondrocyte differentiation through RNA-seq analysis. A and B, the Venn diagrams showing the number of overlapping genes down-regulated (A) and up-regulated (B) in Tbx18Cre/+;Smad4f/f forelimb between E12.5, E13.5, and E14.5. Top biological process GO terms were determined using DAVID for differentially expressed genes in forelimbs at E12.5 (C) and E13.5 (D). X-axis is the p value for the enrichment of a GO term in the input gene list compared with genes in the whole genome. The gene numbers are listed in the dots. E, heat map of cartilage development at E12.5 and E13.5. Red and green denote up-regulated and down-regulated genes, respectively.
Of interest, we found the down-regulated genes at E12.5 and E13.5, including Acan, Col10a1, Col2a1, Fgfr3, Ihh, Lect1, Runx2, Runx3, and Sp7, are highly related to chondrocyte differentiation and hypertrophy (Fig. 5E) (11, 15, 28, 30, 37–40). In agreement with RNA-Seq analysis, qRT-PCR further confirmed that expression of Runx2, Runx3, Ihh, and Sp7 was reduced by 42, 40, 41, and 58% at E12.5, and 53, 45, 55, and 59% at E13.5 in the mutant, respectively (Fig. 6Q). We performed immunofluorescence and RNA in situ hybridization to examine expression of these genes. It showed that Runx2, Runx3, Ihh, and Sp7 were dramatically down-regulated in Smad4-deficient humerus (E12.5–13.5, Fig. 6, A–P), suggesting Runx2, Runx3, Ihh, and Sp7 may regulate hypertrophic chondrocyte differentiation as downstream of Smad4.
Figure 6.

Reduced expression of chondrocyte hypertrophy–related genes on Smad4 mutant mice. A–D, immunostaining of Runx2 on the forelimb sections from the control (A and C) and mutant (B and D) embryos at E12.5. E–P, RNA in situ hybridization staining of Ihh, Runx3, and Sp7 on forelimb sections from control (E, G, I, K, M, and O) and mutant (F, H, J, L, N, and P) embryos at E12.5 and E13.5. Arrows in A–P indicate protein or RNA expression in humerus. Q, Runx2, Runx3, Ihh, and Sp7 RNA expression levels were determined by qRT-PCR at E12.5 and E13.5 in the forelimbs. n = 3, *, p < 0.05.
Smad4 directly binds to Runx2 regulatory elements
RNA-seq analysis of Smad4 CKO forelimb revealed that most genes remain normal at E11.5, and the genes with altered expression at E14.5 are likely secondary and may not reflect direct regulation by Smad4 during limb development. We next performed ChIP-seq with E12.5 and E13.5 forelimbs to determine Smad4 direct downstream targets in chondrocyte hypertrophy development. Chromatin DNA was prepared from the WT forelimb tissues at E12.5 and E13.5 using anti-Smad4 antibody. The purified DNA was amplified for library construction and large-scale sequencing. Smad4 genome-wide occupancy was identified using MACS1.4 as described (41). Peaks within 5 kb of the transcription start site (TSS) were assigned to the nearest genes. Regulatory regions in 1213 genes were identified and potentially bound by Smad4 (Table S6). We performed GO analysis on these genes and found that regulation of chondrocyte differentiation was on the top of the list (asterisk in Fig. 7B, Table S23).
Figure 7.

Genomewide Smad4-binding sites are associated with chondrocyte differentiation. A, Venn diagrams show the number of down-regulated and up-regulated overlapping genes in E12.5 and E13.5 forelimbs analyzed by ChIP-seq and RNA-seq. B, top biological process GO terms were determined by DAVID in Smad4-binding genes. X axis is the p value for the enrichment of a GO term in the input gene list compared with genes in the whole genome. The number of genes is listed in the dots. C-1, Smad4-binding motif in the HOMER database. C-2, ChIP-seq analysis shows Smad4-binding site on the Runx2-regulatory region (binding motif was marked by gray and core sequence was labeled by red). D, ChIP-PCR verification of Smad4 binding on the Runx2-regulatory region. ChIP was performed anti-Smad4 antibody. A positive band was amplified using primers spanning the Smad4-binding site in the Runx2 promoter with input DNA (lane 1) and Smad4-ChIPed DNA (lane 2), but was not amplified with template ChIPed by IgG (lane 3). Negative control is PCR with GAPDH-CNAP1 primers. E, luciferase reporter assays of the Smad4 expression vector with various Runx2 promoter fragments. The putative Smad4-binding site is present in the 5.0-kb fragment, but not in the 3.0- and 4.2-kb fragments. *, p < 0.01.
To determine whether Smad4 directly regulated genes are associated with chondrocyte differentiation, we combined RNA-seq and ChIP-seq data (E12.5–13.5) for further integrative analysis. It revealed that 6 down-regulated genes, including Runx2 and Fgfr3, are related to cartilage development (Fig. 7A, see full list of the genes in Table S24). In contrast, the up-regulated 4 genes seem not to be associated with chondrocyte differentiation (Fig. 7A, Table S25). Consensus core sequences 5′-GTCT-3′ were called and are consistent with previously reported Smad-binding elements (42, 43). Because the Smad4 binding motif is already known in HOMER database (http://homer.ucsd.edu/homer/motif/motifDatabase.html)4 (Fig. 7, C-1), we applied HOMER motif discovery software (44) to screen for all enriched regions, and found the Smad4-binding motif (5′-TTTTGTCTGC-3′) is in the list of enrichment peaks (Fig. 7, C-2). The Smad4-enrichment peak in Runx2 is ∼4 kb upstream of TSS (two Smad-binding elements within the enriched peak), and the motif is 76 bp away from the peak midpoint. The Integrative Genomics Viewer (45) peak upstream of Runx2 TSS was shown in Fig. 7C. To verify direct binding of the Runx2 regulatory element by Smad4, we further carried out ChIP-PCR around this region with chromatin DNA immunoprecipitated by Smad4 antibody in mouse forelimbs (E12.5–13.5), with DNA ChIPed by IgG as control. The Runx2 regulatory region containing the Smad4-binding site was amplified by PCR (Fig. 7D), confirming that Smad4 directly binds to this Runx2 regulatory element during mouse skeletal development at E12.5–13.5. Furthermore, luciferase reporter assays revealed that Smad4 activates the Runx2 promoter fragment containing the Smad4-binding site (5.0 kb), but not fragments without this site (4.2 and 3.0 kb) (Fig. 7E). Taken together, these data suggest that Smad4 directly binds to Runx2 regulatory elements during mouse limb development.
Discussion
Chondrocyte hypertrophic differentiation is a precisely regulated morphogenetic process in which the chondrocytes differentiate into hypertrophic chondrocytes during skeletal development (Fig. 8). Smad4 has been shown to play important roles in the process of chondrocyte differentiation from mesenchymal cells and chondrocyte proliferation (19, 20). In this study, we showed that Smad4 is essential for chondrocyte hypertrophic differentiation. Smad4 CKO mice display limb growth defects with chondrodysplasia characterized by low chondrocyte proliferation and lack of hypertrophy in humerus. We further demonstrated that Smad4 acts as a direct transcriptional activator for Runx2 in chondrocyte differentiation.
Figure 8.
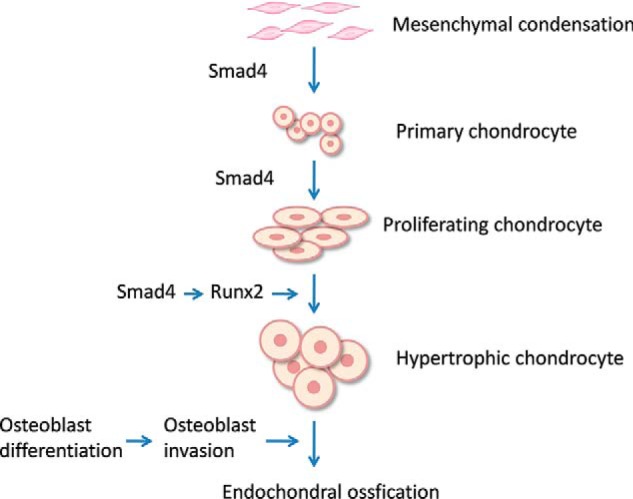
A model for the role of Smad4 in chondrocyte hypertrophy and bone development. Smad4 regulates chondrocyte differentiation and proliferation. Runx2 is required for chondrocyte hypertrophic differentiation. Smad4 controls chondrocyte hypertrophy and bone development through regulation of Runx2.
Smad4 regulates chondrocyte differentiation and hypertrophy
Previous studies showed that ablation of Smad4 with Prx1-Cre or Col2a1-Cre deleter mice resulted in defective limb formation (19, 20). Specifically, Prx1-Cre;Smad4f/f mutant mice displayed a complete loss of limb skeletal elements at E14.5 due to perturbation of mesenchymal cell aggregation, the first step of chondrocyte differentiation (20). Col2a1-Cre;Smad4f/f mutant mice displayed a mild shortened limb at postnatal day 10 with defective proliferation and differentiation of chondrocytes (19). In contrast to defects of these two mutants, limb shortening of Tbx18Cre/+;Smad4f/f mice resulted from the absence of stylopod elements because of chondrocyte hypertrophy failure, the terminal step of chondrocyte differentiation. Phenotype variations of these mutants could be explained by location and timing of distinct Cre-mediated genetic deletions in the developing limbs: Prx1-Cre is expressed in the early limb mesenchyme at E9.5 and it introduces genetic deletions in both chondrocytes and perichondrium (46). Col2a1-Cre is expressed in the differentiating chondrocytes in limbs (47). Tbx18Cre/+ is detected in the limb bud as early as E9.5 and mediates recombination in chondrocytes within the proximal domain of the limb (Fig. S1, C–F). The divergent defects of Smad4 mutants suggest multiple and temporospatially distinct functions of Smad4 in limb formation.
In Smad4 CKO embryos, chondrocyte differentiation is severely perturbed. Cartilage elements (humerus) in the mutant limb were shorter and smaller (Fig. 2, A–F) with decreased Col2a1, Acan, and Lect1 expression (Fig. 2, G–Q). The RNA-seq assay also showed down-regulated Sox9, Sox5, and Sox6 (Fig. 5E). These genes are key transcription factors for chondrocyte differentiation (48–51). Disruption of Sox9 leads to impaired chondrocyte differentiation and endochondral bone formation (50, 52). It was shown that Sox9 directly regulates Col2a1 and Acan and promotes differentiation of mesenchymal cells to chondrocytes (38, 53, 54). Smad4 may activate Col2a1 and Acan via Sox9 during chondrocyte development and this conception needs to be further determined.
Chondrocyte hypertrophy is critical for endochondral ossification. Disruption of Smad4 in Tbx18Cre/+ lineage results in complete loss of chondrocyte hypertrophy (Fig. 3, A–H) and failure of endochondral ossification in humerus (Fig. 1, K and L), suggesting an essential role of Smad4 for chondrocyte hypertrophy during long bone development. This notion was supported by the observation of the absent hypertrophic chondrocytes and missing expression of Col10a1 (specific marker for hypertrophic chondrocytes (30, 31)) and Panx3 (specific marker for pre-hypertrophic chondrocytes and hypertrophic chondrocytes (32, 33)) in Smad4 CKO humerus (Fig. 3, I–V).
Runx2 acts as a crucial mediator of Smad4 signaling in chondrocyte hypertrophy
In this study, we applied RNA-seq and ChIP-seq to decipher how Smad4 controls chondrocyte hypertrophy during mouse limb skeletogenesis. GO analysis of the RNA-seq data showed the down-regulated genes are highly associated with skeletal, cartilage, and bone development at E12.5 and E13.5 (Fig. 5, C and D). RNA-seq revealed that the down-regulated genes at E12.5 and E13.5 included Col10a1, Fgfr3, Ihh, Runx2, Runx3, and Sp7 in Smad4 CKO mice. These down-regulated genes are highly related to chondrocyte hypertrophy. qRT-PCR and immunofluorescence verified these genes (Figs. 3 and 6). Runx2 is a critical mediator of chondrocyte hypertrophy (16, 55–57) and it is expressed in prehypertrophic and hypertrophic chondrocytes during skeletal development. Inactivation of Runx2 causes the absence of ossification and hypertrophic chondrocytes (16, 55, 58, 59), similar to Smad4 CKO mice. Col10a1 is a specific marker for hypertrophic chondrocytes, and Runx2 directly regulates the transcriptional activity of Col10a1 (30, 31). Ihh, a direct target gene of Runx2, is expressed in prehypertrophic chondrocytes and is essential for chondrocyte hypertrophy (10, 11, 15). Sp7 is a zinc finger transcription factor regulating bone formation at the hypertrophic stage in vivo (40). Runx2 specifically binds to the regulatory element on Sp7 promoter (60). Collectively, Runx2, Runx3, Col10a1, Fgfr3, Ihh, and Sp7 may act as key downstream genes of Smad4 signaling involved in chondrocyte hypertrophy.
The integrated RNA-seq and ChIP-seq data analyses identified potential targets of Smad4. Six down-regulated genes highly related to bone formation, including Runx2 and Fgfr3, were uncovered (55, 61). Fgfr3 is a negative regulator for bone growth (61). Fgfr3 was shown to restrain chondrocyte proliferation and limit osteogenesis. Fgfr3−/− mutant mice displayed prolonged endochondral bone growth accompanied by expansion of proliferating and hypertrophic chondrocytes in the cartilaginous growth plate. Given that Fgfr3 controls endochondral ossification by a negative regulatory mechanism, and Fgfr3−/− bones display prolonged defects compared with Smad4 CKO mice (Fig. 1, K–R), the down-regulated Fgfr3 may not be the main cause of Smad4 CKO limb defects, although Smad4/Fgfr3 signaling cascade could play some unknown essential roles for limb skeletal development. In contrast, Runx2 positively regulates chondrocyte hypertrophy. Runx2 overexpression in chondrocytes restores chondrocyte hypertrophy in Runx2-deficient mice (56, 57). Taken together, our data support that Runx2 acts as a key downstream gene of Smad4 given the critical role of Runx2 in the skeletal hypertrophic development.
Runx2 has been suggested as a common downstream gene of TGF-β/BMP signaling in previous studies, although the mechanisms underlying this process were largely unknown before (22, 62). In this regard, our study first demonstrated that Smad4 transcriptionally activates Runx2 expression by directly binding to Runx2 regulatory elements during chondrocyte hypertrophy, suggesting an important role of Runx2 in cartilage growth and bone formation.
Smad4 is required for chondrocyte proliferation
Chondrocyte proliferation is essential for cartilage growth (11, 63, 64). BMP signaling was shown to be important for chondrocyte proliferation (14, 65). Reduced osteoblast and chondrocyte proliferation was detected in Smad4 mutant mice in the previous studies (18, 19). Here we found that ablation of Smad4 with Tbx18Cre/+ also led to a dramatic reduction in chondrocyte proliferation as early as E12.5 in the forelimb (humerus). The reduced chondrocyte proliferation should contribute to the short cartilage elements in Smad4 CKO limbs (Fig. 2, A–F). Smad4 promotes Ihh expression (Figs. 5E and 6, E–H and Q), and Ihh plays a critical role in regulating chondrocyte proliferation during skeletal development (11, 64, 66). Ihh-deficient mice display severe reduction in skeletal growth and decreased chondrocyte proliferation (11). Ectopic activation of Ihh signaling promotes chondrocyte proliferation during cartilage development (64). Given that Ihh was significantly down-regulated in Smad4 CKO humerus, it is potential that Ihh is one of the key downstream effectors of Smad4 signaling pathways in regulating chondrocyte proliferation.
In summary, our study uncovered divergent, crucial roles of Smad4 in chondrocyte differentiation, especially in hypertrophic differentiation during limb development. We demonstrated that Smad4 controls chondrocyte hypertrophic differentiation through direct regulation of Runx2. Ablation of Smad4 within the proximal domain of limbs results in chondrodysplasia due to reduced chondrocyte proliferation, impaired chondrocyte differentiation, lack of chondrocyte hypertrophy, and failure of ossification in humerus. As the study did not use hypertrophic chondrocyte-specific Cre (e.g. Col10a-Cre) to delete Smad4 from hypertrophic chondrocytes in the limb, it is uncertain whether the impaired chondrogenesis also affects chondrocyte hypertrophy in Smad4 CKO mice. ChIP-seq assay uncovered a previously unknown critical role of Runx2 as a direct downstream target of Smad4 signaling in cartilage and bone formation. The Smad4–Runx2 regulatory pathways provide important mechanisms to understand hypertrophic differentiation of limb development and etiology of chondrodysplasia in mammals.
Experimental procedures
Animals
All animal experiments conformed to the United States National Institutes of Health guidelines and were conducted in accordance with the protocol approved by the Institutional Animal Care and Use Committee (IACUC) at Icahn School of Medicine at Mount Sinai (Permit LA09-00494). Smad4-floxed (denoted as Smad4f/f), Rosa26:lacZ (Rosa26lacZ/+), Rosa26:GFP (Rosa26GFP/+), Tbx18:Cre (Tbx18Cre/+), and Tbx18:nlacZ (Tbx18nlacZ/+) mice were described previously (24–27, 67). Mouse tails or yolk sac tissues were collected for genotyping.
Skeletal analysis
Calcified tissue was stained with Alizarin red (Sigma) and cartilage was stained with Alcian blue (Sigma) with a standard procedure (68). For E15.5–18.5 embryos, the skin and internal organs were removed. Skeletons were fixed in 95% ethanol overnight and stained with 0.015% Alcian blue followed by digestion in 1% potassium chloride for 2 days, and then stained with 0.005% Alizarin red for 3 days. E12.5–14.5 embryos were fixed in ethanol overnight, stained with 0.009% Alcian blue for 3 days, and cleared by benzyl alcohol/benzyl benzoate. Images of the stained skeletons and cartilages were taken under a Leica stereomicroscope.
X-Gal staining
Embryos were fixed with 4% paraformaldehyde (PFA) for 30 min at 4 °C. The fixed embryos were washed twice with PBS and then stained with X-Gal staining solution (5 mm potassium ferricyanide, 5 mm potassium ferrocyanide, 2 mm MgCl2, 1 mg/ml X-Gal) for 12 h at room temperature.
H&E staining and Alcian blue staining
Mouse embryos were fixed with 4% PFA overnight at 4 °C and then dehydrated in an ascending ethanol series followed by two changes of 100% xylene. The tissues were embedded in liquid paraffin and placed on a cold plate for solidification. Sections were cut into 6-μm thickness on a microtome and stained with hematoxylin and eosin (H&E). For Alcian blue staining, the sections were stained in 1% Alcian blue solution (in 3% acetic acid) for 20 min and counterstained in nuclear fast red solution (Eng Scientific) for 10 min.
Immunofluorescence
Mouse limbs were fixed in 4% PFA for 30 min and embedded in Optimal Cutting Temperature compound (Tissue-Tek). Frozen samples were cut into 6-μm thickness. Sections were incubated for specific primary antibodies for 1 h at room temperature. The primary antibodies used in this study were: rabbit anti-Smad4 (1:100, Millipore), rabbit anti-Sox9 (1:300, Millipore), and mouse anti-Runx2 (1:200, Abcam). Alexa Fluor 488- or 594-conjugated secondary antibodies (1:500; Invitrogen) were used to detect the corresponding primary antibodies. Sections were then counterstained with 4′,6-diamidino-2-phenylindole, and examined by fluorescence.
RNA in situ hybridization
Whole mount and section RNA in situ hybridization was performed as described previously (69). Col2a1, Col10a1, Ihh, and Runx3 probes were obtained from Dr. John Cobb (University of Calgary, Canada). Fgf4, Fgf8, Shh, Hoxa13, and Hoxd11 probes were from Dr. Brian Harfe (University of Florida). Ezh2, Hoxa11, Hand2, and Gli3 probes were from Dr. Sevan Hopyan (University of Toronto, Canada). Fgf10 probe was obtained from Dr. Juan Jose Sanz-Ezquerro (National Center for Biotechnology, Spain). Raldh2 probe was from Dr. Gregg Duester (Sanford-Burnham Medical Research Institute) and the Cyp26b1 probe was from Dr. Martin Petkovich (Queen's University, Canada). Chondromodulin 1 (Lect1), Osterix (Sp7), and Pannexin 3 (Panx3) cDNA fragments were generated by RT-PCR. Antisense probes were synthesized with RNA polymerases (Promega) and DIG RNA Labeling Mix (Roche Applied Science). Primer sequences for RNA probes are listed in Table S1.
Cell proliferation assay
Click-iT EdU Cell Proliferation Assay Kit (Invitrogen) was used for the cell proliferation assay according to the manufacturer's instructions. Pregnant mice were intraperitoneally injected with 10 mm EdU in PBS (5 mg/100 g body weight). Embryos were collected 4 h after EdU injection and fixed with 4% PFA for 30 min at 4 °C and washed with PBS twice. Samples were embedded in Optimal Cutting Temperature compound and sectioned into 6 μm pieces. Frozen sections were incubated with Click-iT Reaction mixture for 30 min at room temperature. Slides were analyzed under a fluorescence microscope.
qRT-PCR
Total RNA was isolated from the forelimbs of mouse embryos using TRIzol reagent (Invitrogen). First strand cDNA was synthesized with QuantiTect Reverse Transcription Kit (Qiagen). qRT-PCR was carried out using the StepOne Plus PCR system (Applied Biosystems) and SYBR Green (Qiagen). The mRNA expression level was normalized to β-actin. Statistical analysis was performed by t test and p < 0.05 was considered significant. Primer sequences for qRT-PCR are listed in Table S2.
RNA-Seq (RNA-seq)
Total RNA was prepared using TRIzol reagent (Invitrogen) and RNA quality was determined using an Agilent Bioanalyser. RNA-seq Sample Preparation Kit (Illumina) was used to prepare mRNA and cDNA following the manufacturer's protocol. RNA-Seq (RNA-seq) was performed as described previously using a HiSeq 2500 (Illumina) (69).
ChIP-seq and ChIP-PCR
Mouse forelimbs at E12.5 and E13.5 were collected in ice-cold PBS and cross-linked immediately in 1% formaldehyde, PBS at room temperature for 15 min. The forelimbs were washed twice with PBS, and then crushed in Lysis Buffer (50 mm HEPES, pH 8.0, 140 mm NaCl, 1 mm EDTA, 10% glycerol, 0.5% IGEPAL-CA630, 0.25% Triton X-100) with 1 mm PMSF and protease inhibitor mixture (Roche Applied Science) on a Benchmark BeadBug Homogenizer. The chromatin was resuspended in the Shearing Buffer (0.1% SDS, 1 mm EDTA, 10 mm Tris-HCl, pH 8.0) and sonicated to 100–300 bp by a Covaris S220 Focused ultrasonicator. Dynabeads® Magnetic Protein A and G beads (Invitrogen) were pre-blocked with SEA BLOCK Blocking Buffer (Thermo), and the chromatin was pre-cleared with Protein A and G beads. Smad4 ChIP was performed using anti-rabbit Smad4 antibody (04-1033, Millipore) and IgG control antibody (ab136636, Abcam). In brief, 10 μl of Protein A and 10 μl of Protein G beads were incubated with 1 ml of sheared chromatin and 10 μl of antibody overnight at 4 °C. The beads were then washed once with Low Salt Wash Buffer (2 mm EDTA, 20 mm HEPES, pH 8.0, 150 mm NaCl, 0.1% SDS, 1% Triton X-100), High Salt Wash Buffer (2 mm EDTA, 20 mm HEPES, pH 8.0, 500 mm NaCl, 0.1% SDS, 1% Triton X-100), and LiCl Wash Buffer (250 mm LiCl, 1% IGEPAL-CA630, 1% deoxycholic acid sodium salt, 1 mm EDTA, 10 mm Tris, pH 8.0), respectively. The beads were eluted in 100 μl of Elution Buffer (20 mm Tris-HCl, pH 7.5, 5 mm EDTA, 50 mm NaCl, 1% SDS). Chromatin DNA was reverse cross-linked by adding 1 μl of Proteinase K (20 mg/ml) and incubation at 65 °C for 4 h. DNA sample was purified by Qiagen MinElute PCR Purification Kit, and was amplified with an Illumina ChIP-seq DNA Sample Prep Kit (IP-102-1001). DNA libraries were sent for sequencing with an Illumina HiSeq 2500 sequencer. ChIP-PCR was performed as described previously (69). Primers used for ChIP-PCR are listed in Table S1.
Luciferase reporter assay
ATDC5 cells were transfected with a mixture of luciferase reporter plasmid containing Runx2 promoter fragments (200 ng), Smad4 expression vector (200 ng), and Renilla plasmid (5 ng) using Lipofectamine 3000 (Invitrogen) according to the manufacturer's instructions. After 48 h of incubation, cells were lysed and luciferase activity was measured using a dual luciferase reporter assay system (Promega) and normalized to Renilla activity. The pGL3-TK vector (Promega) was used as control. Primers used to generate the 3.0-, 4.2-, and 5.0-kb Runx2 promoter fragments are listed in Table S1.
Author contributions
J. Y. and C.-L. C. conceptualization; J. Y. and C.-L. C. data curation; J. Y., C. W., N. S., X. C., W. Z., and C.-L. C. formal analysis; J. Y. and C.-L. C. supervision; J. Y. and C.-L. C. funding acquisition; J. Y., J. L., J. H., L. Z., C. W., N. S., X. C., W. Z., and C.-L. C. investigation; J. Y. writing-original draft; J. Y. and C.-L. C. writing-review and editing.
Supplementary Material
Acknowledgments
We thank Dr. Chuxia Deng (National Institutes of Health, NIDDK) for providing Smad4f/f mice and Dr. John Cobb for providing RNA in situ probes (University of Calgary, Canada).
This work was supported by National Institutes of Health Grants 1R01HL131735, 1R01HL095810, 1R56HL129807, and 1K02HL094688, American Heart Association Grants 15GRNT25710153 and 0855808D, and March of Dimes Foundation Grant 5-FY07-642 (to C. L. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1 and S2 and Tables S1–S25.
The ChIP-Seq and RNA-Seq data can be accessed through the NCBI Gene Expression Omnibus (GEO) Repository using accession numbers GSE114081 and GSE114079, respectively.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- Runx2
- runt-related transcription factor 2
- CDMP1
- cartilage-derived morphogenetic protein 1
- Ihh
- Indian hedgehog
- BMP
- bone morphogenetic protein
- R-Smads
- receptor-activated Smads
- qRT
- quantitative RT
- GO
- gene ontology
- TSS
- transcription start site
- X-Gal
- 5-bromo-4-chloro-3-indolyl β-d-galactoside
- PFA
- paraformaldehyde
- EdU
- 5-ethynyl-2′-deoxyuridine
- ChIP-seq
- ChIP-sequencing.
References
- 1. Rousseau, F., Bonaventure, J., Legeai-Mallet, L., Pelet, A., Rozet, J. M., Maroteaux, P., Le Merrer, M., and Munnich, A. (1994) Mutations in the gene encoding fibroblast growth factor receptor-3 in achondroplasia. Nature 371, 252–254 10.1038/371252a0 [DOI] [PubMed] [Google Scholar]
- 2. Warman, M. L., Abbott, M., Apte, S. S., Hefferon, T., McIntosh, I., Cohn, D. H., Hecht, J. T., Olsen, B. R., and Francomano, C. A. (1993) A type X collagen mutation causes Schmid metaphyseal chondrodysplasia. Nat. Genet. 5, 79–82 10.1038/ng0993-79 [DOI] [PubMed] [Google Scholar]
- 3. Mundlos, S., Otto, F., Mundlos, C., Mulliken, J. B., Aylsworth, A. S., Albright, S., Lindhout, D., Cole, W. G., Henn, W., Knoll, J. H., Owen, M. J., Mertelsmann, R., Zabel, B. U., and Olsen, B. R. (1997) Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell 89, 773–779 10.1016/S0092-8674(00)80260-3 [DOI] [PubMed] [Google Scholar]
- 4. Thomas, J. T., Lin, K., Nandedkar, M., Camargo, M., Cervenka, J., and Luyten, F. P. (1996) A human chondrodysplasia due to a mutation in a TGF-β superfamily member. Nat. Genet. 12, 315–317 10.1038/ng0396-315 [DOI] [PubMed] [Google Scholar]
- 5. Karsenty, G., and Wagner, E. F. (2002) Reaching a genetic and molecular understanding of skeletal development. Dev. Cell 2, 389–406 10.1016/S1534-5807(02)00157-0 [DOI] [PubMed] [Google Scholar]
- 6. Provot, S., and Schipani, E. (2005) Molecular mechanisms of endochondral bone development. Biochem. Biophys. Res. Commun. 328, 658–665 10.1016/j.bbrc.2004.11.068 [DOI] [PubMed] [Google Scholar]
- 7. de Crombrugghe, B., Lefebvre, V., and Nakashima, K. (2001) Regulatory mechanisms in the pathways of cartilage and bone formation. Curr. Opin. Cell Biol. 13, 721–727 10.1016/S0955-0674(00)00276-3 [DOI] [PubMed] [Google Scholar]
- 8. de Crombrugghe, B., Lefebvre, V., Behringer, R. R., Bi, W., Murakami, S., and Huang, W. (2000) Transcriptional mechanisms of chondrocyte differentiation. Matrix Biol. 19, 389–394 10.1016/S0945-053X(00)00094-9 [DOI] [PubMed] [Google Scholar]
- 9. Mackie, E. J., Tatarczuch, L., and Mirams, M. (2011) The skeleton: a multi-functional complex organ: the growth plate chondrocyte and endochondral ossification. J. Endocrinol 211, 109–121 10.1530/JOE-11-0048 [DOI] [PubMed] [Google Scholar]
- 10. Mak, K. K., Kronenberg, H. M., Chuang, P. T., Mackem, S., and Yang, Y. (2008) Indian hedgehog signals independently of PTHrP to promote chondrocyte hypertrophy. Development 135, 1947–1956 10.1242/dev.018044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. St-Jacques, B., Hammerschmidt, M., and McMahon, A. P. (1999) Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 13, 2072–2086 10.1101/gad.13.16.2072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yoon, B. S., Pogue, R., Ovchinnikov, D. A., Yoshii, I., Mishina, Y., Behringer, R. R., and Lyons, K. M. (2006) BMPs regulate multiple aspects of growth-plate chondrogenesis through opposing actions on FGF pathways. Development 133, 4667–4678 10.1242/dev.02680 [DOI] [PubMed] [Google Scholar]
- 13. Yoon, B. S., Ovchinnikov, D. A., Yoshii, I., Mishina, Y., Behringer, R. R., and Lyons, K. M. (2005) Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc. Natl. Acad. Sci. U.S.A. 102, 5062–5067 10.1073/pnas.0500031102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shu, B., Zhang, M., Xie, R., Wang, M., Jin, H., Hou, W., Tang, D., Harris, S. E., Mishina, Y., O'Keefe, R. J., Hilton, M. J., Wang, Y., and Chen, D. (2011) BMP2, but not BMP4, is crucial for chondrocyte proliferation and maturation during endochondral bone development. J. Cell Sci. 124, 3428–3440 10.1242/jcs.083659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yoshida, C. A., Yamamoto, H., Fujita, T., Furuichi, T., Ito, K., Inoue, K., Yamana, K., Zanma, A., Takada, K., Ito, Y., and Komori, T. (2004) Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev. 18, 952–963 10.1101/gad.1174704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim, I. S., Otto, F., Zabel, B., and Mundlos, S. (1999) Regulation of chondrocyte differentiation by Cbfa1. Mech. Dev. 80, 159–170 10.1016/S0925-4773(98)00210-X [DOI] [PubMed] [Google Scholar]
- 17. Derynck, R., and Zhang, Y. E. (2003) Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 425, 577–584 10.1038/nature02006 [DOI] [PubMed] [Google Scholar]
- 18. Tan, X., Weng, T., Zhang, J., Wang, J., Li, W., Wan, H., Lan, Y., Cheng, X., Hou, N., Liu, H., Ding, J., Lin, F., Yang, R., Gao, X., Chen, D., and Yang, X. (2007) Smad4 is required for maintaining normal murine postnatal bone homeostasis. J. Cell Sci. 120, 2162–2170 10.1242/jcs.03466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang, J., Tan, X., Li, W., Wang, Y., Wang, J., Cheng, X., and Yang, X. (2005) Smad4 is required for the normal organization of the cartilage growth plate. Dev. Biol. 284, 311–322 10.1016/j.ydbio.2005.05.036 [DOI] [PubMed] [Google Scholar]
- 20. Bénazet, J. D., Pignatti, E., Nugent, A., Unal, E., Laurent, F., and Zeller, R. (2012) Smad4 is required to induce digit ray primordia and to initiate the aggregation and differentiation of chondrogenic progenitors in mouse limb buds. Development 139, 4250–4260 10.1242/dev.084822 [DOI] [PubMed] [Google Scholar]
- 21. Salazar, V. S., Zarkadis, N., Huang, L., Norris, J., Grimston, S. K., Mbalaviele, G., and Civitelli, R. (2013) Embryonic ablation of osteoblast Smad4 interrupts matrix synthesis in response to canonical Wnt signaling and causes an osteogenesis-imperfecta-like phenotype. J. Cell Sci. 126, 4974–4984 10.1242/jcs.131953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee, K. S., Kim, H. J., Li, Q. L., Chi, X. Z., Ueta, C., Komori, T., Wozney, J. M., Kim, E. G., Choi, J. Y., Ryoo, H. M., and Bae, S. C. (2000) Runx2 is a common target of transforming growth factor β1 and bone morphogenetic protein 2, and cooperation between Runx2 and Smad5 induces osteoblast-specific gene expression in the pluripotent mesenchymal precursor cell line C2C12. Mol. Cell. Biol. 20, 8783–8792 10.1128/MCB.20.23.8783-8792.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang, Y. W., Yasui, N., Ito, K., Huang, G., Fujii, M., Hanai, J., Nogami, H., Ochi, T., Miyazono, K., and Ito, Y. (2000) A RUNX2/PEBP2α A/CBFA1 mutation displaying impaired transactivation and Smad interaction in cleidocranial dysplasia. Proc. Natl. Acad. Sci. U.S.A. 97, 10549–10554 10.1073/pnas.180309597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yan, J., Zhang, L., Xu, J., Sultana, N., Hu, J., Cai, X., Li, J., Xu, P. X., and Cai, C. L. (2014) Smad4 regulates ureteral smooth muscle cell differentiation during mouse embryogenesis. PLoS ONE 9, e104503 10.1371/journal.pone.0104503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Soriano, P. (1999) Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21, 70–71 10.1038/5007 [DOI] [PubMed] [Google Scholar]
- 26. Madisen, L., Zwingman, T. A., Sunkin, S. M., Oh, S. W., Zariwala, H. A., Gu, H., Ng, L. L., Palmiter, R. D., Hawrylycz, M. J., Jones, A. R., Lein, E. S., and Zeng, H. (2010) A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133–140 10.1038/nn.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang, X., Li, C., Herrera, P. L., and Deng, C. X. (2002) Generation of Smad4/Dpc4 conditional knockout mice. Genesis 32, 80–81 10.1002/gene.10029 [DOI] [PubMed] [Google Scholar]
- 28. Shukunami, C., Iyama, K., Inoue, H., and Hiraki, Y. (1999) Spatiotemporal pattern of the mouse chondromodulin-I gene expression and its regulatory role in vascular invasion into cartilage during endochondral bone formation. Int. J. Dev. Biol. 43, 39–49 [PubMed] [Google Scholar]
- 29. Jeon, J., Oh, H., Lee, G., Ryu, J. H., Rhee, J., Kim, J. H., Chung, K. H., Song, W. K., Chun, C. H., and Chun, J. S. (2011) Cytokine-like 1 knock-out mice (Cytl1−/−) show normal cartilage and bone development but exhibit augmented osteoarthritic cartilage destruction. J. Biol. Chem. 286, 27206–27213 10.1074/jbc.M111.218065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zheng, Q., Zhou, G., Morello, R., Chen, Y., Garcia-Rojas, X., and Lee, B. (2003) Type X collagen gene regulation by Runx2 contributes directly to its hypertrophic chondrocyte-specific expression in vivo. J. Cell Biol. 162, 833–842 10.1083/jcb.200211089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li, F., Lu, Y., Ding, M., Napierala, D., Abbassi, S., Chen, Y., Duan, X., Wang, S., Lee, B., and Zheng, Q. (2011) Runx2 contributes to murine Col10a1 gene regulation through direct interaction with its cis-enhancer. J. Bone Miner. Res. 26, 2899–2910 10.1002/jbmr.504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bond, S. R., Lau, A., Penuela, S., Sampaio, A. V., Underhill, T. M., Laird, D. W., and Naus, C. C. (2011) Pannexin 3 is a novel target for Runx2, expressed by osteoblasts and mature growth plate chondrocytes. J. Bone Miner. Res. 26, 2911–2922 10.1002/jbmr.509 [DOI] [PubMed] [Google Scholar]
- 33. Iwamoto, T., Nakamura, T., Doyle, A., Ishikawa, M., de Vega, S., Fukumoto, S., and Yamada, Y. (2010) Pannexin 3 regulates intracellular ATP/cAMP levels and promotes chondrocyte differentiation. J. Biol. Chem. 285, 18948–18958 10.1074/jbc.M110.127027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li, H., and Durbin, R. (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang, L., Feng, Z., Wang, X., Wang, X., and Zhang, X. (2010) DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26, 136–138 10.1093/bioinformatics/btp612 [DOI] [PubMed] [Google Scholar]
- 36. Huang da, W., Sherman, B. T., and Lempicki, R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- 37. Domowicz, M. S., Cortes, M., Henry, J. G., and Schwartz, N. B. (2009) Aggrecan modulation of growth plate morphogenesis. Dev. Biol. 329, 242–257 10.1016/j.ydbio.2009.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lefebvre, V., Huang, W., Harley, V. R., Goodfellow, P. N., and de Crombrugghe, B. (1997) SOX9 is a potent activator of the chondrocyte-specific enhancer of the proα1(II) collagen gene. Mol. Cell. Biol. 17, 2336–2346 10.1128/MCB.17.4.2336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Iwata, T., Chen, L., Li, C., Ovchinnikov, D. A., Behringer, R. R., Francomano, C. A., and Deng, C. X. (2000) A neonatal lethal mutation in FGFR3 uncouples proliferation and differentiation of growth plate chondrocytes in embryos. Hum. Mol. Genet. 9, 1603–1613 10.1093/hmg/9.11.1603 [DOI] [PubMed] [Google Scholar]
- 40. Nishimura, R., Wakabayashi, M., Hata, K., Matsubara, T., Honma, S., Wakisaka, S., Kiyonari, H., Shioi, G., Yamaguchi, A., Tsumaki, N., Akiyama, H., and Yoneda, T. (2012) Osterix regulates calcification and degradation of chondrogenic matrices through matrix metalloproteinase 13 (MMP13) expression in association with transcription factor Runx2 during endochondral ossification. J. Biol. Chem. 287, 33179–33190 10.1074/jbc.M111.337063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Feng, J., Liu, T., Qin, B., Zhang, Y., and Liu, X. S. (2012) Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 7, 1728–1740 10.1038/nprot.2012.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Qin, H., Chan, M. W., Liyanarachchi, S., Balch, C., Potter, D., Souriraj, I. J., Cheng, A. S., Agosto-Perez, F. J., Nikonova, E. V., Yan, P. S., Lin, H. J., Nephew, K. P., Saltz, J. H., Showe, L. C., Huang, T. H., and Davuluri, R. V. (2009) An integrative ChIP-chip and gene expression profiling to model SMAD regulatory modules. BMC Syst. Biol. 3, 73 10.1186/1752-0509-3-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jonk, L. J., Itoh, S., Heldin, C. H., ten Dijke, P., and Kruijer, W. (1998) Identification and functional characterization of a Smad binding element (SBE) in the JunB promoter that acts as a transforming growth factor-β, activin, and bone morphogenetic protein-inducible enhancer. J. Biol. Chem. 273, 21145–21152 10.1074/jbc.273.33.21145 [DOI] [PubMed] [Google Scholar]
- 44. Heinz, S., Benner, C., Spann, N., Bertolino, E., Lin, Y. C., Laslo, P., Cheng, J. X., Murre, C., Singh, H., and Glass, C. K. (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., and Mesirov, J. P. (2011) Integrative genomics viewer. Nat. Biotechnol 29, 24–26 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Logan, M., Martin, J. F., Nagy, A., Lobe, C., Olson, E. N., and Tabin, C. J. (2002) Expression of Cre recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis 33, 77–80 10.1002/gene.10092 [DOI] [PubMed] [Google Scholar]
- 47. Ovchinnikov, D. A., Deng, J. M., Ogunrinu, G., and Behringer, R. R. (2000) Col2a1-directed expression of Cre recombinase in differentiating chondrocytes in transgenic mice. Genesis 26, 145–146 10.1002/(SICI)1526-968X(200002)26:2<145::AID-GENE14>3.0.CO;2-C [DOI] [PubMed] [Google Scholar]
- 48. Leung, V. Y., Gao, B., Leung, K. K., Melhado, I. G., Wynn, S. L., Au, T. Y., Dung, N. W., Lau, J. Y., Mak, A. C., Chan, D., and Cheah, K. S. (2011) SOX9 governs differentiation stage-specific gene expression in growth plate chondrocytes via direct concomitant transactivation and repression. PLoS Genet. 7, e1002356 10.1371/journal.pgen.1002356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Akiyama, H., Chaboissier, M. C., Martin, J. F., Schedl, A., and de Crombrugghe, B. (2002) The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 16, 2813–2828 10.1101/gad.1017802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bi, W., Deng, J. M., Zhang, Z., Behringer, R. R., and de Crombrugghe, B. (1999) Sox9 is required for cartilage formation. Nat. Genet. 22, 85–89 10.1038/8792 [DOI] [PubMed] [Google Scholar]
- 51. Lefebvre, V., Li, P., and de Crombrugghe, B. (1998) A new long form of Sox5 (L-Sox5), Sox6, and Sox9 are coexpressed in chondrogenesis and cooperatively activate the type II collagen gene. EMBO J. 17, 5718–5733 10.1093/emboj/17.19.5718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Akiyama, H., Stadler, H. S., Martin, J. F., Ishii, T. M., Beachy, P. A., Nakamura, T., and de Crombrugghe, B. (2007) Misexpression of Sox9 in mouse limb bud mesenchyme induces polydactyly and rescues hypodactyly mice. Matrix Biol. 26, 224–233 10.1016/j.matbio.2006.12.002 [DOI] [PubMed] [Google Scholar]
- 53. Bell, D. M., Leung, K. K., Wheatley, S. C., Ng, L. J., Zhou, S., Ling, K. W., Sham, M. H., Koopman, P., Tam, P. P., and Cheah, K. S. (1997) SOX9 directly regulates the type-II collagen gene. Nat. Genet. 16, 174–178 10.1038/ng0697-174 [DOI] [PubMed] [Google Scholar]
- 54. Sekiya, I., Tsuji, K., Koopman, P., Watanabe, H., Yamada, Y., Shinomiya, K., Nifuji, A., and Noda, M. (2000) SOX9 enhances aggrecan gene promoter/enhancer activity and is up-regulated by retinoic acid in a cartilage-derived cell line, TC6. J. Biol. Chem. 275, 10738–10744 10.1074/jbc.275.15.10738 [DOI] [PubMed] [Google Scholar]
- 55. Inada, M., Yasui, T., Nomura, S., Miyake, S., Deguchi, K., Himeno, M., Sato, M., Yamagiwa, H., Kimura, T., Yasui, N., Ochi, T., Endo, N., Kitamura, Y., Kishimoto, T., and Komori, T. (1999) Maturational disturbance of chondrocytes in Cbfa1-deficient mice. Dev. Dyn. 214, 279–290 10.1002/(SICI)1097-0177(199904)214:4<279::AID-AJA1>3.0.CO;2-W [DOI] [PubMed] [Google Scholar]
- 56. Takeda, S., Bonnamy, J. P., Owen, M. J., Ducy, P., and Karsenty, G. (2001) Continuous expression of Cbfa1 in nonhypertrophic chondrocytes uncovers its ability to induce hypertrophic chondrocyte differentiation and partially rescues Cbfa1-deficient mice. Genes Dev. 15, 467–481 10.1101/gad.845101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Enomoto, H., Enomoto-Iwamoto, M., Iwamoto, M., Nomura, S., Himeno, M., Kitamura, Y., Kishimoto, T., and Komori, T. (2000) Cbfa1 is a positive regulatory factor in chondrocyte maturation. J. Biol. Chem. 275, 8695–8702 10.1074/jbc.275.12.8695 [DOI] [PubMed] [Google Scholar]
- 58. Komori, T., Yagi, H., Nomura, S., Yamaguchi, A., Sasaki, K., Deguchi, K., Shimizu, Y., Bronson, R. T., Gao, Y. H., Inada, M., Sato, M., Okamoto, R., Kitamura, Y., Yoshiki, S., and Kishimoto, T. (1997) Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 89, 755–764 10.1016/S0092-8674(00)80258-5 [DOI] [PubMed] [Google Scholar]
- 59. Otto, F., Thornell, A. P., Crompton, T., Denzel, A., Gilmour, K. C., Rosewell, I. R., Stamp, G. W., Beddington, R. S., Mundlos, S., Olsen, B. R., Selby, P. B., and Owen, M. J. (1997) Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 89, 765–771 10.1016/S0092-8674(00)80259-7 [DOI] [PubMed] [Google Scholar]
- 60. Nishio, Y., Dong, Y., Paris, M., O'Keefe, R. J., Schwarz, E. M., and Drissi, H. (2006) Runx2-mediated regulation of the zinc finger Osterix/Sp7 gene. Gene 372, 62–70 10.1016/j.gene.2005.12.022 [DOI] [PubMed] [Google Scholar]
- 61. Deng, C., Wynshaw-Boris, A., Zhou, F., Kuo, A., and Leder, P. (1996) Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell 84, 911–921 10.1016/S0092-8674(00)81069-7 [DOI] [PubMed] [Google Scholar]
- 62. Lee, K. S., Hong, S. H., and Bae, S. C. (2002) Both the Smad and p38 MAPK pathways play a crucial role in Runx2 expression following induction by transforming growth factor-β and bone morphogenetic protein. Oncogene 21, 7156–7163 10.1038/sj.onc.1205937 [DOI] [PubMed] [Google Scholar]
- 63. Terpstra, L., Prud'homme, J., Arabian, A., Takeda, S., Karsenty, G., Dedhar, S., and St-Arnaud, R. (2003) Reduced chondrocyte proliferation and chondrodysplasia in mice lacking the integrin-linked kinase in chondrocytes. J. Cell Biol. 162, 139–148 10.1083/jcb.200302066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Long, F., Zhang, X. M., Karp, S., Yang, Y., and McMahon, A. P. (2001) Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development 128, 5099–5108 [DOI] [PubMed] [Google Scholar]
- 65. Retting, K. N., Song, B., Yoon, B. S., and Lyons, K. M. (2009) BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development 136, 1093–1104 10.1242/dev.029926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Saito, A., Kanemoto, S., Zhang, Y., Asada, R., Hino, K., and Imaizumi, K. (2014) Chondrocyte proliferation regulated by secreted luminal domain of ER stress transducer BBF2H7/CREB3L2. Mol. Cell 53, 127–139 10.1016/j.molcel.2013.11.008 [DOI] [PubMed] [Google Scholar]
- 67. Cai, C. L., Martin, J. C., Sun, Y., Cui, L., Wang, L., Ouyang, K., Yang, L., Bu, L., Liang, X., Zhang, X., Stallcup, W. B., Denton, C. P., McCulloch, A., Chen, J., and Evans, S. M. (2008) A myocardial lineage derives from Tbx18 epicardial cells. Nature 454, 104–108 10.1038/nature06969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. McLeod, M. J. (1980) Differential staining of cartilage and bone in whole mouse fetuses by Alcian blue and Alizarin red S. Teratology 22, 299–301 10.1002/tera.1420220306 [DOI] [PubMed] [Google Scholar]
- 69. Cai, X., Zhang, W., Hu, J., Zhang, L., Sultana, N., Wu, B., Cai, W., Zhou, B., and Cai, C. L. (2013) Tbx20 acts upstream of Wnt signaling to regulate endocardial cushion formation and valve remodeling during mouse cardiogenesis. Development 140, 3176–3187 10.1242/dev.092502 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.