

Abstract
The sequestosome 1 gene encodes the p62 protein and is the major genetic risk factor associated with Paget's disease of bone. In 2004, p62 was reported to up-regulate osteoclast differentiation by activating the transcription factors Nfatc1 and NF-κB. Here, we characterized the osteoclastogenic potential of murine p62−/−-derived cells compared with WT cells. Our data confirmed previous findings indicating that p62 is induced during murine osteoclast differentiation. Surprisingly, an indispensable role for p62 in in vitro osteoclast differentiation was not reproducible because p62-deficient osteoclasts exhibited robust activation of Nfatc1, NF-κB, and osteoclast marker enzymes. Thus, we concluded that in vitro osteoclast differentiation is not negatively influenced by knocking out p62. On the contrary, our results revealed that p62 deficiency accelerates osteoclastogenesis. Differentiation potential, multinucleation status, and soluble receptor activator of NF-κB ligand (sRANKL) sensitivity were significantly elevated in p62-deficient, murine bone marrow–derived stem cells. Moreover, femur ultrastructures visualized by micro-computed tomography revealed pronounced accumulation of adipocytes and trabecular bone material in distal femora of obese p62−/− mice. Increased tartrate-resistant acid phosphatase activity, along with increased trabecular bone and accumulation of adipocytes, was confirmed in both paraffin-embedded decalcified and methyl methacrylate–embedded nondecalcified bones from p62−/− mice. Of note, Paget's disease–like osteolytic lesions and increased levels of the bone turnover markers CTX-I and PINP were also observed in the p62−/− mice. Our results indicate that p62 predominantly suppresses murine in vitro osteoclast differentiation and highlight previously undetected Paget's disease–like phenotypes in p62−/− mice in vivo.
Keywords: osteoclast, bone marrow, osteoporosis, genetic disease, bone, μCT, bone turnover, p62/sequestosome 1, Paget's disease, osteoclastogenesis
Introduction
Paget's disease of bone (PDB),3 which was first described by Sir James Paget as osteitis deformans in 1877 (1), is a severe condition of pathological bone abnormalities that affects about 2–5% of the Caucasian population over 55 years (2). PDB is characterized by elevated number, size, and nucleation of osteoclasts (OCs) (3). Bone turnover rates are abnormal as a consequence of increased OC-related bone resorption, which is coupled with intensive formation of unstable and woven bone material characterized by irregular collagen fibers (2, 3). PDB risk factors include environmental and genetic components (4). Environmental factors consist of diet, toxins, biomechanical overuse, and virus infections (4). Conversely, linkage and genome-wide association studies have uncovered several susceptibility genes for classical PDB, including SQSTM1, CSF1, TNFRSF11A, OPTN, and TM7SF4 (3, 5).
SQSTM1, encoding the 62-kDa protein p62, is by far the most prominent PDB susceptibility gene (PDB3, OMIM number 167250), illustrated by an estimated 40% of PDB patients that carry a mutation in SQSTM1 (3). To date, about 30 different mutations in SQSTM1 have been identified (4). The most frequent mutation, causing a proline to leucine amino acid (AA) substitution at position 392 (P392L), affects the ubiquitin-associated (UBA) domain ranging from AA 386 to 434 (6). Because almost all remaining PDB-related p62 mutations have been detected within this domain, a UBA domain–dependent mechanism of p62-associated PDB was postulated (7–11). The P392L p62 mutation causes total loss of monoubiquitin and a significant reduction in polyubiquitin binding (10).
Despite the broad knowledge regarding PDB-causing p62 mutations and extensive research efforts over the past several years, the etiology and pathomechanisms characterizing PDB still remain unclear. Different p62 knockin (KI) and knockout (KO) mouse models have been established in the past to aid in elucidating these mechanisms. Thus far, however, the genetic ablation of p62 failed to show a bone phenotype under basal conditions but resulted in a reduced number of tartrate-resistant acid phosphatase (TRAP)-positive OCs in tibiae of mice challenged with parathyroid hormone–related protein (12). Because p62 deficiency led to an impaired activation of Nfatc1 and NF-κB and consequently to a defective osteoclastogenesis (OCG) in vitro and in vivo, p62 was defined as a positive regulator of OCG (12). In contrast, Kurihara et al. (13) showed increased OC values and a progressive loss of bone in mice after targeted expression of the human p62 P392L version under the control of a TRAP promoter, pointing rather to a negative role of p62 in OC biology. Two independent groups generated p62 P394L KI mice, covering the murine equivalent of human P392L p62. Hiruma et al. (14) described the hypersensitivity of p62-deficient OC precursors to pro-osteoclastic stimuli like soluble receptor activator of NF-κB ligand (sRANKL) or TNF, but young p62 P394L mice did not show any skeletal abnormalities in vivo. However, Daroszewska et al. (15) published a PDB-like phenotype in p62 P394L mice at 12 months. They found focal bone lesions and an increased bone turnover (15). In support of a potential negative role of p62 for OCs, increased NF-κB signaling and OCG were detected as a consequence of functional inactivation of the UBA domain, e.g. by the P392L mutation (16–18). Overexpression of a p62 UBA domain deletion mutant (Δ371–442) in RAW264.7 cells enhanced OCG through excessive activation of Nfatc1 and NF-κB and produced abnormally large OCs with enhanced resorption activity (19).
Because these previous research projects have yielded partially conflicting results and the detailed contribution of p62 to negative, positive, or dual effects on NF-κB and other signaling pathways, as well as on OC biology in general, has not yet been clarified, further research focusing on the role of p62 for OC differentiation and activity is necessary. Thus, in the present study, we utilized an established p62 KO mouse model for in vitro generation of OCs (20). Activation of Nfatc1 and NF-κB, as well as functional properties of WT and p62-deficient cells, were evaluated. Femoral bone abnormalities were investigated by micro-computed tomography (μCT) and histology of decalcified and nondecalcified bones. Our study confirmed previously published results indicating p62 up-regulation during in vitro OCG (12). However, in contrast to this earlier study, we were able to demonstrate the dispensability of p62 expression and induction for in vitro OCG. p62 KO cells had no general defect in basal activation of Nfatc1 and NF-κB but showed hypersensitivity to sRANKL and a premature onset of OCG. Above all, we detected a yet unknown PDB-like phenotype in p62−/− mice by μCT, histology, and serum measurements. Our results indicate a predominant role of p62 as a negative regulator of in vitro OCG and highlight yet undetected PDB-like phenotypical observations in p62−/− mice in vivo.
Results
In vitro OC differentiation of WT and p62-deficient bone marrow–derived monocytes (BMMs)
To evaluate the role of p62 for in vitro OCG, BMMs were generated after isolation of total bone marrow cells from hind limbs of WT and p62-deficient mice. After 4 days under OCG conditions, mature OCs were identified by multinucleation and TRAP staining (Fig. 1A). Comparable examination of both in vitro OCG and the isolation and replating of mature OCs was guaranteed by applying these standard conditions in both WT and p62-deficient cell cultures (Fig. 1A). To characterize the p62-deficient in vitro OCs, we next determined the multinucleation status of TRAP-positive cells. We detected a significantly increased number of TRAP-positive cells with more than 10 nuclei in p62-deficient OCs at differentiation day 3, which disappeared at day 4, as OCG peaked (Fig. 1B). Together, these results surprisingly indicate a yet unknown dispensability of p62 during in vitro OCG. To further evaluate this hypothesis, we next looked at the time course and kinetics of OCG. To this end, we determined TRAP activity in cell culture supernatants isolated from WT or p62-deficient cells between days 1 and 6 under standard OCG conditions. As shown in Fig. 1C (left), TRAP activity in C57BL/6N-derived cells was hardly detectable at days 1–2 but appeared at day 3 in both cultures. In accordance with an increased multinucleation status of p62-deficient OCs at day 3 (Fig. 1B), supernatants isolated from p62−/−-derived cells also indicated a statistically significant increase in OC activity at day 3 compared with WT cells (Fig. 1C, left). At days 4–6, TRAP activity reached an equal level in both cultures. Similar effects were detectable after backcross of mice to the BALB/c background. As TRAP activity values indicate, OCG was delayed 1 day due to the new mouse background (Fig. 1C, right). At day 4, activity was detectable in both cultures; however, p62−/−-derived cells depicted a statistically significant increase in TRAP activity. TRAP levels were equal at days 5–6. To validate a possible reason for the increased OCG potential of p62-deficient cells, we investigated the sRANKL sensitivity of OC precursors. As shown in Fig. 1D, p62-deficient cells were sensitive to lower doses of sRANKL at day 3 of OCG. These statistically significant effects disappeared at day 4 (Fig. 1D, right). Fig. 1E supports these findings by depicting an elevated number and size of p62-deficient cells at lower sRANKL doses compared with WT-derived cells. Overall, our findings indicate an unexpectedly fast and premature in vitro OCG in p62-deficient cells and that p62 functions as a negative and clearly not as a positive regulator of in vitro OCG.
Figure 1.
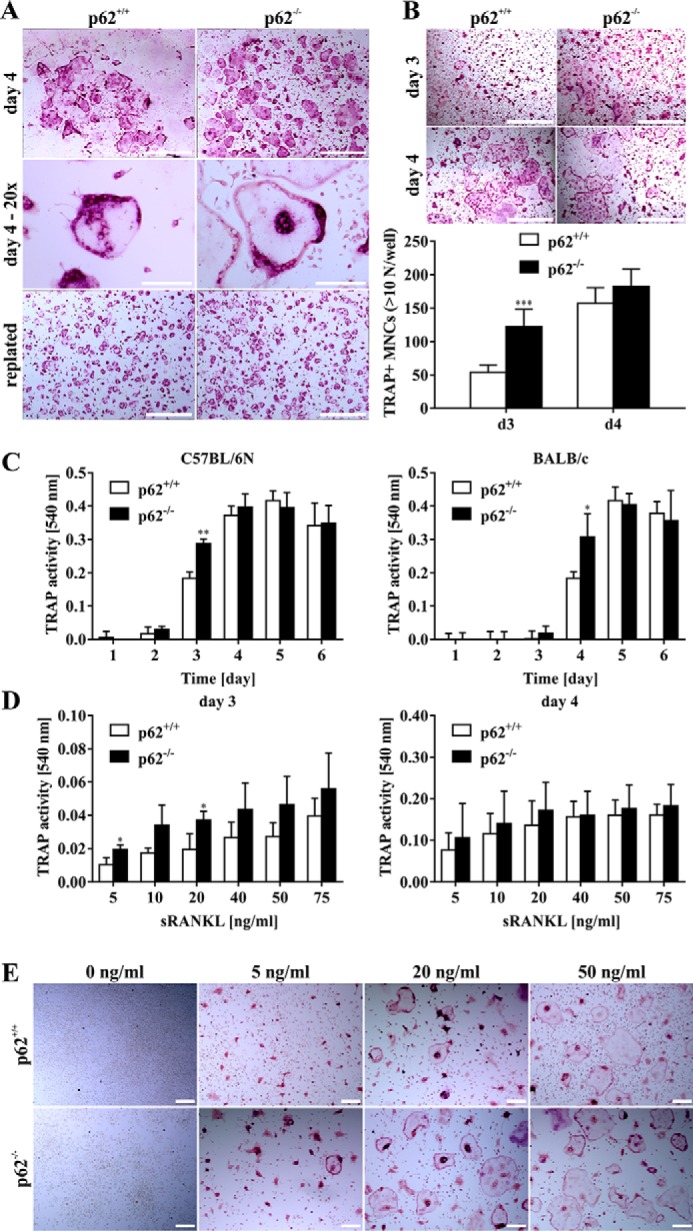
A, representative bright-field microscopy images of p62+/+- and p62−/−-derived TRAP-positive OCs (C57BL/6N) after 4-day M-CSF and sRANKL stimulation of BMMs (in vitro OCG) at low (top; scale bars, 1 mm) and high (middle; scale bars, 150 μm) magnification. Replated mature OCs are presented in the bottom row (scale bars, 1 mm). B, microscopy images of TRAP-stained p62+/+- or p62−/−-derived OCs (C57BL/6N) at days 3 and 4 illustrate the increased size and multinucleation of p62−/− OCs at day 3. The quantification of TRAP-positive multinucleated cells (MNCs) with more than 10 nuclei derived from either p62+/+ or p62−/−BMMs at days 3 and 4 of OC differentiation is shown graphically. Significance is indicated by asterisks (***, p < 0.001, Student's t test). Data are displayed as the mean ± S.D. (error bars); n ≥ 3. Scale bars, 1 mm. C, TRAP activity of cell culture supernatants isolated between days 1 and 6 of OCG. p62−/−-derived OCs show statistically significant elevated TRAP activity at day 3 (C57BL/6N) or 4 (BALB/c) compared with WT-derived cells. Significance is indicated by asterisks (*, p < 0.05; **, p < 0.01, Student's t test). Data are displayed as the mean ± S.D. (error bars); n = 3 independent experiments. D, WT- and p62−/−-derived BMMs (C57BL/6N) were cultured in 96-well plates and stimulated with M-CSF (30 ng/ml) and different concentrations of sRANKL. TRAP activity of supernatants at days 3 (left) and 4 (right) was plotted against sRANKL concentration. Significance is indicated by asterisks (*, p < 0.05, Student's t test). Data are displayed as the mean ± S.D. (error bars); n = 3 independent experiments. E, BMMs from p62+/+ and p62−/− animals (C57BL/6N) were subjected to OCG with M-CSF (30 ng/ml) and varying concentrations of sRANKL. At day 4, cells were formalin-fixed and TRAP-stained. Representative microscopy images show higher sRANKL sensitivity of p62−/− cells. Scale bars, 200 μm.
Induction of p62 during OC differentiation
Durán et al. (12) established p62 as positive regulator of OCG by showing progressive induction of p62 protein level during in vitro OCG. To validate the physiological role of p62 up-regulation during OCG, we produced cell lysates of p62 KO and WT cells between differentiation days 0 and 5. As shown in Fig. 2A and in agreement with the data from Durán et al. (12), p62 protein levels started to increase at day 1 and peaked between days 3 and 4 of OCG. Interestingly, p62 levels in macrophage colony-stimulating factor (M-CSF)–treated macrophage and in interleukin 4 (IL-4)–inhibited OCG samples were significantly increased compared with unstimulated and M-CSF/sRANKL–treated cells. Induction of OC hallmark proteins Nfatc1 and cathepsin K (Ctsk) indicated successful OCG in WT cultures (Fig. 2A). Furthermore, induction and phosphorylation of NF-κB p65 was detectable. Surprisingly, immunoblot (IB) experiments with lysates from p62-deficient cells also showed robust and comparable up-regulation of OC markers Nfatc1 and Ctsk during OCG despite the absence of p62 protein (Fig. 2B). TRAF6 expression, as well as induction and phosphorylation of NF-κB p65, was apparently not disturbed by p62 deficiency (Fig. 2, A and B). In summary, these observations are contrary to previously published data (12) and argue against the postulated indispensable role of p62 as a positive regulator for murine in vitro OCG.
Figure 2.

Expression of p62 and OC marker proteins during OCG is shown by IB analyses of RIPA cell lysates from BMM-derived cells of p62+/+ (A) and p62−/− (B) origin (C57BL/6N). BMMs were stimulated with M-CSF/sRANKL for the indicated time (days (d) 0–5). M-CSF or M-CSF/sRANKL/IL-4 stimulation served as macrophage and negative controls, respectively. Lysates were subjected to IB analysis with Abs against p62, Nfatc1, Ctsk, TRAF6, P-NF-κB p65, NF-κB p65, and β-actin. Induction of Nfatc1 and Ctsk during OCG is independent of p62 expression. Shown IBs are representatives of replicates with comparable results.
Nfatc1 induction and localization in p62-deficient OCs
Because we observed pronounced induction of Nfatc1 in p62-deficient OCs in IB experiments (Fig. 2), we now validated Nfatc1 mRNA expression and activation during OCG. Thus, we performed SYBR® Green-based quantitative RT-PCR (qRT-PCR) analyses with primers covering p62 and Nfatc1. We used the OC-specific genes Acp5 (TRAP) and Ctsk as positive controls for successful OCG. M-CSF/sRANKL treatment of BMMs increased gene expression of p62 in WT cells at days 2–5 (Fig. 3A). Macrophage differentiation (M-CSF) and IL-4–mediated inhibition of OCG led to an even higher induction of p62 (Fig. 3A). p62 deficiency was successfully confirmed in all p62−/− samples analyzed (Fig. 3A). Furthermore, induction of Nfatc1 mRNA expression was present in both WT and p62-deficient cells. Successful OCG was confirmed by significantly elevated amounts of Acp5 and Ctsk mRNA transcripts, which were absent in macrophages and marginal in IL-4–treated samples (Fig. 3A). It is noteworthy to mention that the induction of Nfatc1 and Acp5 transcripts was slightly higher in p62-deficient cells during the first 2 days. In contrast, Nfatc1, Acp5, and Ctsk quantities in p62-deficient cells at day 5 dropped below respective WT values. Fig. 3B summarizes expression data of three independent OCG approaches at days 0 and 4. In summary, our findings show activation of OC-specific genes and a premature onset of the initial phase of in vitro OCG in p62-deficient cells, which again indicate a negative rather than a positive role of p62.
Figure 3.
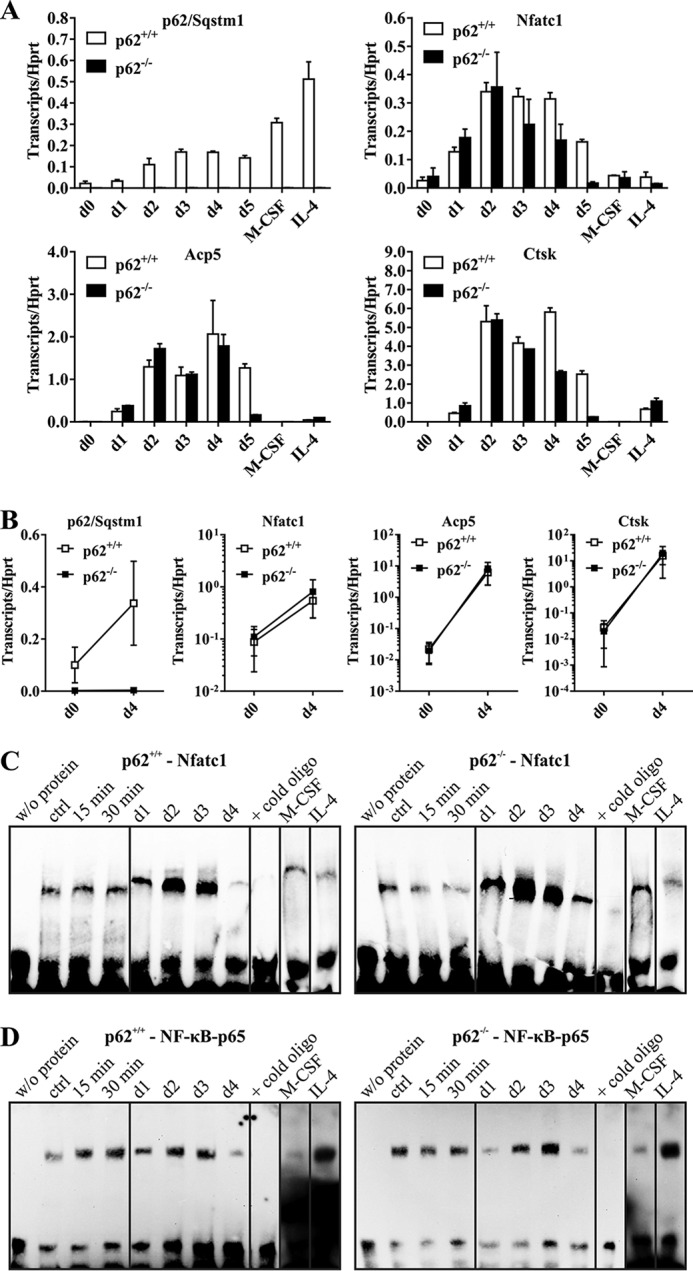
A, p62, Nfatc1, Acp5, and Ctsk gene expression in p62+/+- and p62−/−-derived cells (C57BL/6N) during OCG. cDNAs were obtained from p62+/+- and p62−/−-derived RNA samples isolated from cells at the indicated OCG time points. p62 KO cells produce a significant induction of Nfatc1, Acp5, and Ctsk during OCG despite the verified p62 deficiency. Hprt was used for normalization. Data are mean ± S.D. (error bars) of duplicates. B, gene expression data of at least three independent OCG approaches at days (d) 0 and 4. C and D, EMSA-based verification of Nfatc1 and NF-κB p65 activation during OCG with either p62+/+- or p62−/−-derived OC progenitors (C57BL/6N). BMMs were stimulated with M-CSF/sRANKL for 15 or 30 min (short time) or 1–4 days (long time). Nuclear extracts were analyzed for Nfatc1 (C) and NF-κB p65 (D) activation. The validity of the experiments was confirmed by indicated controls (w/o protein, excess of nonbiotinylated oligonucleotides, M-CSF only, IL-4–mediated inhibition of OCG; ctrl, control). WT and p62−/−-derived OCs show activation of Nfatc1 and NF-κB p65 at days 2 and 3 of M-CSF/sRANKL–induced OCG.
We next characterized nuclear translocation of Nfatc1 and its interaction with target DNA in p62-deficient cells upon M-CSF/sRANKL stimulation via electrophoretic mobility shift assay (EMSA). We used nuclear fractions together with biotinylated primer pairs possessing a specific core sequence for either the Nfatc1 or the NF-κB p65 transcription factor (Table 1). Under the applied conditions, p62+/+ cells showed a significant induction of Nfatc1 and NF-κB p65 as a result of M-CSF/sRANKL stimulation, especially at days 2–3 (Fig. 3, C and D, left). These effects were not negatively affected by p62 deficiency. The association of Nfatc1 and NF-κB p65 with target DNA was actually slightly increased at days 2 and 3 despite the absence of p62 (Fig. 3, C and D, right). Integrity of nuclear and cytosolic fractions used for EMSA was assayed by IB analysis (Fig. S1, A and B) and antibodies (Abs) to Ctsk, Nfatc1, and c-Fos (OC marker) as well as to P-NF-κB p65, NF-κB p65, p62, and β-actin (loading control). Although p62 deficiency in p62−/− cells was confirmed, no differences concerning activation of Nfatc1, Ctsk, c-Fos, or NF-κB-65 were obvious. These results again argue against a role for p62 as positive regulator of OCG.
Table 1.
Primers with specific recognition sequences used for EMSA approaches
TEG, triethylene glycol.
| Protein | Primers with recognition sequence (5′–3′) |
|---|---|
| Nfatc1 | 5′-CGCCCAAAGAGGAAAATTTGTTTCATA-3′-biotin-TEG |
| 5′-CGCCCAAAGAGGAAAATTTGTTTCATA | |
| 5′-TATGAAACAAATTTTCCTCTTTGGGCG | |
| NF-κB p65 | 5′-AGTTGAGGGGACTTTCCCAGGC-3′-biotin-TEG |
| 5′-AGTTGAGGGGACTTTCCCAGGC | |
| 5′-GCCTGGGAAAGTCCCCTCAACT |
Next, we used immunofluorescence to analyze the cellular localization of Nfatc1, Ctsk, and c-Fos during OCG. Ctsk was detectable in the cytosol at day 3 (Fig. 4A) and increased markedly at day 4 (Fig. 4B) in both WT and p62-deficient cell lines. Although c-Fos and Nfatc1 were localized in cytosolic and nuclear compartments at day 3 (Fig. 4A), nuclear translocation was increased in both WT and p62 KO-derived OCs at the transition from day 3 to 4 (Fig. 4B). In summary, nuclear translocation of transcription factors Nfatc1 and c-Fos induced by M-CSF/sRANKL stimulation and thus in vitro OCG are not negatively affected by p62 deficiency.
Figure 4.

Representative immunofluorescence microscopy images of Nfatc1-, cathepsin K–, and c-Fos–associated signals in WT- and p62−/−-derived OCs (C57BL/6N) at day 3 (A) or 4 (B) of stimulation with M-CSF/sRANKL. Both cell lines show comparable expression and nuclear translocation of transcription factors c-Fos and Nfatc1 at days 3 and 4 of OCG. Scale bars, 80 μm.
Analyses of signaling pathways in WT and p62-deficient cells
We subsequently analyzed M-CSF– and sRANKL-associated signaling in WT and p62 KO OC precursors by stimulation with M-CSF, sRANKL, or both cytokines and IB analysis–based validation of expression and phosphorylation of known signaling pathway components (Fig. 5). As assumed, p62 was slightly induced in p62+/+ cells at 120 min and absent in p62−/− cells. Phosphorylation of ERK1/2, which is predominantly associated with M-CSF stimulation, as well as phosphorylation of p38 and JNK1/2, which is triggered by both cytokines, was not negatively affected by p62 deficiency (Fig. 5). Activation of IκBα and its subsequent degradation and de novo synthesis were not influenced by p62 protein expression. Finally, phosphorylation (Fig. 5) and nuclear translocation of NF-κB p65 (Fig. S1, A and B) were not disturbed by p62 deficiency. In summary, M-CSF– and sRANKL-associated signaling pathways, and especially the activation of the NF-κB p65 pathway, are not positively regulated by p62.
Figure 5.

Expression and phosphorylation of signaling pathway proteins in p62+/+- and p62−/−-derived OC precursors (C57BL/6N) detected by IB analysis. BMMs of p62+/+ (left) and p62−/− (right) origin were either left untreated (ctrl) or incubated with M-CSF (30 ng/ml) and sRANKL (50 ng/ml) alone or with a combination of both cytokines for the stated time periods (5–120 min). RIPA cell lysates were subjected to SDS-PAGE and IB analysis with the indicated Abs. Activation of p38, ERK1/2, JNK1/2, IκBα, and NF-κB p65 was tested with Abs against total and phosphorylated protein variants. Anti-p62 Ab was used to confirm p62 deficiency in p62−/−-derived cells. M-CSF/sRANKL–induced activation of ERK, p38, and JNK kinases; phosphorylation of IκBα and NF-κB p65; and IκBα de novo synthesis are not negatively affected by p62 deficiency.
Functional characterization of p62-deficient OCs
For functional characterization of WT and p62-deficient OCs, we focused on bone resorption capacity of in vitro generated OCs. Mature OCs were isolated and plated on bovine bone slices for 2–3 days. Both OC cultures were able to resorb bone as indicated by hematoxylin (Fig. 6A) and toluidine (Fig. 6B) staining of resorption pits. To harmonize the quantification of resorption capacities in vitro, we used a biomimetic calcium phosphate (CaP) assay (21, 22). OCs were differentiated from WT and p62-deficient progenitors for 4 days and subsequently plated on CaP-coated cell culture plates for a further 2 days. Microscopy images of TRAP-positive cells on CaP substrate after formalin fixation revealed actively resorbing OCs of WT and p62 KO origin (Fig. 6, C and D). Resorption areas were visualized by counterstaining with AgNO3 (Fig. 6E). Quantification of areas (Fig. 6F, left) yielded a slightly elevated resorption capacity of p62−/−- (2,927,099 ± 424,871 μm2) compared with WT-derived (2,597,356 ± 239,254 μm2) OCs. In parallel, we determined the number of total TRAP-positive OCs plated on CaP substrate after the initial differentiation step and normalized resorption values to the respective cell numbers (Fig. 6F, right). Because we observed a slightly higher differentiation efficiency of p62-deficient cells, resorption values normalized to total OC numbers were now indistinguishable from WT (WT, 13,524 ± 3,232 μm2 versus p62 KO, 13,301 ± 2,273 μm2). Thus, the increased resorption values of p62-deficient cells (Fig. 6F, right) are likely due to higher total OC numbers and rather to increased resorption capacity in general. Furthermore, we looked at F-actin rings in p62-depleted cells. As shown in Fig. 6G, F-actin ring formation is comparable with WT-derived OCs and is not negatively affected by the loss of p62 protein expression.
Figure 6.

A and B, representative microscopy images of pit formation assays (functional characterization) using mature p62+/+- and p62−/−-derived OCs (C57BL/6N) seeded on bovine bone slices. Resorption areas were visualized by chemical removal of cells and staining with Mayer's hematoxylin (A) or by direct staining with toluidine blue solution (B). Scale bars, 200 μm. C and D, microscopy images of mature OCs plated on CaP-coated cell culture plates after formalin fixation and TRAP staining in combination with AgNO3 at low (C; scale bars, 1 mm) and high (D; scale bars, 100 μm) magnification. Cells reveal intense TRAP staining and CaP resorption activity regardless of p62 expression. E, representative examples of AgNO3-stained CaP-coated cell culture plates after chemical removal of mature OCs. Scale bars, 1 mm. F, graphic illustration of resorption activity of p62+/+- and p62−/−-derived OCs (left). CaP resorption, normalized to cell number (right), of p62-deficient cells is comparable with p62+/+-derived OCs. Data are displayed as the mean ± S.D. (error bars); n = 3 independent experiments. G, F-actin rings in mature OCs are visualized by FITC-phalloidin (green). Nuclei are counterstained by DAPI (blue). p62+/+- and p62−/−-derived OCs are shown at low (top; scale bars, 200 μm) and high (bottom; scale bars, 100 μm) magnification. White arrows in upper row point to heavily stained areas of F-actin rings.
μCT and histological examination of bone phenotype in p62-deficient mice
Because we postulated and in part already validated p62 as a negative regulator of murine OCG in vitro and because it is widely accepted that point mutations in p62 predispose for the development of PDB in humans, we tried to document a possible bone phenotype of p62 KO mice. Fig. 7A depicts three-dimensional (3D) reconstruction images derived from μCT analysis of distal femora isolated from p62 KO or WT mice (C57BL/6N) of the indicated age. Femora of p62 KO mice clearly showed an unexpectedly elevated density of trabecular structures compared with WT bones, especially at 9, 12, and 15 months. WT femora exhibited an age-dependent gradual increase in trabecular separation (Tb.Sp), which reflects the normal aging process (Fig. 7B). However, femora of p62 KO mice maintained a relatively high bone density and thus a low Tb.Sp during aging, indicating a pathogenic transformation in born turnover triggered by p62 deficiency. An interesting finding already published in the literature (23) is that p62 KO mice revealed a gradually increasing body weight, whereas weight of WT mice remained relatively constant until 21 months (Fig. 7C). The quantitative evaluation of μCT data illustrated a statistically significant increased trabecular number (Tb.N) and a decreased Tb.Sp in femora of p62−/− mice compared with WT at 15 months (Fig. 7D). Bone volume fraction (BV/TV) was also higher in p62−/− femora, whereas trabecular thickness (Tb.Th) was not influenced by p62 KO (Fig. 7D). Differences in Tb.N and Tb.Sp between WT and p62 KO femora started to appear around 9 months and only scarcely failed to be statistically significant at 12 months. Furthermore, the evaluation of trabecular structures was also performed in male mice at 12 months to exclude gender-specific differences. As expected, male p62-deficient mice also depicted increased Tb.N, decreased Tb.Sp (Fig. S2, A and B), and higher trabecula-associated TRAP activity (Fig. S2B). Together, these μCT data advocate a yet undetected PDB-like bone phenotype in femora of mature p62 KO mice.
Figure 7.
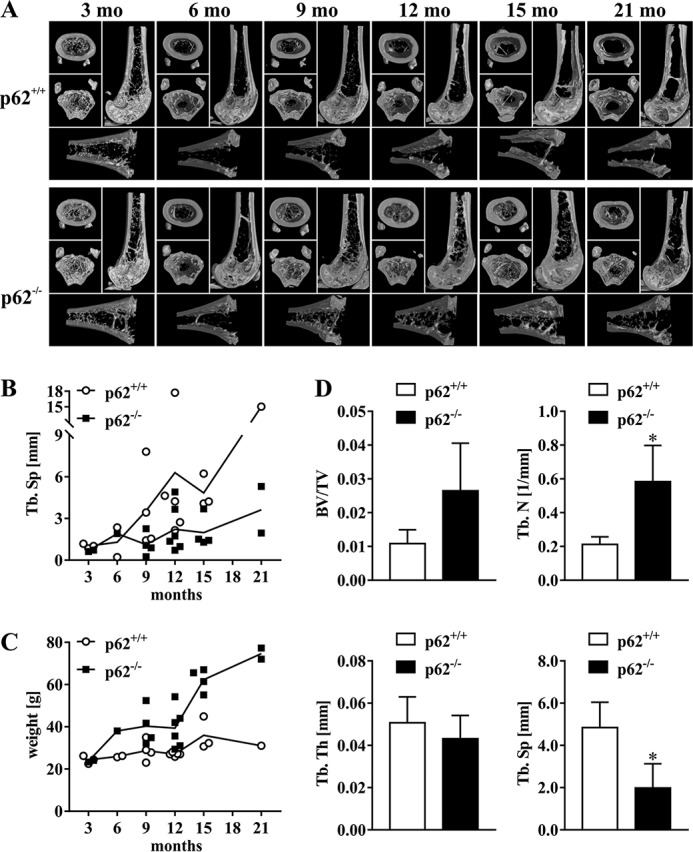
A, representative examples of μCT images (3D reconstruction) from sagittal and cross-sections of distal femora showing trabecular and cortical bone areas in WT and p62−/− mice (C57BL/6N) of the indicated age. Compared with WT, femora of p62−/− mice exhibit an elevated Tb.N, especially at 9, 12, and 15 months (mo). B and C, quantitative presentation of Tb.Sp (B) and body weight (C) data of mice used for μCT as exemplified in A. D, quantitative evaluation of μCT data at 15 months showing statistically significant increased Tb.N and decreased Tb.Sp of p62−/− bones. Values representing BV/TV were higher in p62−/− femora but fell just below statistical significance. In contrast, Tb.Th is not influenced by p62 deficiency. Significance is indicated by asterisks (*, p < 0.05, Student's t test). Data are displayed as the mean ± S.D. (error bars); n = 3 (WT) and n = 4 (p62−/−).
To validate the μCT in vivo data, we performed histological experiments using thin sections of either decalcified paraffin-embedded or nondecalcified methyl methacrylate (MMA)-embedded femora. Thin sections of paraffin-embedded bones subjected to hematoxylin and eosin (H&E) (morphology), TRAP (OCs), or Safranin O (cartilage tissue) staining procedures are summarized in Fig. 8A. At 3 and 6 months, Tb.N was comparable between WT and p62−/− mice. Higher Tb.N and thus lower Tb.Sp of p62−/− femora were visible especially at 12 and 15 months. WT and heterozygous p62+/− (data not shown) distal femora hardly showed any remaining trabecular structures at 21 months, whereas p62-deficient bones revealed a strikingly elaborate trabecular network and excessive TRAP activity, indicating both high osteoblast (OB) and OC activity. Because p62-deficient mice showed age-related obesity (23), lipid accumulation started to develop between 6 and 9 months in p62−/−-derived bones of female mice. However, fat accumulation was no prerequisite for increased Tb.Th because WT mice at the age of 9 and 15 months also showed lipid droplets but almost completely lacked trabecular structures (Fig. 8A) and because male mice at 12 months also had elevated Tb.N but no signs of fat accumulation at all (Fig. S2B). Furthermore, p62 KO animals at 21 months possessed a denser trabecular network in combination with marginal fat accumulation compared with the representative example at 9 months. Increased Tb.N in p62−/−-derived femora at 15 months was also visible in MMA-embedded nondecalcified bone sections after Masson-Goldner trichrome (Fig. 8B) and von Kossa (Fig. 8C) staining.
Figure 8.
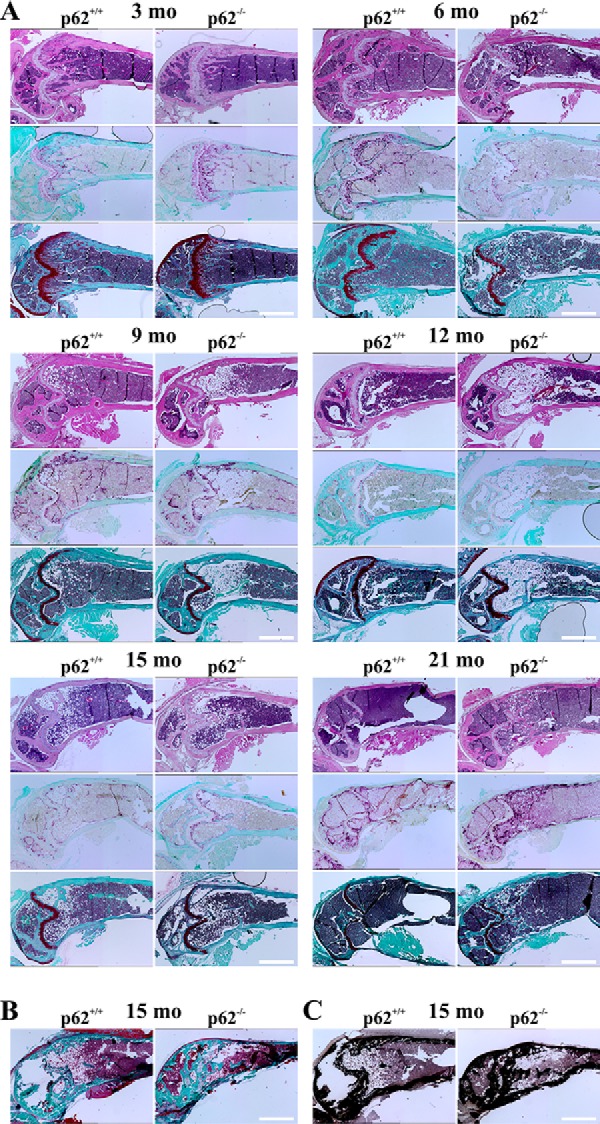
A, representative examples of microscopy images of H&E- (top), TRAP- (middle), and Safranin O (bottom)-stained paraffin sections of decalcified femora with WT or p62−/− origin at 3, 6, 9, 12, 15, or 21 months (mo) (C57BL/6N). Note the age-dependent lipid accumulation in p62-deficient bones. Tb.N and TRAP activity are elevated, and Tb.Sp is reduced in p62−/−-derived bones, especially at 15 and 21 months. B and C, microscopy images presenting sections of MMA-embedded nondecalcified bones (C57BL/6N) after Masson-Goldner trichrome (B) or von Kossa staining (C). Scale bars, 1 mm.
A closer look at TRAP activity at the diaphysis area in femora of 21-month-old p62-deficient female mice also revealed remarkable PDB-like osteolytic lesions, which were not seen at all in age-matched WT and heterozygous p62+/− mice (Fig. 9, A and B). Furthermore, we observed an increased extent of trabecular bone structures accompanied by high bone-associated TRAP staining in these p62−/− femora in the proximal diaphysis, which were obviously not present in WT or p62 heterozygous mice. This indicates an increased bone turnover due to the genetic p62 deficiency (Fig. 9B). Thus, to quantitatively assess changes in bone turnover in p62-deficient animals, we measured CTX-I and PINP levels in serum samples of C57BL/6N mice at 12 months. As depicted in Figs. 9C and D, both CTX-I and PINP were significantly elevated in serum samples of p62−/− mice, indicating the predicted increased bone turnover in p62 KO animals. Finally, we also quantified serum levels of different (pro-inflammatory) cytokines (Fig. S3). TNF, IL-10 and IL-1β were below detection level in WT and p62 KO animals (data not shown), whereas IL-6 was slightly increased in WT, and MCP-1 was slightly increased in p62 KO mice (Fig. S3). In summary, p62−/− animals show an age-related, PDB-like phenotype characterized by both increased bone forming (increased trabecular network and serum PINP) and bone degrading activity (increased TRAP activity and serum CTX-I) but lack any signs of significantly elevated pro-inflammatory cytokines in serum samples.
Figure 9.
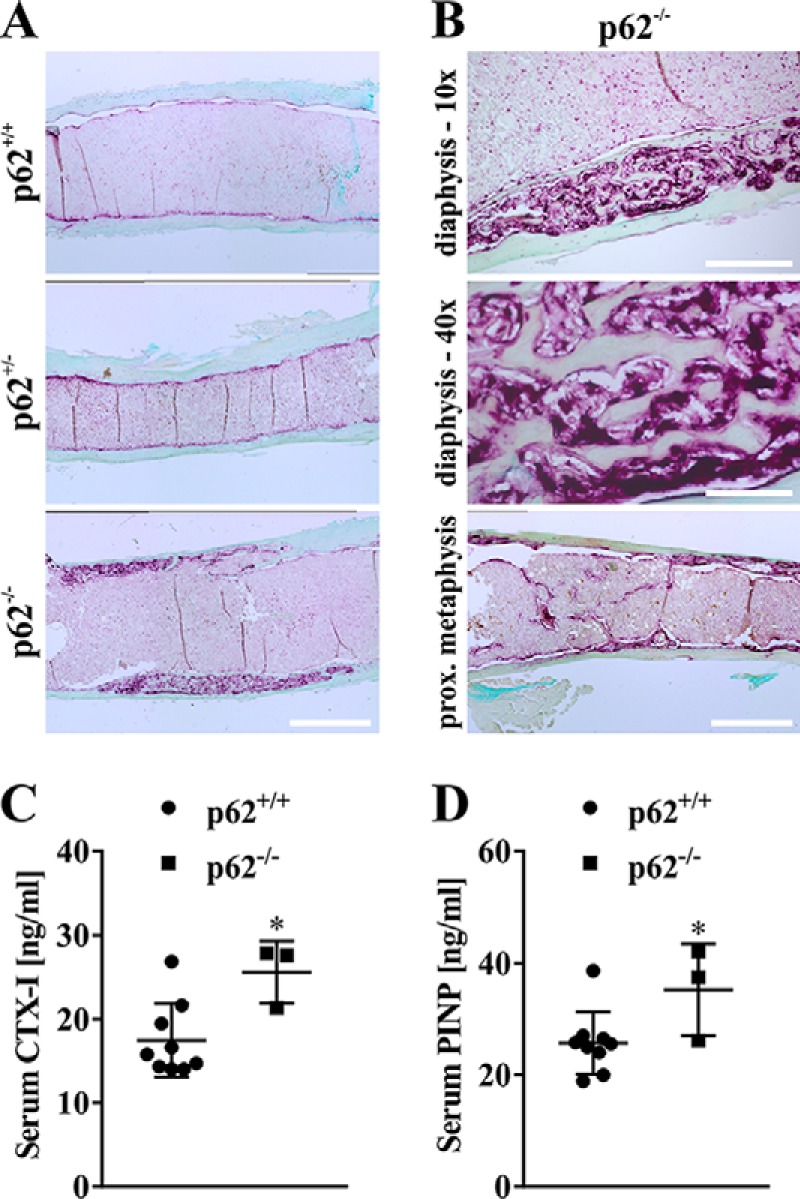
A, representative microscopy images of TRAP-stained bone sections from female p62+/+, p62+/−, and p62−/− mice at 21 months indicate PDB-like osteolytic lesions in femora (distal diaphysis) of p62-deficient animals (C57BL/6N). Scale bars, 1 mm. B, osteolytic lesions and TRAP activity in diaphysis of p62−/−-derived bones from A are shown at higher magnification (scale bars, 300 μm (upper image) and 75 μm (middle image)). Increased trabecular material combined with pronounced TRAP activity exclusively detected in proximal metaphysis of p62 KO female animals is exemplified in the lower image (scale bar, 1 mm). C and D, quantification of bone degradation marker CTX-I (C) and bone formation marker PINP (D) in serum of p62+/+ and p62−/− mice (C57BL/6N). Data are displayed as the mean ± S.D. (error bars). Significance is indicated by asterisks (*, p < 0.05, Student's t test).
Discussion
PDB is characterized by increased activity of OCs and intensive OB-associated formation of disorganized new bone material, resulting in bone pain, deformity, and increased risk of bone fractures (2). Bone turnover runs out of control by hyperactive OCs showing elevated nucleation, size, and RANKL sensitivity combined with increased bone formation (3). Because PDB-associated mutations in p62 are the most common genetic risk factor (3–5), detailed knowledge about normal p62 function and pathological dysfunction by mutations is of critical importance. Also, because the loss of ubiquitin binding caused by p62 mutations was defined as a unifying mechanism in p62-associated PDB (11), it was postulated that the UBA domain deficiency could lead to hyperactive OCs. Thus, defective p62 was shown to disturb the recruitment of Cyld to TRAF6, which prevents deubiquitination and deactivation of RANK signaling cascades (24).
In the present study, we demonstrate that p62 is dispensable for murine OCG in vitro. Surprisingly, p62 deficiency results in premature initiation of OCG, which is highlighted by significant sRANKL hypersensitivity and increased multinucleation status at day 3 of OC differentiation. The obvious discrepancy between presented and previously published data (12) may be a result of the cell numbers used for OCG. In the present work, 3.2 × 104 cells per 24-well plate were defined as optimal for in vitro OCG. However, Durán et al. (12) used 1 × 106 cells. Because previous work has shown that in vitro OCG is critically dependent on seeded cell numbers and excessive cells are inhibitory for OCG (25, 26), accelerated cell death may be a possible reason for unsuccessful OCG in p62−/− cells in this previous work. Moreover, independently generated p62 KO mice were used (12, 20). However, in both p62 gene–deficient mouse models, comparable targeting strategies were chosen, e.g. deletion of exon 1 (12) or exons 1–4 (20), respectively, which resulted in the complete loss of gene product in both cases. Moreover, similar embryonic stem cells (129/SvJ) and backcross strategies to a C57BL/6 background were used (12, 20).
Increased sRANKL sensitivity and premature differentiation of p62−/− cells could, among others, also be the result of elevated receptor surface expression, altered proliferation and apoptosis rates of OC progenitors, or defective intracellular signaling pathways. However, the molecular mechanisms linking p62 point mutations or its complete ablation to Paget's disease in humans or Paget's disease–like observations in mice still remain incompletely understood and, on some points, controversial despite intensive research since the first discovery of p62 mutations in Paget's disease patients about 15 years ago (27).
The increased OCG potential of p62−/− cells detected in the present work may define the enhanced Tb.N seen in vivo as a direct consequence of coupling increased OC activity to OB activity. This may be an explanation for the simultaneously enhanced TRAP activity and Tb.N in p62-deficient femora, which is also underlined by both elevated CTX-I and PINP levels in serum of p62 KO mice. Increased sRANKL sensitivity and OCG have also been seen in two independently generated p62 KI mouse models (14, 15) and by overexpression of a p62 ΔUBA construct in RAW264.7 cells (19), which corresponds perfectly to the negative function of p62 in OCG as shown in the present work using the p62 KO mouse model.
Induction of p62 protein levels during OCG has been published previously (12) and was now confirmed in the present study. Until now, the p62 induction has been interpreted as a positive requirement for successful OCG (12). However, because genetic ablation of p62 did not negatively interfere with OCG in our study and macrophage differentiation and IL-4–inhibited OCG conditions resulted in highest p62 protein levels, a positive role of p62 for OCG is questionable. Furthermore, induction of OC-related proteins, e.g. Cyld or Optn, during OCG has lately been interpreted as a hallmark of negative regulators of OCG, which serve as negative feedback loops to control and deactivate OC-associated signaling cascades (24, 28). This line of argument may thus lead to a new interpretation describing p62 as another negative regulator during OC differentiation.
In the present study, we further used p62-deficient cells to show nuclear translocation of key OC transcription factors Nfatc1, c-Fos, and NF-κB p65 on RNA, protein, and cellular levels. Furthermore, activation of signaling cascades (p38, ERK, JNK, and NF-κB p65) was detectable despite the missing p62 expression. Again, this argues against a predominant positive function of p62 for OCG in vitro.
We also looked at functionality of in vitro OCs, including bone resorption and F-actin ring formation. Thus far, we have not detected any functional difference between mature p62 KO and WT OCs. This is in contrast, for example, to proteins like Src or Ctsk (29, 30), which are important for F-actin ring formation and bone resorption, respectively. Thus, it is likely that p62−/−-associated pathological consequences leading to PDB are linked to differentiation or fusion processes of OC progenitors and are not primarily caused by defects in activity or function of mature OCs.
In vivo characterization of p62 KO mice using μCT and histological examinations revealed significantly elevated Tb.N and decreased Tb.Sp in femora at 15 months, indicating increased formation of new bone material or decreased OC activity, which is comparable with Ctsk-deficient mice (30). In contrast to data from Ctsk KO mice, we also detected a clearly increased TRAP enzyme activity in histological sections of p62 KO bones at 15 and especially at 21 months. The increased bone formation rates thus seem to be coupled directly to elevated OC activities in p62-deficient mice. However, exact TRAP activity levels in bone sections must be further analyzed on a single-cell level in future experiments. Overall, our histological in vivo data nicely correspond to in vitro findings because p62-deficient cells showed increased OCG efficiency during early stages of OC differentiation, providing a convincing explanation for the increased TRAP-positive areas in bone sections of p62 KO compared with WT mice.
We and others (23) have also observed progressive obesity and lipid accumulation in p62-deficient animals, which could be a potential source of femoral abnormalities seen in p62 KO animals. However, because we observed examples of lipid accumulation in younger WT animals without increased Tb.N or missing lipid accumulation in both older female and in 12-month-old male p62 KO mice characterized by high Tb.N, the trabecular phenotype is likely to be p62-associated and independent from fat accumulation. The in vivo PDB-associated femur phenotype presented in detail in the current study has briefly been mentioned in a previous publication as a small increase in Tb.N in 6–8-week-old p62−/− mice (12) and thus seems to be present in both independently generated p62 KO mice. However, it is essential to keep in mind that bone phenotype is critically dependent on background (e.g. BALB/c versus C57BL/6), age (young versus old), and development status of animals. In line with this, characterization of two independent p62 P394L KI mouse lines resulted in conflicting data (14, 15). Although both studies detected increased OCG potential of p62 P394L cells in vitro, which is similar to data presented in the present study, a PDB-associated bone phenotype was only obvious in one study using older mice (15). Daroszewska et al. (15) showed increased OC numbers and concurrently increased bone formation rates in older animals, which clearly coincide with our in vivo data also using older p62-deficient mice in the presented experiments.
In summary, we rebut the postulated positive role of murine p62 during in vitro OCG because genetic ablation of p62 showed no negative effect on OCG. We further define p62 as a negative regulator of OCG because p62−/− OC progenitors, which lack a functional p62 protein, showed significantly increased sensitivity to sRANKL resulting in a premature onset of OCG in vitro. In vivo, we detected an exaggerated bone turnover in p62−/− mice characterized by increased trabecular formation in combination with increased TRAP activity in distal femora and confirmed by increased CTX-I and PINP serum levels. We further observed osteolytic Paget's disease–like lesions in p62−/− mice. Thus, we define p62-deficient mice as a suitable model for studying PDB-associated pathological mechanisms, which will tremendously help to unravel the etiology and pathomechanisms underlying PDB in humans in the future.
Experimental procedures
Reagents and media
Recombinant murine M-CSF, sRANKL, and IL-4 were purchased from Peprotech Inc. (Rocky Hill, NJ). Unless otherwise stated, medium (αMEM) and reagents were from Sigma-Aldrich. Supplements penicillin/streptomycin and l-glutamine were obtained from Pan-Biotech (Aidenbach, Germany). Abs were purchased from Sigma-Aldrich (p62, P0067; β-actin, AC-15), Santa Cruz Biotechnology (Dallas, TX; Nfatc1, 7A6; Ctsk, E-7; TRAF6, H-274; c-Fos, K-25), or Cell Signaling Technology (Boston, MA; P-NF-κB p65, 3033; NF-κB p65, 8242; P-p38, 9211; p38, 9212; P-ERK1/2, 4377; ERK1/2, 9102; P-JNK1/2, 4668; JNK1/2, 9258; P-IκBα, 2859; IκBα, 9242).
Animals
We used the previously published p62/sequestosome 1–deficient mouse line on the C57BL/6N background (20). WT mice for propagation or backcross of p62-deficient mice onto the BALB/c background were purchased from Charles River Laboratories (Wilmington, MA). Mice were kept at the animal facility of the University of Regensburg in a specific pathogen–free environment until sacrificed by CO2 asphyxiation. All procedures involving animals were performed according to the guidelines of the University of Regensburg animal facility and approved by the local veterinary authorities and the ethics committee of the District Government of Upper Palatinate (reference 54-2532.1-38/12).
In vitro OC differentiation using primary BMMs
Murine BMMs were isolated by flushing femora and tibiae of 10–20-week-old mice with complete αMEM. Filtered cells (sieve, 100 μm) were washed with PBS (two times). Cells were immediately centrifuged after a short red blood cell lysis step (150 mm NH4Cl, 0.1 mm Na2EDTA, 10 mm KHCO3, pH 7.2–7.4), resolved in αMEM with M-CSF (30 ng/ml), and cultured in cell culture dishes. After 24 h, nonadherent cells were used as macrophage and OC progenitors. For OCG, cells (density, 4 × 104 cells/ml; 1.6 × 104 cells/cm2) were suspended in αMEM supplemented with M-CSF (30 ng/ml) plus sRANKL (50 ng/ml) and cultured in different cell culture well formats as appropriate.
TRAP staining of supernatants, cells, and paraffin sections
TRAP staining was performed with the TRAP staining kit from B-Bridge International (Cupertino, CA). To quantify TRAP activity in cell culture medium, 30 μl of supernatant were mixed with 60 μl of substrate (or with 40 μl of substrate in the sRANKL sensitivity assay) and incubated at 37 °C for 3 h. Optical density was measured at 540 nm with a microplate reader (Bio-Rad). To detect TRAP-positive OCs, formalin-fixed cells were incubated with TRAP substrate at 37 °C for 30 min. To perform TRAP staining on thin sections derived from paraffin-embedded decalcified bones, sections were deparaffinized, rehydrated, and incubated with TRAP solution in a wet chamber at 37 °C for 1 h. Sections were washed, counterstained with aqueous 0.08% (w/v) Fast Green for 1 min, and mounted using the Roti®-Histokitt (Roth, Karlsruhe, Germany).
Cell lysis and IB analysis
Adherent cells were harvested by scratching or incubation with 0.05% trypsin, 0.02% EDTA (Pan-Biotech) for 5–10 min at 37 °C. Cells were washed with PBS and lysed by incubation with radioimmunoprecipitation assay (RIPA) buffer (50 mm Tris/HCl, pH 7.5, 150 mm NaCl, 1% Nonidet P40, 0.1% SDS, 1% sodium deoxycholate). Lysates were cleared by centrifugation and used for protein quantification with the DCTM Protein Assay kit (Bio-Rad). Samples were boiled in Laemmli buffer (95 °C, 5 min), subjected to SDS-PAGE, and transferred to nitrocellulose membranes according to standard procedures. Membranes were blocked with 5% nonfat milk in TBS-T (0.1% Tween® 20) and probed overnight (4 °C) with primary Abs. Blots were washed with TBS-T and incubated with peroxidase-conjugated secondary Abs (Dianova, Hamburg, Germany). Detection of IB signals was performed with enhanced chemiluminescence (ECL) solution. For reprobing, membranes were incubated with aqueous 0.2 m NaOH solution for 30 min.
Extraction of RNA, cDNA synthesis, and qRT-PCR
RNA was extracted with the RNeasy® Mini kit from Qiagen (Hilden, Germany) according to the manufacturer's suggestions. Reverse transcription (0.5 μg of RNA) was performed with the iScriptTM Advanced cDNA Synthesis kit (Bio-Rad). qRT-PCR analyses were carried out with LightCycler® 480 SYBR Green I Master Mix (Roche Diagnostics) and the ABI PRISM® 7900HT system (Life Technologies). Primer sequences specific for cDNA segments of analyzed genes are indicated in Table 2 (31, 32). Hprt was used as an endogenous control. Cycle threshold (Ct) values and absolute gene expression data were calculated from standard curves of serial plasmid DNA dilutions using SDS 2.4 software.
Table 2.
Primers used for standard, semiquantitative, and quantitative RT-PCR
| Gene | Accession no. | Sequence (5′–3′) | cDNA |
|---|---|---|---|
| bp | |||
| Acp5 | NM_007388.3 | 5′-AAGAACTTGCGACCATTGTTAGC | 91 |
| 5′-CGTTCTCGTCCTGAAGATACTGCA | |||
| Ctsk | NM_007802.4 | 5′-ACGTTACTCCAGTCAAGAACCAGGG | 139 |
| 5′-GTCACACAGTCCACAAGATTCTGGG | |||
| Hprt | NM_013556.2 | 5′-GTTGAATACAGGCCAGACTTTGTTG | 163 |
| 5′-GATTCAACTTGCGCTCATCTTAGGC | |||
| Nfatc1 | NM_001164111.1 | 5′-TCTGGGAGATGGAAGCAAAGACTGA | 82 |
| 5′-TGGTTGCGGAAAGGTGGTATCTCAA | |||
| Sqstm1 | NM_011018.3 | 5′-GAACTCCAGTCTCTACAGAT | 220 |
| 5′-CGATGTCGTAATTCTTGGTC |
Nuclear and cytoplasmic extraction and EMSA
Preparation of nuclear and cytoplasmic extracts was done with NE-PERTM Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific, Waltham, MA) as stated in the manufacturer's protocol. Nuclear extracts (10 μg) were examined using the LightShift® Chemiluminescent EMSA kit (Thermo Fisher Scientific) under standard conditions. Nucleotides used for EMSA (Table 1) have been described previously (33, 34).
Immunofluorescence-based detection of OC marker proteins
Paraformaldehyde (4%)–fixed cells were permeabilized with 0.2% TritonTM X-100 for 10 min, blocked with 1% goat serum for 1 h, and incubated with primary Abs overnight (4 °C). Cells were washed with PBS and incubated with Alexa Fluor® 488- or 594-coupled secondary Abs (Dianova; 1:800). Specimens were washed with PBS, mounted in SlowFade® Gold antifade mountant (Life Technologies), and examined with a BZ-9000 microscope (Keyence, Osaka, Japan).
Isolation and transfer of mature OCs
For functional characterization of mature OCs, differentiation was performed in cell culture dishes as described above. Cells were washed with PBS, and nonadherent cells were dissociated by 0.05% trypsin, 0.02% EDTA (Pan-Biotech) incubation (1 min) at 37 °C. Adherent cells (multinucleated OCs) were isolated from dishes by a second (5–10-min) incubation with trypsin/EDTA. Cells were suspended with medium plus M-CSF (30 ng/ml) and sRANKL (50 ng/ml) and seeded for functional assays (resorption activity and F-actin ring).
Pit formation assay using bovine bone slices
Mature OCs were isolated from 100-mm dishes as described above and replated on bovine bone slices (Immunodiagnostic Systems GmbH, Frankfurt am Main, Germany). After incubation for 48 h, bone slices were washed with PBS, and cells were either detached by 10-min incubation with aqueous NH4OH solution (1 m) or left untreated. Resorption pits were visualized by staining with Mayer's hematoxylin (Sigma-Aldrich) for 1 min or aqueous 0.1% (w/v) toluidine blue solution for 7 min. Stained slices were washed, air-dried, and examined by bright-field microscopy.
Biomimetic CaP coatings and quantification of resorption
CaP coatings were prepared as described previously (21, 22). Mature OCs were isolated from 100-mm dishes as described above and subsequently incubated on CaP-coated plates at 37 °C for 48 h. Cells were washed with PBS and removed from plates with NaCl (1 m in 0.2% Triton X-100). Culture plates were washed with water (two times), stained with 5% aqueous AgNO3 solution in combination with UV light treatment for 30 min, and washed again with water (three times). Resorption areas were quantified as described previously (31).
Detection of F-actin ring formation
To verify F-actin ring formation, replated mature OCs were cultured for 48 h, washed with PBS, and fixed with 4% paraformaldehyde for 10 min. Following permeabilization with 0.5% Triton X-100 for 5 min, cells were incubated with FITC-phalloidin (Sigma-Aldrich; 1:1,000) for 30 min. After three washing steps with PBS, coverslips were mounted with SlowFade Gold antifade mountant and examined by fluorescence microscopy (Keyence).
μCT and histological analysis of murine femora
The following parameters were used for μCT analysis of murine femora with the V-Tome-X S 240 system (GE Healthcare): 10× magnification, 20-μm voxel size, 35-kV X-ray voltage, 500-μA X-ray current, and 333-ms integration time. 3D reconstructions of bones and quantification of trabecular bone parameters were done using VGStudio MAX 2.2.3 software (Volume Graphics, Heidelberg, Germany). For histological analysis, murine hind limbs were dissected, fixed in neutral buffered formalin (10%) for 2 days, and decalcified with EDTA solution (25%; two changes; 5 days in total). Specimens were then dehydrated with an ethanol series, treated twice with xylene, and embedded in paraffin. Bone sections (7 μm) were treated with H&E, TRAP, or Safranin O staining solution according to standard protocols. Nondecalcified murine hind limbs were fixed in neutral buffered formalin (10%) for 2 days, dehydrated with an ethanol series, and embedded in plastic resin using the Technovit® 9100 system (Heraeus Kulzer, Hanau, Germany) as stated in the manufacturer's recommendations. Sections (7 μm) were produced with a tungsten carbide knife and stained with von Kossa and Masson-Goldner trichrome staining protocols.
CTX-I, PINP, and Luminex multiplex immunoassays
RatLapsTM CTX-I and rat/mouse PINP enzyme immunoassays were purchased from Immunodiagnostic Systems GmbH and used according to the manufacturer's recommendations to quantify CTX-I and PINP bone turnover markers in murine serum samples. The ProcartaPlex® multiplex immunoassay (IL-1β, IL-6, IL-10, MCP-1, and TNF) was purchased from eBioscience (Vienna, Austria) and performed with murine serum samples according to the manufacturer's protocol. Raw data were obtained with the Luminex® xMAP 100 system (Luminex Corp., Austin, TX), and sample concentrations were calculated with a five-parameter logistic curve fit using Liquichip Analyzer software (Qiagen).
Statistical analyses
Data are shown as the mean ± S.D. Statistical analysis was performed using Student's t test. Results were considered statistically significant at p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***).
Author contributions
F. Z., F. P., and A. G. conceptualization; F. Z., F. P., A. M., and A. G. formal analysis; F. Z., F. P., and A. G. supervision; F. Z., F. P., A. M., and A. G. validation; F. Z., F. P., and A. M. investigation; F. Z., F. P., and A. M. visualization; F. Z., F. P., A. M., and A. G. methodology; F. Z. writing-original draft; F. Z. and A. G. project administration; F. P. and A. G. writing-review and editing; A. G. resources; A. G. data curation; A. G. software; A. G. funding acquisition.
Supplementary Material
Acknowledgments
We thank Martina Toelge, Nicole Ritter, Nadja Reul, Markus Rueckl, Prof. Dr. Matthias Evert (UKR Regensburg), Dr. Birgit Striegl, and Prof. Dr. Lars Krenkel (OTH Regensburg) for excellent experimental support.
This work was supported by Bayerische Forschungsstiftung Grant AZ 1070-13 (to A. G.) and Deutsche Forschungsgemeinschaft Grants GE671/14-2 (to A. G.) and INST 102/11-1 FUGG (to Prof. Dr. Lars Krenkel). The authors declare that they have no conflicts of interest with the contents of this article.
This article was selected as one of our Editors' Picks.
This article contains Figs. S1–S3.
- PDB
- Paget's disease of bone
- OC
- osteoclast
- AA
- amino acid
- UBA
- ubiquitin-associated
- KI
- knockin
- TRAP
- tartrate-resistant acid phosphatase
- OCG
- osteoclastogenesis
- sRANKL
- soluble receptor activator of NF-κB ligand
- μCT
- micro-computed tomography
- BMM
- bone marrow–derived monocyte
- Ctsk
- cathepsin K
- IB
- immunoblot
- qRT-PCR
- quantitative RT-PCR
- Ab
- antibody
- CaP
- calcium phosphate
- Tb.Sp
- trabecular separation
- Tb.N
- trabecular number
- Tb.Th
- trabecular thickness
- BV/TV
- bone volume fraction
- MMA
- methyl methacrylate
- OB
- osteoblast
- RIPA
- radioimmunoprecipitation assay
- P-
- phospho-
- MEM
- minimum essential medium
- CTX-I
- C-terminal telopeptides of type I collagen
- PINP
- procollagen type I N-terminal propeptide.
References
- 1. Paget, J. (1877) On a form of chronic inflammation of bones (osteitis deformans). Med. Chir. Trans. 60, 37–64.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Galson, D. L., and Roodman, G. D. (2014) Pathobiology of Paget's disease of bone. J. Bone Metab. 21, 85–98 10.11005/jbm.2014.21.2.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tan, A., and Ralston, S. H. (2014) Paget's disease of bone. QJM 107, 865–869 10.1093/qjmed/hcu075 [DOI] [PubMed] [Google Scholar]
- 4. Vallet, M., and Ralston, S. H. (2016) Biology and treatment of Paget's disease of bone. J. Cell. Biochem. 117, 289–299 10.1002/jcb.25291 [DOI] [PubMed] [Google Scholar]
- 5. Albagha, O. M. (2015) Genetics of Paget's disease of bone. Bonekey Rep. 4, 756 10.1038/bonekey.2015.125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rea, S. L., Walsh, J. P., Layfield, R., Ratajczak, T., and Xu, J. (2013) New insights into the role of sequestosome 1/p62 mutant proteins in the pathogenesis of Paget's disease of bone. Endocr. Rev. 34, 501–524 10.1210/er.2012-1034 [DOI] [PubMed] [Google Scholar]
- 7. Ciani, B., Layfield, R., Cavey, J. R., Sheppard, P. W., and Searle, M. S. (2003) Structure of the ubiquitin-associated domain of p62 (SQSTM1) and implications for mutations that cause Paget's disease of bone. J. Biol. Chem. 278, 37409–37412 10.1074/jbc.M307416200 [DOI] [PubMed] [Google Scholar]
- 8. Hocking, L. J., Lucas, G. J., Daroszewska, A., Cundy, T., Nicholson, G. C., Donath, J., Walsh, J. P., Finlayson, C., Cavey, J. R., Ciani, B., Sheppard, P. W., Searle, M. S., Layfield, R., and Ralston, S. H. (2004) Novel UBA domain mutations of SQSTM1 in Paget's disease of bone: genotype phenotype correlation, functional analysis, and structural consequences. J. Bone Miner. Res. 19, 1122–1127 10.1359/JBMR.0403015 [DOI] [PubMed] [Google Scholar]
- 9. Layfield, R., Ciani, B., Ralston, S. H., Hocking, L. J., Sheppard, P. W., Searle, M. S., and Cavey, J. R. (2004) Structural and functional studies of mutations affecting the UBA domain of SQSTM1 (p62) which cause Paget's disease of bone. Biochem. Soc. Trans. 32, 728–730 10.1042/BST0320728 [DOI] [PubMed] [Google Scholar]
- 10. Cavey, J. R., Ralston, S. H., Hocking, L. J., Sheppard, P. W., Ciani, B., Searle, M. S., and Layfield, R. (2005) Loss of ubiquitin-binding associated with Paget's disease of bone p62 (SQSTM1) mutations. J. Bone Miner. Res. 20, 619–624 10.1359/JBMR.041205 [DOI] [PubMed] [Google Scholar]
- 11. Cavey, J. R., Ralston, S. H., Sheppard, P. W., Ciani, B., Gallagher, T. R., Long, J. E., Searle, M. S., and Layfield, R. (2006) Loss of ubiquitin binding is a unifying mechanism by which mutations of SQSTM1 cause Paget's disease of bone. Calcif. Tissue Int. 78, 271–277 10.1007/s00223-005-1299-6 [DOI] [PubMed] [Google Scholar]
- 12. Durán, A., Serrano, M., Leitges, M., Flores, J. M., Picard, S., Brown, J. P., Moscat, J., and Diaz-Meco, M. T. (2004) The atypical PKC-interacting protein p62 is an important mediator of RANK-activated osteoclastogenesis. Dev. Cell 6, 303–309 10.1016/S1534-5807(03)00403-9 [DOI] [PubMed] [Google Scholar]
- 13. Kurihara, N., Hiruma, Y., Zhou, H., Subler, M. A., Dempster, D. W., Singer, F. R., Reddy, S. V., Gruber, H. E., Windle, J. J., and Roodman, G. D. (2007) Mutation of the sequestosome 1 (p62) gene increases osteoclastogenesis but does not induce Paget disease. J. Clin. Investig. 117, 133–142 10.1172/JCI28267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hiruma, Y., Kurihara, N., Subler, M. A., Zhou, H., Boykin, C. S., Zhang, H., Ishizuka, S., Dempster, D. W., Roodman, G. D., and Windle, J. J. (2008) A SQSTM1/p62 mutation linked to Paget's disease increases the osteoclastogenic potential of the bone microenvironment. Hum. Mol. Genet. 17, 3708–3719 10.1093/hmg/ddn266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Daroszewska, A., van't Hof, R. J., Rojas, J. A., Layfield, R., Landao-Basonga, E., Rose, L., Rose, K., and Ralston, S. H. (2011) A point mutation in the ubiquitin-associated domain of SQSMT1 is sufficient to cause a Paget's disease-like disorder in mice. Hum. Mol. Genet. 20, 2734–2744 10.1093/hmg/ddr172 [DOI] [PubMed] [Google Scholar]
- 16. Rea, S. L., Walsh, J. P., Ward, L., Yip, K., Ward, B. K., Kent, G. N., Steer, J. H., Xu, J., and Ratajczak, T. (2006) A novel mutation (K378X) in the sequestosome 1 gene associated with increased NF-κB signaling and Paget's disease of bone with a severe phenotype. J. Bone Miner. Res. 21, 1136–1145 10.1359/jbmr.060405 [DOI] [PubMed] [Google Scholar]
- 17. Chamoux, E., Couture, J., Bisson, M., Morissette, J., Brown, J. P., and Roux, S. (2009) The p62 P392L mutation linked to Paget's disease induces activation of human osteoclasts. Mol. Endocrinol. 23, 1668–1680 10.1210/me.2009-0066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sundaram, K., Shanmugarajan, S., Rao, D. S., and Reddy, S. V. (2011) Mutant p62P392L stimulation of osteoclast differentiation in Paget's disease of bone. Endocrinology 152, 4180–4189 10.1210/en.2011-1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yip, K. H., Feng, H., Pavlos, N. J., Zheng, M. H., and Xu, J. (2006) p62 ubiquitin binding-associated domain mediated the receptor activator of nuclear factor-κB ligand-induced osteoclast formation: a new insight into the pathogenesis of Paget's disease of bone. Am. J. Pathol. 169, 503–514 10.2353/ajpath.2006.050960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Komatsu, M., Waguri, S., Koike, M., Sou, Y. S., Ueno, T., Hara, T., Mizushima, N., Iwata, J., Ezaki, J., Murata, S., Hamazaki, J., Nishito, Y., Iemura, S., Natsume, T., Yanagawa, T., et al. (2007) Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131, 1149–1163 10.1016/j.cell.2007.10.035 [DOI] [PubMed] [Google Scholar]
- 21. Patntirapong, S., Habibovic, P., and Hauschka, P. V. (2009) Effects of soluble cobalt and cobalt incorporated into calcium phosphate layers on osteoclast differentiation and activation. Biomaterials 30, 548–555 10.1016/j.biomaterials.2008.09.062 [DOI] [PubMed] [Google Scholar]
- 22. Maria, S. M., Prukner, C., Sheikh, Z., Mueller, F., Barralet, J. E., and Komarova, S. V. (2014) Reproducible quantification of osteoclastic activity: characterization of a biomimetic calcium phosphate assay. J. Biomed. Mater. Res. B Appl. Biomater. 102, 903–912 10.1002/jbm.b.33071 [DOI] [PubMed] [Google Scholar]
- 23. Rodriguez, A., Durán, A., Selloum, M., Champy, M. F., Diez-Guerra, F. J., Flores, J. M., Serrano, M., Auwerx, J., Diaz-Meco, M. T., and Moscat, J. (2006) Mature-onset obesity and insulin resistance in mice deficient in the signaling adapter p62. Cell Metab. 3, 211–222 10.1016/j.cmet.2006.01.011 [DOI] [PubMed] [Google Scholar]
- 24. Jin, W., Chang, M., Paul, E. M., Babu, G., Lee, A. J., Reiley, W., Wright, A., Zhang, M., You, J., and Sun, S. C. (2008) Deubiquitinating enzyme CYLD negatively regulates RANK signaling and osteoclastogenesis in mice. J. Clin. Investig. 118, 1858–1866 10.1172/JCI34257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ishida, N., Hayashi, K., Hoshijima, M., Ogawa, T., Koga, S., Miyatake, Y., Kumegawa, M., Kimura, T., and Takeya, T. (2002) Large scale gene expression analysis of osteoclastogenesis in vitro and elucidation of NFAT2 as a key regulator. J. Biol. Chem. 277, 41147–41156 10.1074/jbc.M205063200 [DOI] [PubMed] [Google Scholar]
- 26. Ikeda, K., and Takeshita, S. (2016) The role of osteoclast differentiation and function in skeletal homeostasis. J. Biochem. 159, 1–8 10.1093/jb/mvv112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Laurin, N., Brown, J. P., Morissette, J., and Raymond, V. (2002) Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am. J. Hum. Genet. 70, 1582–1588 10.1086/340731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Obaid, R., Wani, S. E., Azfer, A., Hurd, T., Jones, R., Cohen, P., Ralston, S. H., and Albagha, O. M. (2015) Optineurin negatively regulates osteoclast differentiation by modulating NF-κB and interferon signaling: implications for Paget's disease. Cell Rep. 13, 1096–1102 10.1016/j.celrep.2015.09.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Destaing, O., Sanjay, A., Itzstein, C., Horne, W. C., Toomre, D., De Camilli, P., and Baron, R. (2008) The tyrosine kinase activity of c-Src regulates actin dynamics and organization of podosomes in osteoclasts. Mol. Biol. Cell 19, 394–404 10.1091/mbc.E07-03-0227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Saftig, P., Hunziker, E., Wehmeyer, O., Jones, S., Boyde, A., Rommerskirch, W., Moritz, J. D., Schu, P., and von Figura, K. (1998) Impaired osteoclastic bone resorption leads to osteopetrosis in cathepsin-K-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 95, 13453–13458 10.1073/pnas.95.23.13453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zach, F., Mueller, A., and Gessner, A. (2015) Production and functional characterization of murine osteoclasts differentiated from ER-Hoxb8-immortalized myeloid progenitor cells. PLoS One 10, e0142211 10.1371/journal.pone.0142211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xia, M., Gonzalez, P., Li, C., Meng, G., Jiang, A., Wang, H., Gao, Q., Debatin, K. M., Beltinger, C., and Wei, J. (2014) Mitophagy enhances oncolytic measles virus replication by mitigating DDX58/RIG-I-like receptor signaling. J. Virol. 88, 5152–5164 10.1128/JVI.03851-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pang, X., and Sun, N. L. (2009) Calcineurin-NFAT signaling is involved in phenylephrine-induced vascular smooth muscle cell proliferation. Acta Pharmacol. Sin. 30, 537–544 10.1038/aps.2009.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gerlag, D. M., Ransone, L., Tak, P. P., Han, Z., Palanki, M., Barbosa, M. S., Boyle, D., Manning, A. M., and Firestein, G. S. (2000) The effect of a T cell-specific NF-κB inhibitor on in vitro cytokine production and collagen-induced arthritis. J. Immunol. 165, 1652–1658 10.4049/jimmunol.165.3.1652 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.