

Abstract
The bacterial nucleoid-associated protein H-NS is a DNA-binding protein, playing a major role in gene regulation. To regulate transcription, H-NS silences genes, including horizontally acquired foreign genes. Escherichia coli H-NS is 137 residues long and consists of two discrete and independent structural domains: an N-terminal oligomerization domain and a C-terminal DNA-binding domain, joined by a flexible linker. The N-terminal oligomerization domain is composed of two dimerization sites, dimerization sites 1 and 2, which are both required for H-NS oligomerization, but the exact role of dimerization site 2 in gene silencing is unclear. To this end, we constructed a whole set of single amino acid substitution variants spanning residues 2 to 137. Using a well-characterized H-NS target, the slp promoter of the glutamic acid–dependent acid resistance (GAD) cluster promoters, we screened for any variants defective in gene silencing. Focusing on the function of dimerization site 2, we analyzed four variants, I70C/I70A and L75C/L75A, which all could actively bind DNA but are defective in gene silencing. Atomic force microscopy analysis of DNA–H-NS complexes revealed that all of these four variants formed condensed complexes on DNA, whereas WT H-NS formed rigid and extended nucleoprotein filaments, a conformation required for gene silencing. Single-molecule stretching experiments confirmed that the four variants had lost the ability to form stiffened filaments. We conclude that dimerization site 2 of H-NS plays a key role in the formation of rigid H-NS nucleoprotein filament structures required for gene silencing.
Keywords: Escherichia coli (E. coli), gene silencing, dimerization, transcription factor, gene regulation, bacterial chromatin, DNA compaction, H-NS, nucleoid-associated protein, transcription repression
Introduction
A group of nucleoid-associated proteins (NAPs)6 are involved in the regulation of transcription (1–5). H-NS, originally referred to as histone-like protein H1, is one of the major core NAPs. In Escherichia coli, more than 10,000 molecules of H-NS exist, mostly associated with the genome (5, 6). Recent super-resolution imaging and single-particle tracking experiments determined that 95% of H-NS protein was bound to DNA (7). Genomic systematic evolution of ligands by exponential enrichment, ChIP sequencing, and transcriptome analyses have identified around 1000 sites associated with H-NS, which regulates 5% of the genome (8, 9), including horizontally transferred foreign genes (10, 11).
H-NS recognizes and binds to intrinsically curved, AT-rich DNA, often located near promoters (12, 13). Promoter-associated DNA curvature is believed to provide H-NS with an initial contact site for transcriptional silencing (14). After initial binding, H-NS binds cooperatively to form a rigid nucleoprotein filament along the target genes (15, 16). A filamentous structure of DNA–H-NS complexes is required for gene silencing (4, 17). In E. coli and Salmonella, H-NS–mediated silencing can be relieved by a set of positive regulators such as Fis (18), CRP (18, 19), LeuO (8, 20, 21), Ler (22), Lrp (21), SsrB (17, 23), and SlyA (24).
Molecular analysis of truncated H-NS proteins led to predictions that E. coli H-NS (137-amino acid residues) consists of an N-terminal oligomerization domain (residues 1–79) (7) and a C-terminal DNA-binding domain (residues 95–137) (7, 25). The two domains are joined by a flexible linker (residues 80–94) that has no secondary structure between the two domains (Fig. 1A). Recent analysis of the linker identified a role for the five charged residues in initial engagement with DNA, indicating that it is much more than a passive tether (7). The oligomerization and DNA-binding domains and the linker are highly conserved among H-NS family members (7, 26). Numerous single amino acid substitutions have been identified that were unable to repress promoter activities in vivo (27–30), based on the repression patterns of some model promoters (bglG, fimB, and proV). Ten substitutions were located within the oligomerization domain, and 25 mapped within the DNA-binding domain (Fig. 1, A–C). Most substitutions in the DNA-binding domain mapped within the core DNA-binding motif (TWTG-GR-P) between residues 108–116 (Fig. 1, A and C).
Figure 1.

The domain organization and structure of H-NS with mutated positions previously identified by random mutagenesis. A, arrows indicate the amino acid residue positions of H-NS substitutions previously identified with secondary structures (27–29). At the bottom, the substituted positions of H-NS, which did not silence the slp promoter (identified in this study) are represented. Substituted amino acids are colored as the follows: light green, a previously identified residue; red, a residue isolated from this study; yellow, a residue identified by both previous studies and this work. B, the identified amino acid residues shown in A are colored light green in the three-dimensional dimer structure of the E. coli N-terminal truncated H-NS (PDB code 3NR7) (24). C, the substituted amino acid residues shown in A are colored in the three-dimensional structure of the E. coli C-terminal truncated H-NS (PDB code 1HNR) (25). D, the amino acid sequences of dimerization site 2 (residues 57–79) were aligned using ClustalW2 (https://www.ebi.ac.uk/Tools/msa/clustalo/) (48). (Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party–hosted site.) The completely conserved amino acid residues are shown in bold. The H-NS homologues are as follows: H-NSs from E. coli, Shigella flexneri, Salmonella enterica serovar Typhimurium, Serratia marcescens, Yersinia enterocolitica, Erwinia chrysanthemi, Proteus vulgaris, Haemophilus influenzae, StpA from E. coli; and VicH from Vibrio cholerae. Amino acid residues highlighted in gray are mutants isolated in this study, and those in red were studied in detail.
Recent findings with Salmonella H-NS have identified two dimerization sites, dimerization site 1 (amino acids 1–42) and dimerization site 2 (residues 57–79), which serve as oligomerization interfaces in head-to-head and tail-to-tail oligomers (25). Dimerization site 1 is required for gene silencing. At least three substitutions in site 1 (R15E, L26P, and L30P) fail to form a rigid nucleoprotein filament (14, 31, 32), resulting in loss of gene silencing in vivo. Dimerization site 1 in Salmonella H-NS also interacts with Hha (33). Hha is also a NAP and is partially required for H-NS silencing. In E. coli, Ueda et al. (34) identified some genes that are co-silenced by H-NS, Hha, and/or YdgT (Cnu). In contrast, dimerization site 2 has not been characterized, although amino acids in dimerization site 2 are also conserved among H-NS homologues (Fig. 1D). L65P, a mutant in site 2, is defective in silencing in vivo, but further analysis was not performed (30). In a recent paper, a double mutant (D68V and D71V) in this region showed enhanced H-NS oligomerization and transcriptional repression in vitro (35), although the underlying mechanistic basis of this behavior was not examined. Unfortunately, this study referred to this region as the linker (35), which generated confusion in the field. In H-NS family members, formation of a rigid nucleoprotein filament is essential for gene silencing (14, 31, 32), and mutants in the oligomerization or DNA-binding domain of H-NS family proteins that are incapable of gene silencing are also incapable of forming rigid nucleoprotein filaments (16, 20). Thus, we were inspired to re-examine the role of dimerization site 2 in H-NS function.
We created an entire set of H-NS mutants, each carrying a single cysteine substitution from residues 2 to 137. We then examined the ability of these mutants to silence a well-characterized target, the slp promoter of the glutamic acid-dependent acid resistance (GAD) cluster promoters (36, 37). Two substitutions in dimerization site 2, I70C and L75C, resulted in loss of gene silencing. We then examined the mode of DNA binding using atomic force microscopy (AFM) and transverse magnetic tweezers. Our results indicated that the mutants were capable of binding to DNA and forming compact DNA structures but were unable to form higher-ordered nucleoprotein filaments. Thus, the second dimerization site coordinates N-terminal oligomerization and C-terminal DNA binding. Amino acid substitutions can disrupt this site-dependent domain–domain communication, abrogating gene silencing. The Cys-scanned H-NS series constructed in this study will provide a useful tool to analyze H-NS mutants by cysteine modification.
Results
Construction of an entire set of single Cys-substituted H-NS mutants
The GAD system is a major acid resistance system of E. coli, in which the transcription factor GadE plays a key role in regulating genes involved in acid resistance, including slp, dctR, yhiD, hdeB, hdeA, hdeD, gadE, mdtE, mdtF, gadW, gadX, and gadA (37). These genes are organized into eight transcription units: slp-dctR, hdeAB-yhiD, hdeD, gadE, mdtEF, gadW, gadX, and gadAX (38). Because most of the GAD cluster genes are the targets of silencing by H-NS (34), we used the GAD cluster promoters as representative of H-NS-mediated gene silencing and examined the effect of H-NS cysteine substitution on its silencing ability.
For detection of promoter activity, we employed the lux reporter system. For this purpose, the target promoters pgadA, pgadE, pgadW, phdeA, phdeD, and pslp, were inserted into the vector pLUX (38). The lux fusion plasmids were introduced into both the parent and the hns-deficient strains. Transformants were harvested at logarithmic phase (OD600 = 0.3), and then luciferase activity was measured. The activity of three promoters, pgadA, pgadW, and pslp, increased more than 10-fold in the hns-deficient strain compared with the parent strain (data not shown). Among these, the slp promoter was selected for systematic analysis of H-NS in E. coli because its activity was the highest in the hns-deficient mutant.
To examine the structural and functional roles of H-NS, we constructed an entire set of single Cys-substituted mutants from residues 2 to 136 of H-NS. For this purpose, the lone Cys residue at position 21 in WT H-NS was first converted to Ser, and the resulting C21S mutant hns gene was cloned into pQE80hns-C21S (see “Experimental procedures”). Transformants harboring pLUXslp and pQE80Lhns or pQE80Lhns-C21S, were grown in 96-well plates at 37 °C, and luciferase activity was measured. In E. coli cells, Cys substitutions did not affect cell growth (data not shown). The activity of pslp increased in the hns-deficient strain compared with the parent strain. This enhancement was suppressed by complementation with pQE80Lhns and pQE80Lhns-C21S (Fig. 2A). Using pQE80Lhns-C21S as a template, we next tried to construct 136 single Cys-substituted H-NS mutants at all positions. Except for the A18C mutant, the entire set of single Cys-substituted mutants was obtained. These Cys mutants will be useful for future site-specific modification experiments.
Figure 2.

Single Cys substitutions in H-NS that cannot silence slp promoter activity. A, hns plasmids (pQE80Lhns and pQE80Lhns-C21S) or an empty plasmid (a vector, pQE80L) were introduced into the WT or a Δhns strain harboring pLUXslpp. Transformants were grown in a 96-well plate containing LB medium with 10 μm IPTG at 37 °C overnight, and then the promoter activity was calculated as described under “Experimental procedures.” B, an entire set of single Cys-substituted hns-expressing plasmids (Table S2) was used for each promoter activity in Δhns strains as described above. The horizontal axis in the right panel indicates the position of H-NS amino acid residues. The A18C mutant was not measured. Seven mutants lost silencing activity (stars). C, hns plasmids (pQE80Lhns and pQE80LhnsN1) or an empty plasmid (a vector, pQE80L) were introduced into the WT, a Δhns strain, a Δhha strain, a ΔydgT strain, or a ΔhhaΔydgT strain harboring pLUXslpp. Transformants were grown in a 96-well plate containing LB medium with 10 μm IPTG at 37 °C overnight, and then the promoter activity was calculated as described under “Experimental procedures.”
Identification of hns mutants that were unable to silence genes
To determine the influence of Cys substitution on the silencing activity of H-NS, the pslp reporter plasmid was introduced into an entire array of hns mutants (Table S2). The level of lux reporter expression increased for a subset of H-NS mutants, indicating an inability to silence, as shown in Fig. 2B. The disruption in silencing activity of the H-NS mutants was essentially the same for all three promoters tested. Cys substitution resulted in a marked loss of silencing activity at seven positions in H-NS: four within the DNA-binding domain (T97C, W109C, G111C, and G113C) and three within dimerization site 2 (A67C, I70C, and L75C) (Figs. 1 and 2). Mutants in dimerization site 1 rarely affected GAD promoters, although others reported that R12C and R54C derepressed proV (28).
We further examined the effects of site 1 on the GAD promoter. A truncated H-NS, lacking N-terminal residues 1–46 (containing dimerization site 1), could not silence the slp promoter (Fig. 2C). We suspected that additional factors were involved in H-NS function for slp silencing because Hha and YdgT (Cnu) are modulators that interact with oligomerization site 1 of H-NS (33). The slp promoter activity increased in an hha and ydgT double mutant strain as well as an hns null strain (Fig. 2C), indicating that silencing of the slp promoter requires H-NS, Hha, and YdgT. Therefore, site 1 mutants might prevent slp silencing by Hha and/or YdgT activity (see “Discussion”).
As Ile70 and Leu75 are completely conserved among orthologs and paralogs of E. coli H-NS (Fig. 1D), these mutants displayed the largest defect in silencing pslp. We focused on them for a detailed analysis of H-NS structure and function. As an aside, the expression levels of I70C and L75C H-NS were similar to or higher than the WT (Fig. S1), indicating that the loss of silencing was attributable to a loss of function and not due to a reduced level of protein.
H-NS Cys substitutions in dimerization site 2 bind DNA
The loss of the silencing phenotype observed above could be due to an inability to bind slp promoter DNA. To examine this possibility, the DNA binding activity of I70C, I70A, L75C, and L75A mutants was analyzed using an electrophoretic mobility assay (EMSA). WT and the substituted proteins were purified from E. coli. To confirm the secondary structure of the mutant proteins, CD spectrometry analysis was performed, showing that purified H-NS–C21S–I70C displayed two negative maxima at ∼208 and 222 nm and a positive maximum at ∼192 nm and was similar to the WT H-NS and H-NS–C21S (Fig. S2). Estimation of the secondary structure indicated that all of the H-NS WT and mutant proteins had a similar α helix content (92%) (Table S1).
We next examined the DNA binding of the H-NS mutants in detail by EMSA. In the presence of WT H-NS, shifted complexes were first apparent at 120 nm (Fig. 3A, lane 4). The pattern of complex formation of the H-NS-C21S protein was similar to the WT but required >2.5-fold higher protein, indicating that the binding property of H-NS–C21S differs from WT H-NS. The two single Cys-substituted mutants, H-NS–C21S–I70C and H-NS–C21S–L75C, also formed protein–DNA complexes (Fig. 3, C and D), as did the Ala-substituted mutants (Fig. 3, E and F), although the binding pattern was different between Cys- and Ala-substituted H-NS (see “Discussion”). These results indicate that the loss of silencing activity of these two H-NS mutants was not due to an inability to bind DNA.
Figure 3.
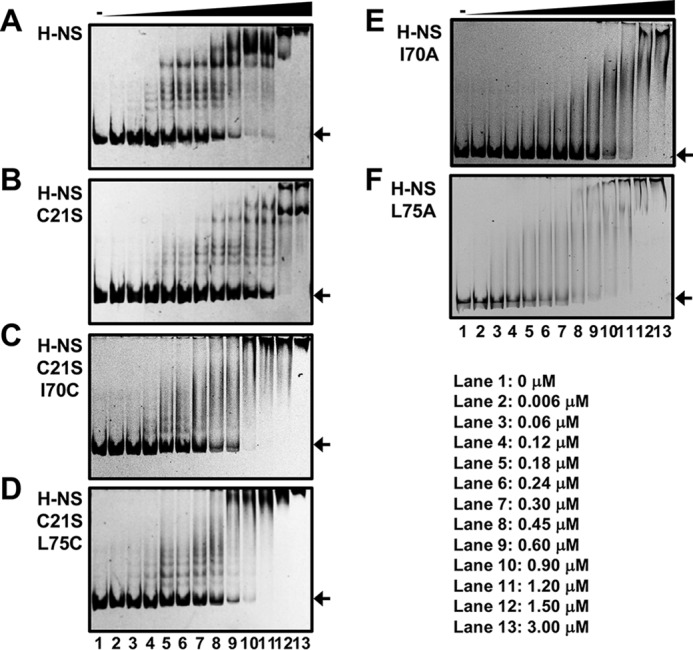
EMSA of H-NS binding to slp promoters. A–F, the DNA probe was amplified by PCR using the reporter plasmids as templates and the following primer pairs: pLUXslpp and a pair of SLP-F-2 and Lux-R-FITC. The FITC- labeled probe (0.033 μm) was incubated with H-NS (A), H-NS–C21S (B), H-NS–C21S-I70C (C), H-NS–C21S–L75C (D), H-NS–I70A (E), or H-NS–L75A (F) protein with 0 μm (lane 1), 0.006 μm (lane 2), 0.06 μm (lane 3), 0.12 μm (lane 4), 0.18 μm (lane 5), 0.24 μm (lane 6), 0.3 μm (lane 7), 0.45 μm (lane 8), 0.6 μm (lane 9), 0.9 μm (lane 10), 1.2 μm (lane 11), 1.5 μm (lane 12), and 3 μm (lane 13). The DNA–H-NS complex was analyzed by native PAGE. In the gel, nonspecific amplifying DNA was also found in both DNA probes. The arrows indicate the free DNA.
Dimerization site 2 mutants fail to form stiffened filaments
To understand the molecular basis of the silencing defect of the mutants in dimerization site 2, we examined DNA binding using AFM. In low-Mg2+ buffers, H-NS forms a rigid filament on DNA, and this filament is the basis for gene silencing (4, 15, 16). In the absence of protein, the naked DNA formed random coils (Fig. 4A). At 600 nm of H-NS or H-NS–C21S (2 monomers/bp), the DNA–protein complexes formed extended, thick filaments (Fig. 4, B, C, and H), indicating that both H-NS and H-NS–C21S were capable of forming stiffened H-NS nucleoprotein filaments. In contrast, H-NS–C21S–I70C (Fig. 4, D and H), H-NS–C21S–L75C (Fig. 4, E and H), H-NS–I70A (Fig. 4, F and H), and H-NS–L75A (Fig. 4, G and H) formed condensed DNA–protein complexes that were distinct from the nucleoprotein filaments formed by WT H-NS and H-NS–C21S (Fig. 4H). The quantification of AFM images by the radius of gyration illustrated that Cys- or Ala-substituted H-NS were similar in pattern to naked DNA, whereas H-NS and H-NS–C21S formed nucleoprotein filaments (Fig. 4H). In addition, formation of condensed structures did not differ between Cys– and Ala–H-NS (Fig. 4H).
Figure 4.

AFM imaging of DNA-H-NS complexes. A–G, the AFM imaging experiments were performed on glutaraldehyde-coated mica surfaces. slp promoter DNA (0.2 ng/μl) was incubated with 600 nm purified H-NS protein for 15 min in a test tube in 10 mm Tris-HCl (pH 7.4) and 50 mm KCl at room temperature. All AFM images were 0.7 μm × 0.7 μm. The color gauge represents the height of molecules in the AFM image. H, quantification of AFM imaging by measuring the radius of gyration.
The size of the nucleoid in cells expressing H-NS, H-NS–C21S, H-NS–C21S–I70C, H-NS–C21S–L75C, H-NS–I70A, or H-NS–L75A were quantified in cells stained with 4′,6-diamidino-2-phenylindole using structured illumination microscopy as described previously (7). The average nucleoid area of cells expressing H-NS or H-NS–C21S was 0.32 ± 0.12 μm2 or 0.38 ± 0.14 μm2 (n = 108), respectively. In contrast, that of H-NS–C21S–I70C, H-NS–C21S–L75C, H-NS–I70A, and H-NS–L75A was 0.36 ± 0.13 μm2 (n = 101), 0.36 ± 0.13 μm2 (n = 101), 0.35 ± 0.13 μm2 (n = 105), and 0.35 ± 0.11 μm2 (n = 107), respectively (Fig. S4). Thus, the site 2 substitution between Cys and Ala does not change DNA compaction in vitro.
The extended, stiffened DNA–protein filament formed by H-NS provides structural rigidity, which can be probed by single-molecule stretching experiments (15, 16). AFM imaging showed that dimerization site 2 mutants formed compact structures (Fig. 4). We next performed a force-jumping procedure to measure the force extension curve of DNA (see “Experimental procedures” for details), which minimizes interference from DNA folding during the measurement (4, 15, 16). In the absence of H-NS, naked DNA gradually extended with increased pulling force (Fig. 5, solid line). The extension of DNA complexed with H-NS or H-NS–C21S was significantly increased compared with naked DNA at the same force (Fig. 5, squares and circles), indicating that their binding increased the apparent DNA bending rigidity. In contrast, the extension of DNA complexed with H-NS–C21S–I70C or H-NS–C21S–L75C was similar to naked DNA at the same force (Fig. 5, triangles). Similar observations were also found for H-NS–I70A and H-NS–75A. The effects of protein binding on the apparent DNA bending rigidity can be quantified from such single-DNA stretching experiments (Fig. 5). The elastic behavior of a DNA polymer under tension can be modeled using the worm-like chain model, where the DNA bending rigidity is represented by a quantity with the dimension of length referred to as the persistence length. For naked DNA, the value of the bending persistence length has been determined to be ∼50 nm (39, 40). The persistence length of a DNA can be quantified in single-DNA stretching experiments by fitting the measured force extension curve using the Marko–Siggia formula (39, 40). Using this method, DNA fully coated with H-NS and H-NS–C21S had persistence lengths of 1130.29 ± 40.53 nm and 852.21 ± 53.34 nm, respectively (Table 1), which indicated significant DNA stiffening compared with the naked DNA with a persistence length of 54.58 ± 1.25 nm. The persistence lengths of complexed H-NS–C21S–I70C or H-NS–C21S–L75C were 47.06 ± 5.33 nm and 34.67 ± 1.36 nm, respectively. The persistence lengths of complexed H-NS–I70A and H-NS–L75A were 40.61 ± 4.82 nm and 46.63 ± 3.06 nm, respectively. These values were similar to naked DNA, indicating the absence of DNA stiffening. These results confirm the formation of a rigid nucleoprotein filament when DNA is complexed with H-NS or H-NS–C21S and the absence of a filament in the dimerization site 2 mutants.
Figure 5.
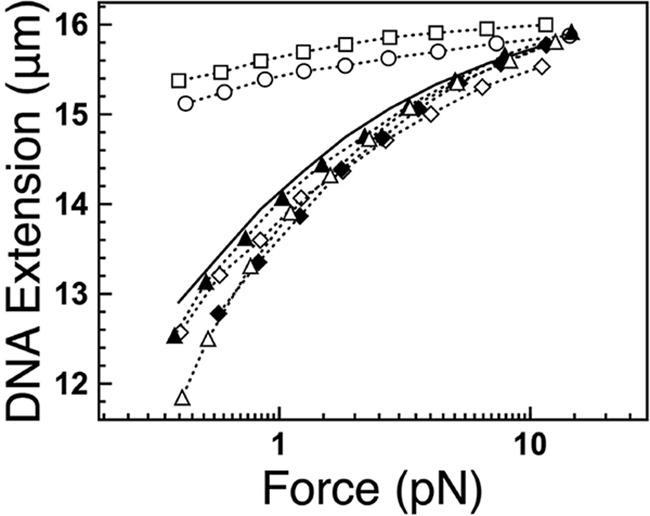
DNA stretching assay of DNA–H-NS complexes. Dotted lines and solid line, force extension curves of λ-DNA incubated with (dotted lines) or without (solid line) 600 nm H-NS proteins using force jumping (described under “Experimental procedures”). Shown are H-NS WT (open circles), H-NS-C21S (open squares), H-NS–C21S–I70C (filled triangles), H-NS–C21S–L75C (filled diamonds), H-NS–I70A (open triangles), and H-NS-L75A (open diamonds).
Table 1.
The value of bending rigidity of the DNA–H-NS complex
The values are indicated using the average of three independent DNAs.
| Contour length | Persistence length | |
|---|---|---|
| nm | nm | |
| Naked DNA | 16,365 ± 53 | 54.58 ± 1.25 |
| H-NS | 16,151 ± 25 | 1,130.29 ± 40.53 |
| H-NS–C21S | 15,956 ± 33 | 852.21 ± 53.34 |
| H-NS–C21S–I70C | 16,265 ± 75 | 47.06 ± 5.33 |
| H-NS–C21S–L75C | 16,552 ± 41 | 34.67 ± 1.36 |
| H-NS–I70A | 16,542 ± 11 | 40.61 ± 4.82 |
| H-NS–L75A | 16,533 ± 24 | 46.63 ± 3.06 |
Discussion
H-NS is composed of two discrete domains, an N-terminal oligomerization consisting of dimerization sites 1 and 2 and a C-terminal DNA-binding domain connected by a flexible linker. Because of the difficulty in crystallizing intact H-NS, structural analyses have been performed on the isolated N-terminal oligomerization and C-terminal DNA binding domains (25, 41, 42). Starting from the entire set of single Cys substitutions of H-NS, we identified two mutants in dimerization site 2, I70C and L75C, that suppressed the silencing activity of H-NS. These amino acid residues in dimerization site 2, I70 and L75, were completely conserved among orthologs and paralogs of E. coli H-NS (Fig. 1D).
The crystal structure of the N-terminal oligomerization domain of truncated Salmonella H-NS (residues 1–83) suggests a role for dimerization sites 1 and 2 in forming higher-order oligomers (25). In this model, the N terminus forms a higher-order structure in tandem by interactions between upper sites (for head to head), referred to as dimerization site-1, and between lower sites (for tail to tail), referred to as dimerization site 2 (Fig. 1B). Impairment of one of these two dimerization sites is expected to result in the disruption of high-order oligomers. Substitution at dimerization site 1 also abrogates gene silencing as well as the ability to form nucleoprotein filaments (16). The phenotypes of site 1 mutants that fail to silence target genes are well-known for the presence of DNA-binding ability even though they lack nucleoprotein filament formation. However, the role of site 2 has not been characterized. Recently, van der Valk et al. (43) showed that the E. coli H-NS mutant Y61D-M64D does not form a multimeric structure and stiffer nucleoprotein filament in vitro. Hence, site 2 seems to have an important role for H-NS function. Therefore, we re-examined site 2 function in detail.
Our findings with I70C/A and L75C/A shed light on the role of the H-NS dimerization site 2 in gene silencing. EMSA and AFM indicated the ability of DNA binding in both mutants but changed the binding patterns between Cys and Ala mutants (Fig. 3). This was evident at low H-NS concentrations (120∼240 nm, Fig. 3, lanes 4–6), suggesting that initial binding to DNA differs between Cys- and Ala-substituted H-NS. In addition, the difference between binding patterns might be affected by polar or structural changes of Cys-substituted H-NS compared with Ala substitution. On the other hand, formation of condensed structures was similar between Cys and Ala mutants (Fig. 4H). As the DNA complex in vitro, nucleoid formation in vivo did not differ between Cys and Ala substitutions (Fig. S4 and Table S2). Thus, only H-NS does not play a significant role in overall DNA compaction in the cell. Furthermore, the effects of protein binding on the apparent DNA bending rigidity can be quantified from such single-DNA stretching experiments (Fig. 5 and Table 1). DNA fully coated with H-NS and H-NS–C21S showed significant DNA stiffening compared with naked DNA. On the other hand, H-NS–C21S–I70C, H-NS–C21S–L75C, H-NS–I70A, and H-NS–L75A showed absence of DNA stiffening. These results demonstrate that silencing-defective mutants were unable to stiffen DNA upon binding. Recently, the high-resolution structure based on solid-state NMR spectra from full-length H-NS in E. coli has been determined (44), in which Arg-54 and Lys-57 were identified to contact Glu-74 and Asp-68, respectively, strongly supporting the involvement of dimerization site 2 in the molecular interplay of H-NS dimers forming long filaments needed for gene silencing. As this region is linked to the C-terminal DNA binding domain, this interaction is potentially important for tail-to-tail linkage of H-NS molecules as well as the position and direction of the C-terminal DNA-binding domain in an H-NS binding unit. Thus, we conclude that the phenotype of dimerization site 2 mutants is similar to that of site 1 in vitro; DNA binding ability is present but nucleoprotein filaments are absent. However, the roles of sites 1 and 2 seem to be different in vivo. Our tested promoter, slp, was suppressed by site 2 mutants, whereas the site 1 mutants showed minor effects on this promoter (Fig. 2). We suspect that these effects are due to other NAPs in vivo because the Hha binding site is located in site 1 (33). Hha is a NAP, but it does not have the DNA binding domain. Therefore, Hha cannot bind to DNA alone. Previously, Ueda et al. (34) indicated that the GAD region including slp was densely covered by H-NS and that H-NS distribution was decreased in the hha-deleted strain. Accordingly, the slp promoter is under the control of both H-NS and Hha. Our result confirms that H-NS silences the slp promoter with Hha and YdgT (Fig. 2C). On the other hand, a typical promoter, such as bglG and proV in previous studies, had minor effects on Hha in vivo (34). Site 1 mutants of Salmonella H-NS, I11A, and R12H, disrupt H-NS–Hha and H-NS–YdgT interaction without affecting DNA binding in vitro, resulting in a decrease in hilA and ssrB silencing in vivo (33). In addition, the NMR-based structural model of the complex between Hha and the truncated Salmonella H-NS (residues 1–46) reveals the formation of a three-protein charge zipper with an interdigitated complementary charged residues from Hha and the two units of the H-NS dimer (45). Our comprehensive mutant assay showed that I11C slightly suppressed the silencing activity of H-NS (Fig. 2B) because a cysteine substituted for isoleucine at the 11th residue in site 1 might partially affect electric interactions between H-NS and Hha. In summary, our findings implicate the conserved dimerization site 2 of H-NS as playing an important role in gene silencing and nucleoprotein filament formation in bacteria.
Experimental procedures
E. coli strains, plasmids, and growth media
The E. coli strains and plasmids used in this study are listed in Table S2. Luria–Bertani (LB) or LB agar (LB supplemented with 1.5% w/v bacto agar (pH 7)) was used for standard cloning procedures.
Construction of H-NS expression plasmids
pQE80Lhns for expression of His6–H-NS was kindly provided by Dr. Taku Oshima. To construct pQE80LhnsN1, the truncated hns gene was amplified by PCR using a pair of primers, H-NS–F-47 and H-NS–R, and W3110 typeA genome as the template. The resulting PCR fragment was digested by SalI and SphI and ligated into pQE80L at the corresponding sites. To construct a whole set of single Cys-substituted hns mutants, the original cysteine residue at position 21 was substituted with serine by site-directed mutagenesis. In a round of PCR cycles, a pair of primers, hnsC21S-F and hnsC21S-R2 (Table S2), annealed to the template DNA pQE80Lhns, replicating the plasmid DNA with the mutation. The resulting DNA pool (mutant and parental) was treated with DpnI to destroy the parental methylated DNA, leaving the newly synthesized unmethylated mutant DNA intact to transform E. coli cells. Thus, the plasmid for the original Cys-21-substituted H-NS, named pQE80Lhns-C21S, was prepared. Similar to H-NS–C21S, a set of cysteine-scanned hns mutants was constructed using pQE80Lhns-C21S as the template DNA and a pair of complementary primers with a mutation (Table S2). In addition, a set of alanine-scanning hns mutants for Ile-70 to Leu-75 was constructed using pQE80Lhns and pQE80Lhns-C21S as the template DNA and a pair of complementary primers with a mutation (Table S2). All of the plasmids were confirmed by DNA sequencing with pQE forward and pQE reverse primers for pQE80L derivatives.
Measurement of luciferase activity in E. coli
LB medium supplemented with 50 μg/ml kanamycin and 100 μg/ml ampicillin was used for the luciferase assay. A single colony of a strain carrying a lux reporter plasmid (Kmr) and an hns-expressing plasmid (Apr) was grown overnight at 37 °C with reciprocal shaking. The overnight culture was diluted 100-fold into the medium containing 10 μm IPTG to express lac-inducible H-NS. The culture was again grown overnight, and luciferase activity was measured as described by Yamanaka et al. (46). For identification of H-NS mutants affecting silencing, 96-well plates were used for incubation. Assays were performed in triplicate with three independent colonies for each strain, including that carrying the vector pQE80L in place of an hns-expressing plasmid, to obtain the mean with the standard deviation of luciferase activity relative to that of the WT strain.
Purification of H-NS proteins
To purify H-NS proteins, each pQE80L derivative plasmid was introduced into E. coli BL21 (DE3). In a typical procedure (47), a single colony of transformant was grown to OD600 = 0.6 at 30 °C with shaking in LB medium supplemented with 100 μg/ml ampicillin. His6–H-NS was induced with 0.5 mm IPTG at 30 °C for 3 h with shaking. Cells were isolated by centrifugation and resuspended in lysis buffer (1 m NaCl, 50 mm Tris-HCl (pH 8.0), and 1 mm DTT) containing 2% Triton X-100. Cells were treated with lysozyme and then subjected to sonication. The lysate was centrifuged, and the supernatant was mixed with 0.5 ml of nickel-nitrilotriacetic acid–agarose resin (Qiagen) and loaded onto a column. The column was washed with lysis/2% Triton X-100 buffer and then washed with lysis/2% Triton X-100 buffer containing 25 mm imidazole. Proteins were eluted with each elution buffer (lysis/2% Triton X-100 buffer with 0.1 m, 0.2 m, 0.3 m, 0.4 m, or 0.5 m imidazole), and peak fractions of H-NS were pooled and dialyzed against a storage buffer (1 m NaCl, 50 mm Tris-HCl (pH 8.0), 1 mm DTT, and 50% glycerol). The protein purity was then analyzed on SDS-PAGE.
Western blot analysis
E. coli cells grown in LB medium were harvested by centrifugation and resuspended in lysis buffer containing 8 m urea and sonicated. After centrifugation, the same volume of supernatant was subjected to 18% SDS-PAGE and blotted onto polyvinylidene difluoride membranes using an iBlot semidry transfer apparatus (Invitrogen). Membranes were first immunodetected with anti–H-NS serum (8) and horseradish peroxidase–conjugated anti-rabbit IgG (Nacalai Tesque) antibodies and then developed with a chemiluminescence kit (Nacalai Tesque). The image was analyzed with a LAS-4000 IR multicolor imager (Fuji Film).
CD spectroscopy
CD spectra of H-NS were measured using a J-820 spectropolarimeter (Jasco). The CD measurements were carried out in a wavelength range between 190 and 250 nm in a cell with a path length of 0.2 cm (volume, 400 μl) at 25 °C in binding buffer (10 mm Tris-HCl (pH 7.4) and 50 mm KCl). The spectra are the average of two or three independent measurements of five scans, each recorded in 0.5-nm increments at a scan speed of 20 nm/min. Estimation of secondary structure content was performed using a Spectra Manager (Jasco).
EMSA
Probes were amplified by PCR using pLUXslpp as a template, with a pair of primers: a specific primer and a FITC-labeled primer (Table S2). PCR products with FITC at their termini were purified using the QIAquick PCR purification kit (Qiagen). For EMSA, the FITC-labeled probes (∼700 bp) were each incubated with purified H-NS protein at room temperature for 15 min in the binding buffer. After addition of a DNA dye solution, the mixture was directly subjected to 5% PAGE. Fluorescently labeled DNA in gels was detected using LAS-4000 (Fuji Film).
AFM
The AFM imaging experiments were performed on glutaraldehyde-coated mica surfaces following Winardhi et al. (22). The slp promoter DNA (∼700 bp) was amplified by PCR using the W3110 genome as a template, with a pair of primers (Table S2). The PCR product was purified using the QIAquick PCR purification kit (Qiagen). The promoter DNA fragment was incubated with 600 nm of each H-NS protein for 15 min in a test tube in binding buffer at room temperature. Following this, the DNA or protein–DNA complexes were deposited for surface fixation on glutaraldehyde-coated mica for 15 min. The sample was then gently washed with deionized water, dried with N2 gas, and imaged. AFM imaging was performed using a Dimension FastScan AFM (Bruker Corp.) using tapping mode with a silicon nitride probe (FastScan-A, Bruker Corp.). Images were acquired with a resolution of 1024 × 1024 pixels and processed with Gwyddion software. Radius of gyration distributions were obtained by application of the threshold for image segmentation between the DNA or protein–DNA complexes and the background, followed by calculation of the radius of gyration for each of the distinct objects.
Transverse magnetic tweezers
The magnetic tweezers used in this study were similar to previous studies (15, 16, 22). λ-DNA labeled with biotin on both ends was tethered between a streptavidin-coated glass coverslip edge and streptavidin-coated paramagnetic beads. A pair of permanent magnets was used to stretch the DNA along the focal plane. The pulling force was controlled by adjusting the distance between the magnet and the magnetic bead. The force extension data were obtained by using the force-jumping procedure, which was carried out as follows. A single DNA was initially held at a high force (∼10 pN) and then jumped to a series of lower forces for around 2 s for extension measurement. Following each force jump to lower forces, the force was jumped back to ∼10 pN to ensure that protein-induced DNA folding was minimal (approximately <300 nm below that of naked DNA at a force of ∼10 pN) and to unfold the nucleoprotein complex, if any, before the measurement resumed. The force extension curve is thus obtained with minimal contribution from DNA folding.
Author contributions
Y. Y. and I. K. resources; Y. Y., R. S. W., E. Y., S. N., Y. S., J. Y., and K. Y. data curation; Y. Y., R. S. W., E. Y., S. N., Y. S., J. Y., I. K., and K. Y. formal analysis; Y. Y., R. S. W., E. Y., S. N., Y. S., J. Y., I. K., A. I., and K. Y. investigation; Y. Y., R. S. W., E. Y., S. N., Y. S., and J. Y. methodology; Y. Y., R. S. W., J. Y., I. K., A. I., and K. Y. writing-original draft; Y. Y., R. S. W., S. N., Y. S., J. Y., I. K., A. I., and K. Y. writing-review and editing; J. Y., I. K., A. I., and K. Y. supervision; J. Y. and K. Y. funding acquisition; I. K., A. I., and K. Y. conceptualization; K. Y. project administration.
Supplementary Material
Acknowledgments
We thank Prof. Linda J. Kenney (Mechanobiology Institute) for assistance with planning experiments, interpreting the results, and comments and editing of the manuscript. The set of single Cys substitution mutant H-NS expression plasmids was constructed during experimental training in the Department of Frontier Bioscience of Hosei University. We also thank Dr. Taku Oshima, Biotechnology Research Center and Department of Biotechnology, Toyama Prefectural University, for providing the Δhns strain and the hns-expressing pQE80Lhns plasmid. We also thank Kayoko Yamada, Hiroki Watanabe, Sho Watarai, and Eri Arita for technical support.
This work was supported by an RCE in Mechanobiology, the National Institutes of Health, and the Alcohol and Education Research Council. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4, Tables S1 and S2, Experimental procedures, and references.
- NAP
- nucleoid-associated protein
- GAD
- glutamic acid–dependent acid resistance
- AFM
- atomic force microscopy
- EMSA
- electrophoretic mobility shift assay
- LB
- Luria–Bertani
- IPTG
- isopropyl 1-thio-β-D-galactopyranoside.
References
- 1. Dorman, C. J. (2009) Regulatory integration of horizontally-transferred genes in bacteria. Front. Biosci. (Landmark Ed.) 14, 4103–4112 [DOI] [PubMed] [Google Scholar]
- 2. Dillon, S. C., and Dorman, C. J. (2010) Bacterial nucleoid-associated proteins, nucleoid structure and gene expression. Nat. Rev. Microbiol. 8, 185–195 10.1038/nrmicro2261 [DOI] [PubMed] [Google Scholar]
- 3. Ali, S. S., Xia, B., Liu, J., and Navarre, W. W. (2012) Silencing of foreign DNA in bacteria. Curr. Opin. Microbiol. 15, 175–181 10.1016/j.mib.2011.12.014 [DOI] [PubMed] [Google Scholar]
- 4. Winardhi, R. S., Yan, J., and Kenney, L. J. (2015) H-NS regulates gene expression and compacts the nucleoid: insights from single-molecule experiments. Biophys. J. 109, 1321–1329 10.1016/j.bpj.2015.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ali Azam, T., Iwata, A., Nishimura, A., Ueda, S., and Ishihama, A. (1999) Growth phase-dependent variation in the protein composition of Escherichia coli nucleoid. J. Bacteriol. 181, 6361–6370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ishihama, A., Kori, A., Koshio, E., Yamada, K., Maeda, H., Shimada, T., Makinoshima, H., Iwata, A., and Fujita, N. (2014) Intracellular concentrations of transcription factors in Escherichia coli: 65 species with known regulatory functions. J. Bacteriol. 196, 2718–2727 10.1128/JB.01579-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gao, Y., Foo, Y. H., Winardhi, R. S., Tang, Q., Yan, J., and Kenney, L. J. (2017) Charged residues in the H-NS linker drive DNA binding and gene silencing in single cells. Proc. Natl. Acad. Sci. U.S.A. 114, 12560–12565 10.1073/pnas.1716721114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shimada, T., Bridier, A., Briandet, R., and Ishihama, A. (2011) Novel roles of LeuO in transcription regulation in E. coli: genome antagonistic interplay with the universal silencer H-NS. Mol. Microbiol. 82, 376–397 [DOI] [PubMed] [Google Scholar]
- 9. Oshima, T., Ishikawa, S., Kurokawa, K., Aiba, H., and Ogasawara, N. (2006) Escherichia coli histone-like protein H-NS preferentially binds to horizontally acquired DNA in association with RNA polymerase. DNA Res. 13, 141–153 10.1093/dnares/dsl009 [DOI] [PubMed] [Google Scholar]
- 10. Göransson, M., Sonden, B., Nilsson, P., Dagberg, B., Forsman, K., Emanuelsson, K., and Uhlin, B. E. (1990) Transcriptional silencing and thermoregulation of gene expression in Escherichia coli. Nature 344, 682–685 10.1038/344682a0 [DOI] [PubMed] [Google Scholar]
- 11. Navarre, W. W., Porwollik, S., Wang, Y., McClelland, M., Rosen, H., Libby, S. J., and Fang, F. C. (2006) Selective silencing of foreign DNA with low GC content by the H-NS protein in Salmonella. Science 313, 236–238 10.1126/science.1128794 [DOI] [PubMed] [Google Scholar]
- 12. Bracco, L., Kotlarz, D., Kolb, A., Diekmann, S., and Buc, H. (1989) Synthetic curved DNA sequences can act as transcriptional activators in Escherichia coli. EMBO J. 8, 4289–4296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jáuregui, R., Abreu-Goodger, C., Moreno-Hagelsieb, G., Collado-Vides, J., and Merino, E. (2003) Conservation of DNA curvature signals in regulatory regions of prokaryotic genes. Nucleic Acids Res. 31, 6770–6777 10.1093/nar/gkg882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gordon, B. R., Li, Y., Cote, A., Weirauch, M. T., Ding, P., Hughes, T. R., Navarre, W. W., Xia, B., and Liu, J. (2011) Structural basis for recognition of AT-rich DNA by unrelated xenogeneic silencing proteins. Proc. Natl. Acad. Sci. U.S.A. 108, 10690–10695 10.1073/pnas.1102544108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu, Y., Chen, H., Kenney, L. J., and Yan, J. (2010) A divalent switch drives H-NS/DNA-binding conformations between stiffening and bridging modes. Genes Dev. 24, 339–344 10.1101/gad.1883510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lim, C. J., Lee, S. Y., Kenney, L. J., and Yan, J. (2012) Nucleoprotein filament formation is the structural basis for bacterial protein H-NS gene silencing. Sci. Rep. 2, 509 10.1038/srep00509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Walthers, D., Li, Y., Liu, Y., Anand, G., Yan, J., and Kenney, L. J. (2011) Salmonella enterica response regulator SsrB relieves H-NS silencing by displacing H-NS bound in polymerization mode and directly activates transcription. J. Biol. Chem. 286, 1895–1902 10.1074/jbc.M110.164962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Caramel, A., and Schnetz, K. (2000) Antagonistic control of the Escherichia coli bgl promoter by Fis and CAP in vitro. Mol. Microbiol. 36, 85–92 10.1046/j.1365-2958.2000.01827.x [DOI] [PubMed] [Google Scholar]
- 19. Shimada, T., Fujita, N., Yamamoto, K., and Ishihama, A. (2011) Novel roles of cAMP receptor protein (CRP) in regulation of transport and metabolism of carbon sources. PLoS ONE 6, e20081 10.1371/journal.pone.0020081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Navarre, W. W., McClelland, M., Libby, S. J., and Fang, F. C. (2007) Silencing of xenogeneic DNA by H-NS-facilitation of lateral gene transfer in bacteria by a defense system that recognizes foreign DNA. Genes Dev. 21, 1456–1471 10.1101/gad.1543107 [DOI] [PubMed] [Google Scholar]
- 21. Ishihama, A., Shimada, T., and Yamazaki, Y. (2016) Transcription profile of Escherichia coli: Genomic SELEX search for regulatory targets of transcription factors. Nucleic Acids Res. 44, 2058–2074 10.1093/nar/gkw051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Winardhi, R. S., Gulvady, R., Mellies, J. L., and Yan, J. (2014) Locus of enterocyte effacement-encoded regulator (Ler) of pathogenic Escherichia coli competes off histone-like nucleoid-structuring protein (H-NS) through noncooperative DNA binding. J. Biol. Chem. 289, 13739–13750 10.1074/jbc.M113.545954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Desai, S. K., Winardhi, R. S., Periasamy, S., Dykasm, M. M., Jiem, Y., and Kenney, L. J. (2016) The horizontally-acquired response regulator SsrB drives a Salmonella lifestyle switch by relieving biofilm silencing. eLife 10.7554/eLife.10747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stoebel, D. M., Free, A., and Dorman, C. J. (2008) Anti-silencing: overcoming H-NS-mediated repression of transcription in Gram-negative enteric bacteria. Microbiology 154, 2533–2545 10.1099/mic.0.2008/020693-0 [DOI] [PubMed] [Google Scholar]
- 25. Arold, S. T., Leonard, P. G., Parkinson, G. N., and Ladbury, J. E. (2010) H-NS forms a superhelical protein scaffold for DNA condensation. Proc. Natl. Acad. Sci. U.S.A. 107, 15728–15732 10.1073/pnas.1006966107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shindo, H., Iwaki, T., Ieda, R., Kurumizaka, H., Ueguchi, C., Mizuno, T., Morikawa, S., Nakamura, H., and Kuboniwa, H. (1995) Solution structure of the DNA binding domain of a nucleoid-associated protein, H-NS, from Escherichia coli. FEBS Lett. 360, 125–131 10.1016/0014-5793(95)00079-O [DOI] [PubMed] [Google Scholar]
- 27. Dorman, C. J. (2004) H-NS: A Universal regulator for a dynamic genome. Nat. Rev. Microbiol. 2, 391–400 10.1038/nrmicro883 [DOI] [PubMed] [Google Scholar]
- 28. Williams, R. M., Rimsky, S., and Buc, H. (1996) Probing the structure, function, and interactions of the Escherichia coli H-NS and StpA proteins by using dominant negative derivatives. J. Bacteriol. 178, 4335–4343 10.1128/jb.178.15.4335-4343.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ueguchi, C., Suzuki, T., Yoshida, T., Tanaka, K., and Mizuno, T. (1996) Systematic mutational analysis revealing the functional domain organization of Escherichia coli nucleoid protein H-NS. J. Mol. Biol. 263, 149–162 10.1006/jmbi.1996.0566 [DOI] [PubMed] [Google Scholar]
- 30. Donato, G. M., and Kawula, T. H. (1999) Phenotypic analysis of random hns mutations differentiate DNA-binding activity from properties of fimA promoter inversion modulation and bacterial motility. J. Bacteriol. 181, 941–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Qu, Y., Lim, C. J., Whang, Y. R., Liu, J., and Yan, J. (2013) Mechanism of DNA organization by Mycobacterium tuberculosis protein Lsr2. Nucleic Acids Res. 41, 5263–5272 10.1093/nar/gkt249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Winardhi, R. S., Fu, W., Castang, S., Li, Y., Dove, S. L., and Yan, J. (2012) Higher order oligomerization is required for H-NS family member MvaT to form gene-silencing nucleoprotein filament. Nucleic Acids Res. 40, 8942–8952 10.1093/nar/gks669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ali, S. S., Whitney, J. C., Stevenson, J., Robinson, H., Howell, P. L., and Navarre, W. W. (2013) Structural insights into the regulation of foreign genes in Salmonella by the Hha/H-NS complex. J. Biol. Chem. 10; 288, 13356–13369 10.1074/jbc.M113.455378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ueda, T., Takahashi, H., Uyar, E., Ishikawa, S., Ogasawara, N., and Oshima, T. (2013) Functions of the Hha and YdgT proteins in transcriptional silencing by the nucleoid proteins, H-NS and StpA, in Escherichia coli. DNA Res. 20, 263–271 10.1093/dnares/dst008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Giangrossi, M., Wintraecken, K., Spurio, R., and de Vries, R. (2014) Probing the relation between protein-protein interactions and DNA binding for a linker mutant of the bacterial nucleoid protein H-NS. Biochim. Biophys. Acta 1844, 339–345 10.1016/j.bbapap.2013.11.010 [DOI] [PubMed] [Google Scholar]
- 36. Krin, E., Danchin, A., and Soutourina, O. (2010) Decrypting the H-NS-dependent regulatory cascade of acid stress resistance in Escherichia coli. BMC Microbiol. 10, 273 10.1186/1471-2180-10-273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mates, A. K., Sayed, A. K., and Foster, J. W. (2007) Products of the Escherichia coli acid fitness island attenuate metabolite stress at extremely low pH and mediate a cell density-dependent acid resistance. J. Bacteriol. 189, 2759–2768 10.1128/JB.01490-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Burton, N. A., Johnson, M. D., Antczak, P., Robinson, A., and Lund, P. A. (2010) Novel aspects of the acid response network of E. coli K-12 are revealed by a study of transcriptional dynamics. J. Mol. Biol. 401, 726–742 10.1016/j.jmb.2010.06.054 [DOI] [PubMed] [Google Scholar]
- 39. Bustamante, C., Marko, J. F., Siggia, E. D., and Smith, S. (1994) Entropic elasticity of λ-phage DNA. Science 265, 1599–1600 10.1126/science.8079175 [DOI] [PubMed] [Google Scholar]
- 40. Marko, J., and Siggia, E. (1995) Stretching DNA. Macromol. 28, 8759–8770 10.1021/ma00130a008 [DOI] [Google Scholar]
- 41. Esposito, D., Petrovic, A., Harris, R., Ono, S., Eccleston, J. F., Mbabaali, A., Haq, I., Higgins, C. F., Hinton, J. C., Driscoll, P. C., and Ladbury, J. E. (2002) H-NS oligomerization domain structure reveals the mechanism for high order self-association of the intact protein. J. Mol. Biol. 324, 841–850 10.1016/S0022-2836(02)01141-5 [DOI] [PubMed] [Google Scholar]
- 42. Bloch, V., Yang, Y., Margeat, E., Chavanieu, A., Augé, M. T., Robert, B., Arold, S., Rimsky, S., and Kochoyan, M. (2003) The H-NS dimerization domain defines a new fold contributing to DNA recognition. Nat. Struct. Biol. 10, 212–218 10.1038/nsb904 [DOI] [PubMed] [Google Scholar]
- 43. van der Valk, R. A., Vreede, J., Qin, L., Moolenaar, G. F., Hofmann, A., Goosen, N., and Dame, R. T. (2017) Mechanism of environmentally driven conformational changes that modulate H-NS DNA-bridging activity. eLife 6, e27369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Renault, M., García, J., Cordeiro, T. N., Baldus, M., and Pons, M. (2013) Protein oligomers studied by solid-state NMR: the case of the full-length nucleoid-associated protein histone-like nucleoid structuring protein. FEBS J. 280, 2916–2928 10.1111/febs.12297 [DOI] [PubMed] [Google Scholar]
- 45. Cordeiro, T. N., García, J., Bernadó, P., Millet, O., and Pons, M. (2015) A three-protein charge zipper stabilizes a complex modulating bacterial gene silencing. J. Biol. Chem. 290, 21200–21212 10.1074/jbc.M114.630400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yamanaka, Y., Oshima, T., Ishihama, A., and Yamamoto, K. (2014) Characterization of the YdeO regulon in Escherichia coli. PLoS ONE 9, e111962 10.1371/journal.pone.0111962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yamamoto, K., Hirao, K., Oshima, T., Aiba, H., Utsumi, R., and Ishihama, A. (2005) Functional characterization in vitro of all two-component signal transduction systems from Escherichia coli. J. Biol. Chem. 280, 1448–1456 10.1074/jbc.M410104200 [DOI] [PubMed] [Google Scholar]
- 48. Li, W., Cowley, A., Uludag, M., Gur, T., McWilliam, H., Squizzato, S., Park, Y. M., Buso, N., and Lopez, R. (2015) The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 43, W580–W584 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.