

Abstract
Kidney- and brain-expressed protein (KIBRA), a multifunctional scaffold protein with around 20 known binding partners, is involved in memory and cognition, organ size control via the Hippo pathway, cell polarity, and membrane trafficking. KIBRA includes tandem N-terminal WW domains, a C2 domain, and motifs for binding atypical PKC and PDZ domains. A naturally occurring human KIBRA variant involving residue changes at positions 734 (Met-to-Ile) and 735 (Ser-to-Ala) within the C2 domain affects cognitive performance. We have elucidated 3D structures and calcium- and phosphoinositide-binding properties of human KIBRA C2 domain. Both WT and variant C2 adopt a canonical type I topology C2 domain fold. Neither Ca2+ nor any other metal ion was bound to WT or variant KIBRA C2 in crystal structures, and Ca2+ titration produced no significant reproducible changes in NMR spectra. NMR and X-ray diffraction data indicate that KIBRA C2 binds phosphoinositides via an atypical site involving β-strands 5, 2, 1, and 8. Molecular dynamics simulations indicate that KIBRA C2 interacts with membranes via primary and secondary sites on the same domain face as the experimentally identified phosphoinositide-binding site. Our results indicate that KIBRA C2 domain association with membranes is calcium-independent and involves distinctive C2 domain–membrane relative orientations.
Keywords: nuclear magnetic resonance (NMR); crystallography; phosphoinositide; molecular dynamics; analytical ultracentrifugation; calcium-binding protein; Alzheimer disease; α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPA receptor, AMPAR); Hippo pathway; membrane trafficking; C2 domain; human cognition; KIBRA; WWC protein family
Introduction
Kidney- and brain-expressed protein (KIBRA2; also called WW- and C2 domain–containing protein 1 (WWC1)) is a large (1119-, 1118-, and 1113-amino acid isoforms), multifunctional scaffold protein that has two N-terminal WW domains, a putative nuclear localization signal (1), putative coiled-coil regions, a C2 domain, a glutamic acid–rich motif, an atypical PKC–binding region, and a C-terminal PDZ-binding motif (see Fig. 1). KIBRA has about 20 reported binding partners (2) and disease links, including dementia (3–5), kidney disease (6), Tourette disorder (7), and some cancers (8, 9). Among its multiple functions, KIBRA is an upstream component of the Hippo pathway (8, 10), a central mechanism of organ size control and cellular homeostasis. KIBRA also has a role in cell polarity and migration, functioning as a link between polarity proteins and cytoskeleton components (2, 11–14). In addition, KIBRA functions in membrane trafficking via regulation of vesicular transport (12, 15–17).
Figure 1.
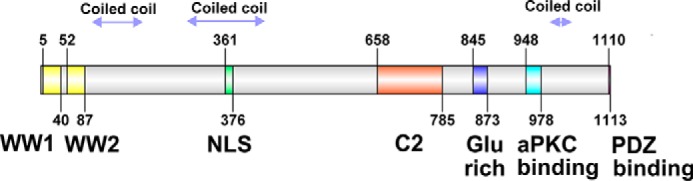
Schematic structure of human KIBRA protein showing locations of motifs and domains. The nuclear localization signal (NLS) and coiled-coil regions are predicted rather than experimentally demonstrated. aPKC, atypical PKC.
KIBRA is linked to memory, cognition, and neurological disorders in humans and in rodent models (for a review, see Ref. 2). A single nucleotide polymorphism (SNP), rs17070145, in the ninth intron of KIBRA, for example, has been implicated in human cognition (15), a finding corroborated by numerous subsequent studies, including a meta-analysis (16). rs17070145 is associated with Alzheimer's disease (AD) (3, 17). KIBRA, moreover, has additive and epistatic interactions with APOE (18), the ϵ4 allele of which is the strongest genetic risk factor for sporadic AD. The effect of SNP rs17070145 on memory and AD may arise due to differential activation of the MAPK pathway (19), important for memory and learning processes. In human, rat, and mouse brains, KIBRA is mainly expressed in memory-related regions, including hippocampus and cortex, as well as cerebellum and hypothalamus (15, 20, 21).
KIBRA acts in the same pathway as protein kinase Mζ (PKMζ), a brain-specific kinase thought to be involved in long-term memory storage through its control of neuroreceptor trafficking, particularly trafficking of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, the major excitatory neurotransmitter receptors in the brain (22). KIBRA binds, colocalizes with, and is phosphorylated by PKMζ (11, 23), and KIBRA is involved in AMPA receptor trafficking (24). KIBRA counteracts proteasomal degradation of PKMζ (25). In addition, KIBRA binds to dendrin (26) and synaptopodin (13, 27), postsynaptic cytoskeleton organizers important for synaptic transmission and cognition. Aberrant acetylation of tau, linked to cognitive deterioration in dementia, disrupts postsynaptic function by reducing postsynaptic KIBRA (4, 28). Consistent with these accumulated observations, KIBRA overexpression and knockdown exert positive and negative effects, respectively, on key molecular and cellular processes in neurons (29). It has been shown by immunoprecipitation that KIBRA can form homodimers (20) and heterodimers with the two other WWC protein family members, WWC2 and WWC3 (10), although the functional significance of this remains unknown.
Here, we characterize the calcium- and phosphoinositide-binding properties of KIBRA C2 domain (Fig. 1), including a variant that arises from two SNPs (rs3822660G/T and rs3822659T/G) in human KIBRA that result in substitution of two adjacent residues (M734I,S735A) (30). Although the molecular consequences of these exonic SNPs have not been fully elucidated, they affect cognitive performance, and there is almost complete linkage disequilibrium between the aforementioned intronic SNP rs17070145 (15) and rs3822660G/T and rs3822659T/G (30).
The C2 domain is found in more than 125 different human proteins (31, 32). Although most C2 domains are Ca2+-dependent membrane association domains, some C2 domains do not bind Ca2+, and some mediate protein–protein rather than protein–lipid interactions (31, 32). Our experimental and computational investigations of WT and variant KIBRA C2 domains indicate that KIBRA C2 is a noncanonical C2 domain that in vitro both binds phosphoinositides and associates with membranes in an atypical, calcium-independent manner.
Results
WT and variant KIBRA C2 adopt a typical C2 fold
Our 2.6-Å resolution crystal structure (Protein Data Bank code 6FD0) shows that variant C2 (varC2) adopts a typical eight-stranded β-sandwich C2 domain fold. The crystal form comprises a parallel dimer (i.e. the two monomers are parallel to each other) formed via an intermonomer disulfide bond involving Cys-771, the fifth and only unpaired cysteine in KIBRA C2 (Fig. 2A). The previously available crystal structure of WT KIBRA C2 (Protein Data Bank code 2Z0U) involves a similar parallel, Cys-771 disulfide-linked dimer. According to analytical ultracentrifugation (AUC) measurements (Fig. S1 and Table S2), WTC2 and varC2 samples both comprise a mixture of monomer and dimer in solution.
Figure 2.
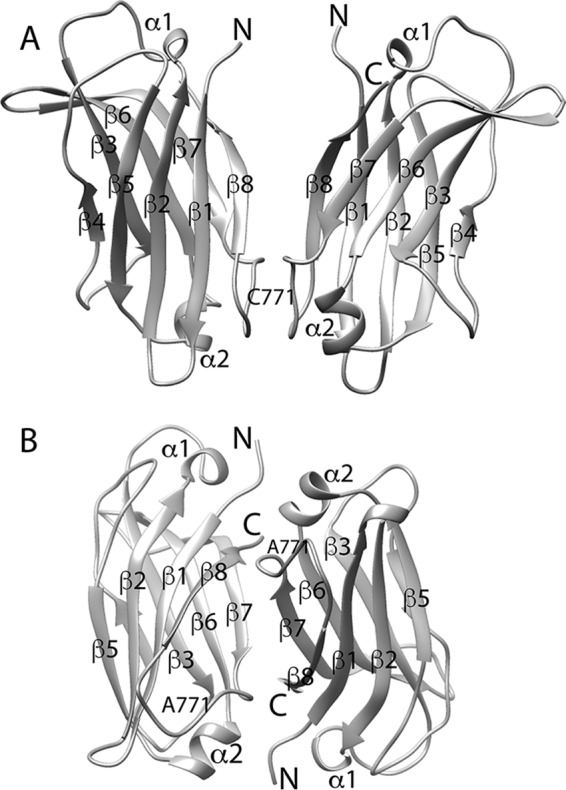
Crystal structures of variant and mutant (C771A) KIBRA C2. A, varC2 parallel dimer (Protein Data Bank code 6FD0). B, C2(C771A) antiparallel dimer (Protein Data Bank code 6FB4). Termini, β-strands, α-helices, and residue 771 are labeled. In A, the Cys-771 label corresponds to the position of the intermonomer disulfide bond, which mediates parallel dimer formation in the WTC2 and varC2 crystal structures.
Conversely, AUC and nuclear magnetic resonance (NMR) relaxation measurements (Figs. S1 and S2 and Tables S2 and S3) indicate that the C771A mutant C2 (C2(C771A)) and variant C771A mutant C2 (varC2(C771A)) are monomeric in solution. NMR samples of C2(C771A) and varC2(C771A) were homogeneous and stable, permitting backbone resonance assignments to be made using standard NMR methods (Fig. S3). Although AUC and NMR indicate that C2(C771A) is monomeric in solution, its crystal structure (Protein Data Bank code 6FB4; 2.5-Å resolution) comprises an antiparallel dimer (Fig. 2B), most likely formed due to crystal packing.
The backbone 1H, 13C, and 15N NMR chemical shifts of C2(C771A) and varC2(C771A) match closely the corresponding chemical shifts back-calculated using SPARTA+ (33) from the C2(C771A) crystal structure (Protein Data Bank code 6FB4). Correspondingly, the TALOS+ (34) and SPARTA+ (33) predictions are that the secondary structure compositions of C2(C771A) and varC2(C771A) in solution are very similar to the C2(C771A) crystal structure (Fig. S4). The β-sheet NOE patterns of C2(C771A) and varC2(C771A), moreover, are as expected based on the C2 topology I fold, allowing for peak overlap and absence. NMR data therefore indicate that C2(C771A) and varC2(C771A) in solution are conformationally very similar to each other (Fig. S5) and to C2 monomers in C2(C771A), varC2 (Protein Data Bank codes 6FB4 and 6FD0, respectively) and WTC2 (Protein Data Bank code 2Z0U) crystal structures.
Because KIBRA dimerization has been indicated by cell-based data (10, 20), MD simulations were conducted to compare conformational stabilities of the C2 dimers observed in crystal structures. Both parallel dimers (WTC2/2Z0U and varC2/6FD0) exhibited significant changes in relative monomer position (Fig. 3) with the changes varying between repeat simulations and force fields. In WTC2 dimer simulations, the mean r.m.s.d. value after three repeat simulations was 0.52 ± 0.04 and 0.49 ± 0.05 nm for GROMOS and OPLS force fields, respectively. The corresponding values in varC2 simulations were 0.51 ± 0.08 nm and 1.4 ± 0.23 nm. In equivalent simulations, in contrast, the C2(C771A) antiparallel dimer (Protein Data Bank code 6FB4) did not undergo significant changes in relative monomer position. There were no significant conformational changes within C2 monomers in any simulation (Fig. 3).
Figure 3.
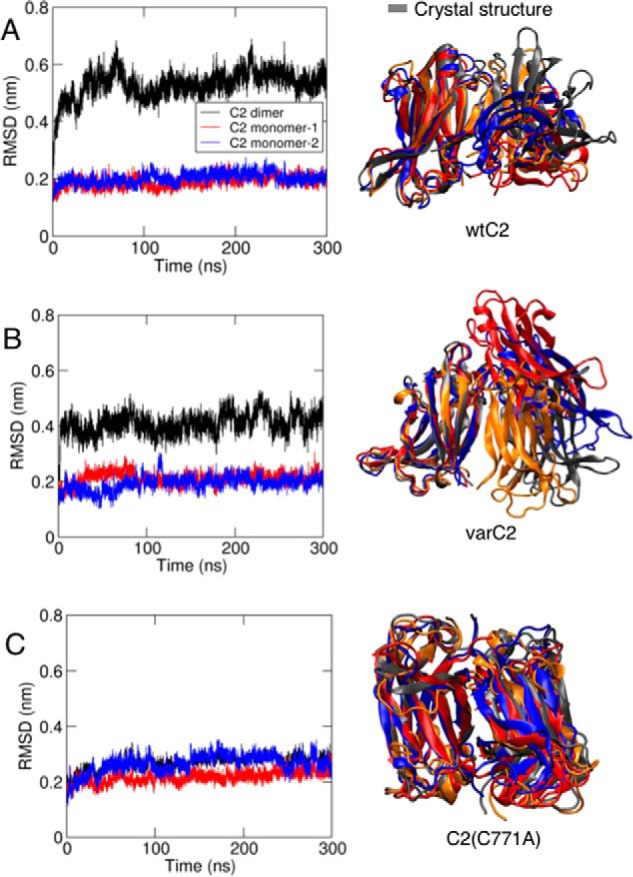
MD simulation of KIBRA C2 dimer conformation in solution. r.m.s.d. for WTC2 (A), varC2 (B), and varC2(C771A) (C) with r.m.s.d. for the monomers in blue and red and for the dimer in black is shown for one of the simulations of each system. Final structures from the three repeat simulations for each system are also shown with one monomer superimposed, in blue, red, and orange, with the corresponding crystal structure in gray.
NMR and X-ray diffraction data indicate that KIBRA C2–Ca2+ interaction is very low-affinity and nonspecific
Ca2+ binding was investigated by NMR, X-ray crystallography (XRC), and simulation. In numerous 1H-15N HSQC-monitored titrations with C2(C771A) and one with WTC2, calcium chloride addition to 10–20 mm and sometimes higher concentrations (up to 76 mm) produced no significant reproducible chemical shift change with the largest composite 1H-15N changes around 0.05 ppm. Such small changes may be due to a very weak Ca2+ ion interaction (Kd > 10 mm), possibly nonspecific Ca2+ binding to oxygen-rich surface clusters. Such a nonspecific interaction is supported by simulation: when eight Ca2+ ions were initially positioned randomly around KIBRA C2, the final Ca2+ positions in eight simulations included the calcium-binding regions (CBRs) plus six other locations around the domain (Fig. S6).
It is also possible that the domain scavenged Ca2+ during expression and purification and was therefore Ca2+-bound prior to the experiments, although this seems unlikely because particular effort was made during some C2 NMR sample preparations to exclude Ca2+. Initial 1H-15N HSQC spectra were very similar, furthermore, irrespective of whether or not measures were taken to exclude Ca2+ from NMR samples.
In several crystal structures, including WTC2 (Protein Data Bank code 2Z0U) and our C2(C771A) and varC2 structures, no bound Ca2+ has been observed, although the protein for 2Z0U was produced by cell-free synthesis and Protein Data Bank entry for 2Z0U does not mention any attempt to introduce metal ions. The crystal structures determined here used Escherichia coli–expressed protein with no attempt to exclude Ca2+, however, so the C2 domains presumably encountered Ca2+ and other divalent metal ions during expression and purification. Several times C2 crystals were soaked in solutions containing divalent metal ions, including 20 mm CaCl2, 100 mm CaCl2, and 100 mm MnSO4. Soaking did not cause visible disintegration of the crystals. Diffraction after soaking was still good, but these experiments yielded only one metal-bound structure, which had one Ca2+ located in the intermonomer interface of a C2 dimer rather than in any of the CBRs.
Overall, our NMR and XRC data are consistent with KIBRA C2 domain having very low (Kd > 10 mm), nonspecific affinity for Ca2+. Together with Protein Data Bank code 2Z0U, moreover, our NMR and XRC data show that lack of bound Ca2+ does not disrupt KIBRA C2 from adopting a typical C2 domain fold.
KIBRA C2 exhibits an unusual phosphoinositide interaction mode
NMR and XRC studies were conducted to define the phosphoinositide-binding site location(s) on KIBRA C2. Three NMR titrations were conducted by recording 1H-15N HSQC spectra as a function of increasing concentration of phosphoinositide and subsequently calcium chloride. These titrations, involving C2(C771A) with phosphatidylinositol 3-phosphate (PI(3)P), C2(C771A) with phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2), and varC2 with PI(4,5)P2, produced very similar results in which phosphoinositide-induced chemical shift changes were observed for numerous peaks, almost all located in the β-sheet comprising β-strands 5, 2, 1, and 8 (Fig. 4). Leu-666 (β1), Ile-677 (β2), and Leu-678 (β2) backbone 1H-15N peaks exhibited the largest chemical shift perturbations with changes observed for around 20 further backbone 1H-15N peaks plus two pairs of unassigned side-chain NH2 peaks (Fig. 4). The Kd for C2(C771A)–PI(3)P binding was determined as a representative case using 13 perturbed residues (Fig. S7); the average Kd was 1 mm. After phosphoinositide titration, CaCl2 was titrated into the same NMR samples to establish whether Ca2+ ions influence C2–phosphoinositide interaction or vice versa; there was no further spectral change upon CaCl2 addition.
Figure 4.
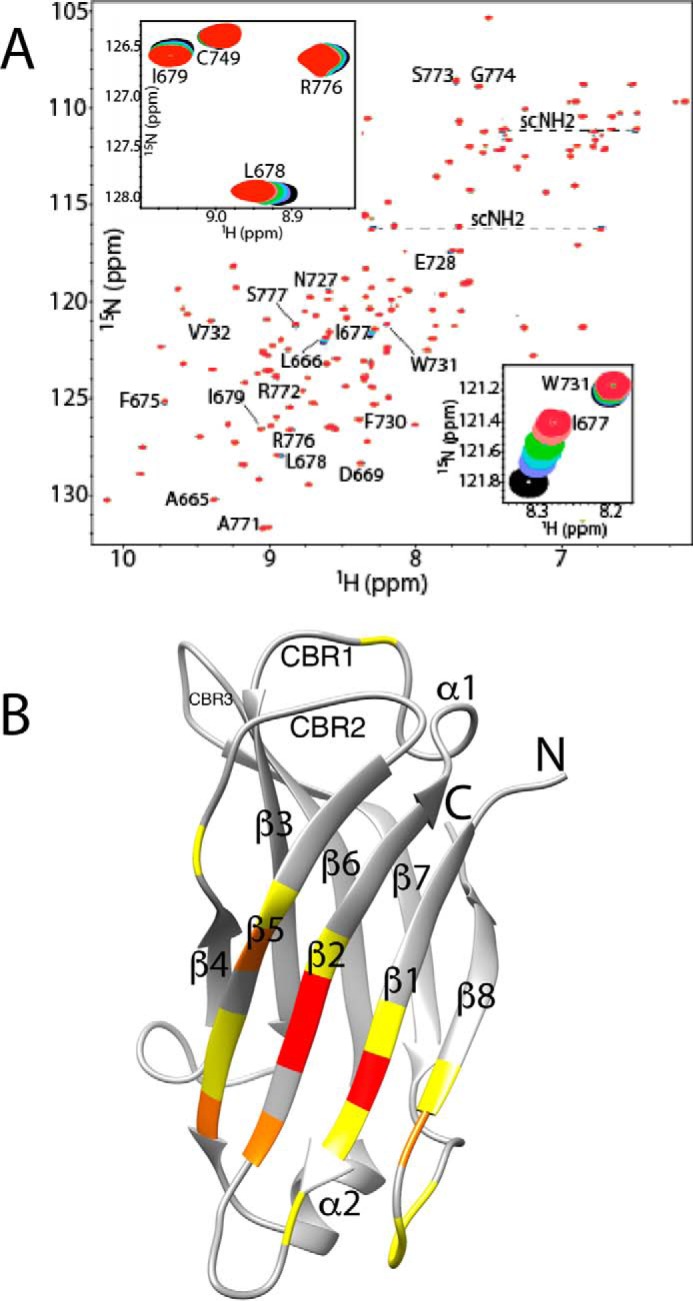
C2(C771A)–PI(3)P interaction monitored by NMR. A, overlay of C2(C771A) 1H-15N HSQC spectra as a function of increasing PI(3)P concentration. Perturbed backbone NH peaks are labeled, and the two perturbed but unassigned side-chain NH2 peak pairs are connected by dashed lines and denoted as “scNH2.” The insets highlight the chemical shift changes of backbone NH peaks of Ile-677 (strand β2) and Trp-731 (β5) and of Leu-678 (β2), Ile-679 (β2), and Arg-776 (β7-β8 loop). Peak colors and corresponding C2(C771A):PI(3)P ratios are as follows: red, 1:0; pink, 1:0.25; green, 1:0.5; cyan, 1:0.75; purple, 1:1; black, 1:1.5. B, perturbed residues mapped onto a ribbon representation of C2(C771A). Yellow represents residues with Δδav (ppm) ≤ 0.01, orange represents 0.01–0.03, and red represents >0.03 where Δδav (ppm) = ((ΔδH2 + ΔδN2(γN/γH))/2)0.5. The secondary structure elements (β-strands and α-helices) are labeled. Also, for ease of comparison with other C2 domains, the locations of canonical CBR1, CBR2, and CBR3 are indicated.
Cocrystallization experiments involving C2(C771A) and varC2 with PI(4,5)P2 or phosphatidylinositol 3,4,5-trisphosphate (PI(3,4,5)P3) yielded crystals of C2(C771A) with bound PI(4,5)P2 and PI(3,4,5)P3 in ProPlex HT-96 condition G1 (Protein Data Bank codes 6FJD and 6FJC; 2.6- and 2.9-Å resolution, respectively). Both PI(4,5)P2 and PI(3,4,5)P3 bound to monomer A of an antiparallel C2(C771A) dimer with polar interactions between Arg-776 (β7-β8 loop) and negatively charged oxygen atoms of phosphoinositide phosphates (Fig. 5). Leu-678 (β2) and Val-729 (β5) side chains and main chains of Leu-666 (β1) and Ile-677 (β2) interact with the phosphoinositide inositol/aliphatic chains. The C2(C771A)–PI(4,5)P2 and C2(C771A)–PI(3,4,5)P3 crystal structure binding site is consistent with the phosphoinositide-induced spectral changes in the 1H-15N HSQC titrations; the backbone NH groups of Leu-666, Ile-677, and Leu-678, for example, showed the largest chemical shift changes. Phosphoinositide binding occurs with small changes in C2 structure, e.g. a typical overall r.m.s.d. of 0.28 Å between unbound and phosphoinositide-bound structures.
Figure 5.

X-ray diffraction structure of the C2(C771A)–PI(3,4,5)P3 complex. Shown are three views of the complex (Protein Data Bank code 6FJC) in which rotation of view A about the vertical axis through ∼90° gives view B. PI(3,4,5)P3 binds to chain A of an antiparallel C2(C771A) dimer as does PI(4,5)P2. C, a closer view of the C2(C771A)–PI(3,4,5)P3 interaction in the same orientation as A with electron density obtained in a simulated annealing omit map around the PI(3,4,5)P3-binding site shown in blue mesh, contoured at 1.1σ. PI(3,4,5)P3 is shown mainly in magenta. Some of the C2 residues that interact with PI(3,4,5)P3 are shown (hydrophobic residues in yellow, polar residues in cyan, and positively charged residues in blue), including the side chains of Leu-678, Gln-681, Asn-727, and Arg-776 and backbone of Leu-666 and Ile-677. Oxygen and nitrogen atoms are shown in red and dark blue, respectively.
Coarse-grained MD simulations were performed to further examine KIBRA C2 monomer and dimer association with phosphoinositides and membranes. In these simulations, which can predict the binding modes of peripheral proteins to model membrane (35, 36), the three experimentally observed forms of KIBRA C2 (monomer, antiparallel dimer, and parallel dimer, all without Ca2+) were displaced away from a preformed phosphoinositide-containing bilayer. In the primary (more frequent) binding mode observed with a C2 monomer, C2 interaction with phosphoinositides involves mainly Arg-661, Lys-671, Arg-772, Arg-776, and Arg-779 (Fig. 6), slightly shifted from the crystal structure/NMR titration binding site. In a small number of simulations, the C2–phosphoinositide interaction involves mainly Lys-667 and Arg-776, coincident with the crystal structure/NMR titration binding site (Figs. 4 and 5). The parallel dimer (WTC2/2Z0U) shows two binding modes with one phosphoinositide often located close to the crystal structure/NMR titration binding site (Fig. 7). The antiparallel dimer (C2(C771A)) exhibits a primary binding mode in which the main interactions with the membrane occur via one of the two C2 domains (Fig. 8). In this orientation, a phosphoinositide is observed near the crystal structure/NMR titration binding site. In all cases, KIBRA C2 domain binding to the bilayer causes a degree of phosphoinositide clustering around the domain. Phosphoinositides interact mainly with lysines and arginines that face the bilayer.
Figure 6.
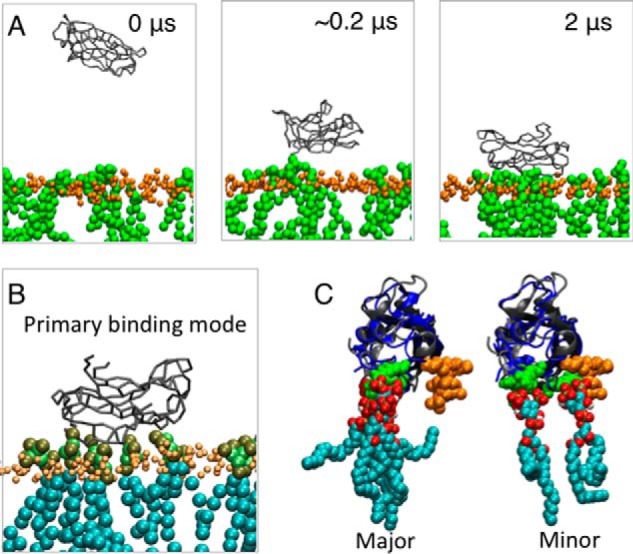
Coarse-grained MD simulation of KIBRA C2 monomer association with membrane. A, coarse-grained simulation setup. Snapshots are shown from one of the simulations at 0, ∼0.2, and 2 μs. KIBRA C2 domain is shown in gray, lipid phosphate atoms are shown in orange, and phosphoinositide lipids are shown in green. B, a snapshot of one of the simulations showing the preferred orientation of the C2 domain relative to the bilayer. C, alignment of C2 domain from the simulations (gray) with PI(3,4,5)P3-bound C2 crystal structure (blue). The phosphoinositides bound to C2 in the simulations are also shown. Oxygen atoms on lipids are shown in red, hydrogen atoms are shown in white, and carbon atoms are shown in cyan.
Figure 7.
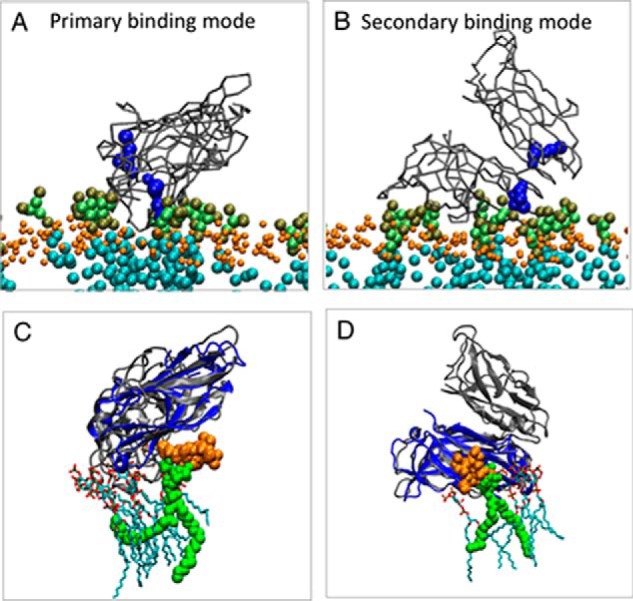
Coarse-grained MD simulation of KIBRA C2 parallel dimer association with membrane. A and B, primary (A) and secondary (B) binding modes of the KIBRA C2 parallel dimer from simulation. KIBRA C2 domain is shown in gray, lipid phosphate atoms are shown in orange, and phosphoinositide lipids are shown in green. Residues Lys-667 and Arg-776 are shown in blue. C and D, alignment of C2 domain from the simulations (gray) with the PI(3,4,5)P3-bound C2 crystal structure (blue). The phosphoinositides bound to C2 in the simulations are also shown. The phosphoinositide molecule that is close to the phosphoinositide-binding site observed by XRC and NMR is shown in green. Oxygen atoms on lipids are shown in red, hydrogen atoms are shown in white, and carbon atoms are shown in cyan.
Figure 8.
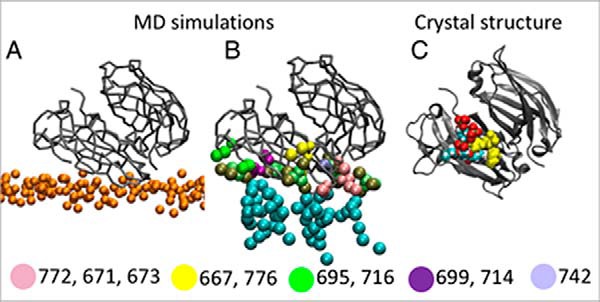
Coarse-grained MD simulation of KIBRA C2 antiparallel dimer association with membrane. A, primary orientation of the varC2(C771A) antiparallel dimer relative to the bilayer. The C2 dimer is shown in gray, and lipid phosphate atoms are shown in orange. B, phosphoinositide-binding sites on KIBRA C2 resulting from simulation. The residues in each site are shown in a different color. C, the varC2(C771A) crystal structure is shown in the same orientation as in A and B. Residues Lys-667 and Arg-776 that interact with the phosphoinositide analogue are shown as yellow spheres. Oxygen atoms on lipids are shown in red, hydrogen atoms are shown in white, and carbon atoms are shown in cyan.
Discussion
KIBRA C2 structure
We have elucidated structural and functional characteristics of four forms of KIBRA C2 domain: WT, naturally occurring variant, and C771A mutants of WT and variant. Variant KIBRA, involving two amino acid changes in the C2 domain (M734I,S735A), affects human cognitive performance and is in almost complete linkage disequilibrium (30) with a previously identified intronic SNP that affects cognition (15, 16). WTC2 and varC2 adopt a type I topology C2 domain fold with slightly different dimerization properties in solution (Fig. S1), indicating a possible effect of M734I,S735A residue differences on domain behavior. The M734I substitution has previously been calculated to reduce C2 stability (30), although WTC2 and varC2 adopt very similar conformations in both crystal structures and in solution (Figs. S3 and S5).
It has been shown previously that full-length WT KIBRA forms dimers in mammalian cells (20). Overexpression of FLAG-tagged KIBRA in HEK293 cells, moreover, resulted in large KIBRA-containing clusters (20). KIBRA also forms heterodimers with the two other WWC protein family members, WWC2 and WWC3 (10). Yeast two-hybrid mapping indicated that KIBRA dimerizes in an antiparallel orientation with the C2 domain and one or more regions N-terminal of C2 required for this interaction (20). Whether or not KIBRA can dimerize in mammalian cells via intermolecular disulfide bonding remains open to question. Because redox potentials, and therefore likelihood of intermolecular disulfide bonding, vary according to cell type, cell status, and cell compartment, a mixture of noncovalent and covalent WWC protein dimerization modes is possible in vivo. The degree of KIBRA dimerization and/or oligomerization in the cell could, for example, fine-tune KIBRA function as a hub for multiprotein complex assembly. Different dimerization tendencies of WTC2 and varC2 could then provide an indication of the molecular mechanism(s) underpinning the link between variant KIBRA C2 and improved cognition (30). Other components of the Hippo pathway are modulated by dimerization; indeed, YAP2L and TAZ form disulfide-mediated dimers that are more stable and more oncogenic than the corresponding monomers (37). Finally, because oxidative stress can promote disulfide bond formation in cytoplasmic proteins (38), the extent of disulfide-mediated KIBRA dimerization, and hence KIBRA function, could be regulated by oxidative stress. It is potentially relevant in this context that KIBRA-associated pathways can be redox-modulated (39) and that oxidative stress is a hallmark of AD (40).
Calcium binding
Any KIBRA C2–Ca2+ interaction is low-affinity (Kd > 10 mm): we did not observe bound metal ions in any of our six C2 crystal structures with or without crystal soaking with Ca2+ or other divalent metal ions or bound phosphoinositide, and there is no bound Ca2+ in the previous WTC2 structure (Protein Data Bank code 2Z0U). In NMR-monitored Ca2+ titrations, moreover, the C2(C771A) 1H-15N HSQC spectrum did not change significantly. Unlike KIBRA C2, most C2 domains bind Ca2+ with sub-mm affinity. For example, synaptotagmin I C2B domain has a Ca2+ affinity of about 500 μm (41), perforin C2 has a Ca2+ affinity of ∼200 μm (42), and rabphilin-3A C2B domain has a particularly high Ca2+ affinity with Kd values of 7 and 11 μm (43) explained by contributions from C2-flanking residues (44). Like KIBRA C2, however, some C2 domains show very weak or no residual calcium binding; e.g. rat synaptotagmin 4 C2A domain binds Ca2+ with a Kd around 10 mm, and rat synaptotagmin 4 C2B domain essentially cannot bind Ca2+ despite the lack of an obvious sequence reason (45). The very low Ca2+ affinity of KIBRA C2 could at least partly arise from suboptimal sequences for Ca2+ binding in CBR1 and CBR3 (Fig. S8).
Phosphoinositide binding and membrane association
C2 domains have been shown to bind phospholipids via the Ca2+-binding regions (31, 46) where the phospholipid contributes to the Ca2+ coordination sphere. C2 domains also bind phospholipids and phosphoinositides via a β3-β4 lysine-rich cluster (31, 47, 48). Our XRC data involving C2(C771A), however, indicate that KIBRA C2 domain binds phosphoinositide in a novel way involving residues from strands β1, β2, and β8 (Fig. 5; Protein Data Bank codes 6FJC and 6FJD). Given this unusual binding mode, we checked whether C2 dimerization and/or crystal packing hinders phosphoinositide binding at the previously identified C2-phosphoinositide-binding sites: antiparallel C2 domain dimerization with strands β7 and β8 forming the intermonomer interface, as observed in our C2(C771A) crystal structures, obscures neither CBRs nor the β3-β4 lysine-rich cluster. Similarly, phosphoinositide interaction at the lysine-rich cluster is not prevented by crystal packing: the cluster is not occluded by symmetry-related molecules. Hence, steric factors do not explain why the previously observed phospholipid-binding modes were not observed for KIBRA C2.
Essentially the same phosphoinositide-binding site was also observed in NMR titrations of two phosphoinositides, PI(3)P and PI(4,5)P2, with C2(C771A) (monomeric), and of PI(4,5)P2 with varC2 (parallel dimer in crystal structure; Fig. 2), although the binding is relatively low-affinity. The NMR-observed binding involved residues in β5 as well as β1, β2, and β8 (Fig. 4). Hence, the observed β1, β2, β5, and β8 phosphoinositide-binding site persisted, irrespective of whether the particular version of KIBRA C2 is predominantly monomer, antiparallel dimer, or parallel dimer.
Possible reasons why phosphoinositide interaction with KIBRA C2 does not occur at previously identified C2 phosphoinositide–binding sites include the fact that KIBRA C2 does not conform to the consensus sequence of the β3-β4 lysine-rich cluster (Fig. S9) and may not have sufficient positively charged residues in the vicinity to support phosphoinositide binding. The lack of a lysine-rich cluster may reflect the fact that KIBRA is not required to promote membrane fusion and hence does not need to induce membrane curvature (49). Also, clashes occur when C2(C771A) and PI(4,5)P2-bound rabphilin 3A structures are overlaid; the side chain of KIBRA residue Glu-757, for example, lies between phosphates 4 and 5 of PI(4,5)P2 and apparently would repel these phosphate groups of the phosphoinositide; a glutamate occurs at this position in the C2 domains of all three WWC proteins, but an asparagine occurs here in seven of the eight other aligned C2 domains (Fig. S9). In addition, our observation that KIBRA C2 binds calcium very weakly, or perhaps not at all, with consequent lack of electrostatic bridging between KIBRA C2 negatively charged side chains and negatively charged phosphoinositides, helps to explain why phosphoinositide binding is not observed at the CBRs in KIBRA C2. Although the phosphoinositide headgroup is normally expected to be the key moiety, it is also worth considering whether the type of nonpolar tail influences binding: we used diC4 forms of phosphoinositides for reasonable solubility while retaining an aliphatic tail. Previous C2 studies have used a range of phosphoinositides, including just the PI(4,5)P2 headgroup (50, 51), diC8 tails (49), and seemingly full-length tails (47), all resulting in typical C2 phosphoinositide–binding sites. It seems unlikely therefore that phosphoinositide tail type is a significant factor in our observation of an unusual phosphoinositide-binding site in KIBRA C2.
MD simulations indicate that, when associated with membranes, KIBRA C2 can bind to multiple phosphoinositides. As observed for other membrane-bound proteins (52), this results in clustering of phosphoinositide lipids around the protein with phosphoinositides binding to lysines and arginines that face the membrane. Such phosphoinositide lipid clustering around KIBRA C2 is expected to change the local lipid environment. This, together with the fact that in some simulations we observed secondary orientations of KIBRA C2 relative to the membrane, could influence partner protein recruitment and consequently processes such as trafficking of receptors involved in learning and memory.
In summary, experimental and simulation data indicate that KIBRA C2 is a nonclassical C2 domain that can associate with membranes in a distinctive side-on, Ca2+-independent manner. Further investigation is required to tease out the detailed functional significance of this unusual mode of membrane association and consequently its implications for molecular mechanisms of learning and memory, organ size control, and major diseases.
Finally, we caution that we have observed this interaction mode using an isolated C2 domain in vitro. Although we have identified several factors that explain the observed binding mode, in the cell it is possible that other factors such as protein-binding partners and membrane composition/structure alter the KIBRA C2–membrane association mode.
Experimental procedures
Cloning, protein expression, and protein purification
Sequences encoding residues 658–785 of WT and variant (M734I/S735A) human KIBRA and C771A mutants of these were PCR-amplified and inserted into pQE30 (Qiagen). Unless otherwise stated, the forms of KIBRA C2 used are as follows: WTC2, varC2, C2(C771A), and varC2(C771A) (details in Table S1). The resulting proteins, with the sequence MRGSHHHHHHGS N-terminal to Gly-658 of human KIBRA, were expressed using BL21(DE3) with isopropyl 1-thio-β-d-galactopyranoside induction and purified as described previously (53). 15N- and 15N/13C-labeled C2 domain samples for NMR were produced by expression in M9 minimal medium supplemented with 1 g/liter 15NH4Cl as the sole nitrogen source or 1 g/liter 15NH4Cl and 2 g/liter [13C]glucose as the sole nitrogen and carbon sources, respectively. For NMR and AUC, C2 was exchanged into 10–50 mm MES, pH 6.5, 50–150 mm NaCl. For crystallization screens, C2 was exchanged into 10 mm MES, pH 6.5, 75 mm NaCl.
NMR
NMR data for resonance assignment (Fig. S3) were acquired at 35 °C on a 600-MHz Varian Unity INOVA spectrometer with an ambient temperature probe at University of Bath or on cryoprobe-equipped 700-MHz Bruker or 800-MHz Varian/Agilent spectrometers at the Medical Research Council Biomedical NMR Centre, Mill Hill. Protein concentration used for 3D NMR experiments was in the range 0.5–0.8 mm. NMR data were processed using NMRPipe/NMRDraw (54) and analyzed using CCPN Analysis (55). Backbone chemical shifts for C2(C771A) and varC2(C771A) were deposited in Biological Magnetic Resonance Bank (BMRB) under accession numbers 27429 and 27430, respectively. TALOS+ (34) was used to predict C2 domain secondary structure from the assigned NMR chemical shifts. For comparison, secondary structure was predicted from crystal structures using SPARTA+ (33) to convert the coordinates to chemical shifts and then using those to predict secondary structure.
Numerous 1H-15N HSQC-monitored titrations of C2(C771A) and one of WTC2 with CaCl2 were conducted, including attempts to produce NMR samples that were initially calcium-free, which involved buffer solutions made with commercial calcium-free water and treatment with EGTA. Typical initial sample conditions for these experiments were 50 mm MES, pH 6.5, 50 mm NaCl, and 10 mm MES, pH 6.5, 150 mm NaCl, sometimes with EGTA (1 or 2 mm) as an additional measure to try to ensure that C2 was not Ca2+-bound to begin with. Final CaCl2 concentrations in some of these titrations exceeded 15 mm, in one case reaching 76 mm.
Titrations of C2(C771A) with PI(3)P and PI(4,5)P2 were monitored via 1H-15N HSQC spectra recorded at Mill Hill (700-MHz cryoprobe system), and titration of varC2 with PI(4,5)P2 was monitored via 1H-15N HSQC spectra recorded in-house (600-MHz ambient temperature probe). Protein concentration used for these titrations was in the range 0.3–0.5 mm. DiC4 versions of phosphoinositides (Echelon Biosciences) were used in these NMR experiments and in XRC (described below). The molar ratio of C2 to diC4-phosphoinositide at each titration point was 1:0, 1:0.25, 1:0.50, 1:0.75, 1:1, and 1:1.5 (phosphoinositide cost-prohibited higher phosphoinositide levels); a 1H-15N-HSQC spectrum was recorded at each titration point. CaCl2 was then titrated into the same NMR sample with ratios of C2 to CaCl2 of 1:0.25, 1:0.50, 1:0.75, and 1:1; a 1H-15N-HSQC spectrum was recorded after each CaCl2 addition. 1H-15N chemical shift changes are reported as Δδav (ppm) where Δδav (ppm) = ((ΔδH2 + ΔδN2(γN/γH))/2)0.5 (56).
Analytical ultracentrifugation
Sedimentation velocity scans were recorded for four (varC2) or five (WTC2, C2(C771A), and varC2(C771A)) concentrations of each construct; concentrations are listed in each panel of Fig. S1. All experiments were performed at 50,000 rpm using a Beckman XL-I analytical ultracentrifuge with an An-50Ti rotor. Data were recorded using the absorbance (at 280 nm) and interference optical detection systems. The density and viscosity of the buffers were measured using a DMA 5000M densitometer equipped with a Lovis 200ME viscometer module. The partial specific volume for each protein was calculated using Sednterp from the amino acid sequence. Data were processed using SEDFIT (57), fitting to the c(s) model (Table S2).
X-ray crystallography
Crystallization was performed using ProPlex and JCSG+ screens (Molecular Dimensions), utilizing the hanging drop vapor diffusion method at 18 °C. VarC2 crystallized in 0.15 m (NH4)2SO4, 0.1 m MES, pH 6.0, 15% (w/v) PEG 4000; C2(C771A) (no ligand) crystallized in 0.1 m Na-HEPES, pH 7.5, 0.8 m NaH2PO4, 0.8 m KH2PO4; and for the lipid-bound studies, the NMR samples containing ligand were crystallized in 0.1 m Tris, pH8.0, 1.5 m (NH4)2SO4. Glycerol was used as a cryoprotectant at 25% (v/v) for all but the C2(C771A) (no ligand) crystals where 5% was sufficient. Diffraction data were collected using a Rigaku MicroMax 007HF with a Saturn 944+ charge-coupled device detector. Data were processed with D*Trek or HKL-2000 (58). Molecular replacement was carried out with BALBES (59), and the model was refined using Coot (60) and PHENIX (61). The resultant structures were evaluated using MolProbity (62, 63). Data collection and processing statistics are given in Table S4.
Coarse-grained molecular dynamics (CG-MD) simulations
CG-MD simulations were performed using the Martini 2.1 force field (64, 65) and GROMACS (66). In the CG-MD simulations, the protein (in monomeric and dimeric forms) was displaced away from a preformed bilayer. Protein orientation relative to the bilayer was different at the beginning of each repeat simulation. 25 repeat simulations of 2 μs each were conducted. The bilayer consisted of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (∼73%), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine (∼20%), PI(4,5)P2 (∼5%), and PI(3,4,5)P3 (∼2%). All systems were energy-minimized and subsequently equilibrated (for 500 ns) with the protein backbone particle restrained. For the production simulation, the time step was 20 fs, the pressure was 1 bar, and the temperature was 323 K. Berendsen's algorithm (67) was used to control the pressure and temperature. An elastic network model was applied to all backbone particles with a cutoff distance of 0.7 nm (68). The LINCS algorithm was used to constrain bond lengths (69), and the Lennard-Jones interactions were shifted to zero between 0.9 and 1.2 nm. Coulombic interactions were shifted to zero between 0 and 1.2 nm.
Atomistic MD simulations
Atomistic MD simulations were run at 310 K using GROMACS and two different force fields: the GROMOS96 43a1 force field (70) was used with SPC water molecules, and the OPLS-AA force field was used with TIP4P water molecules. The velocity rescaling method (71) was used to control the temperature, and the Parrinello-Rahman barostat (72) was used for pressure control. Isotropic pressure coupling was used. Bond lengths were constrained to equilibrium lengths using the LINCS method, and the particle mesh Ewald method was used to model the electrostatic interactions. The time step was 2 fs. WTC2, varC2, and varC2(C771A) structures were used for these simulations. Two simulations of 100 ns and one simulation of 300 ns were performed for each force field. We also performed simulations conducted with monomeric KIBRA C2 domain in solution in which eight calcium ions were randomly added. Eight repeat 100-ns simulations were performed, each starting from different initial configurations, using the OPLS-AA force field with TIP4P water molecules.
Author contributions
M. G. P., A. U., R. I., A. C. K., G. H., S. J. C., and S. B. formal analysis; M. G. P., A. U., R. I., A. C. K., G. H., S. J. C., and S. B. investigation; M. G. P., A. U., R. I., A. C. K., and S. B. methodology; M. G. P., A. U., R. I., A. C. K., G. H., J. K., M. S. S., S. J. C., and S. B. writing-review and editing; J. K. resources; M. S. S. software; M. S. S., S. J. C., and S. B. supervision; S. B. conceptualization; S. B. funding acquisition; S. B. writing-original draft; S. B. project administration.
Supplementary Material
Acknowledgments
We are very grateful to Geoff Kelly and Alain Oregioni, Medical Research Council Biomedical NMR Centre, Mill Hill (now Crick Institute) and Christina Redfield, Department of Biochemistry, University of Oxford, for kind assistance with NMR data acquisition.
This work was supported by Biotechnology and Biological Sciences Research Council Grant BB/J008176/1. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S9 and Tables S1–S4.
The atomic coordinates and structure factors (codes 6FB4, 6FD0, 6FJD, and 6FJC) have been deposited in the Protein Data Bank (http://wwpdb.org/).
NMR chemical shift data have been deposited in the Biological Magnetic Resonance Bank (BMRB) under accession numbers 27429 and 27430.
- KIBRA
- kidney- and brain-expressed protein
- AD
- Alzheimer's disease
- AUC
- analytical ultracentrifugation
- CBR
- calcium-binding region
- MD
- molecular dynamics
- C2(C771A)
- C771A mutant of KIBRA C2 domain
- PI(3)P
- phosphatidylinositol 3-phosphate
- PI(4,5)P2
- phosphatidylinositol 4,5-bisphosphate
- PI(3,4,5)P3
- phosphatidylinositol 3,4,5-trisphosphate
- r.m.s.d.
- root mean square deviation
- varC2
- naturally occurring variant (M734I,S735A) KIBRA C2 domain
- varC2(C771A)
- C771A mutant of variant KIBRA C2 domain
- WTC2
- wildtype KIBRA C2 domain
- WWC
- WW- and C2 domain–containing protein
- XRC
- X-ray crystallography
- PKMζ
- protein kinase Mζ
- HSQC
- heteronuclear single quantum coherence
- CG-MD
- coarse-grained molecular dynamics.
References
- 1. Rayala, S. K., den Hollander, P., Manavathi, B., Talukder, A. H., Song, C., Peng, S., Barnekow, A., Kremerskothen, J., and Kumar, R. (2006) Essential role of KIBRA in co-activator function of dynein light chain 1 in mammalian cells. J. Biol. Chem. 281, 19092–19099 10.1074/jbc.M600021200 [DOI] [PubMed] [Google Scholar]
- 2. Zhang, L., Yang, S., Wennmann, D. O., Chen, Y., Kremerskothen, J., and Dong, J. (2014) KIBRA: in the brain and beyond. Cell. Signal. 26, 1392–1399 10.1016/j.cellsig.2014.02.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Corneveaux, J. J., Liang, W. S., Reiman, E. M., Webster, J. A., Myers, A. J., Zismann, V. L., Joshipura, K. D., Pearson, J. V., Hu-Lince, D., Craig, D. W., Coon, K. D., Dunckley, T., Bandy, D., Lee, W., Chen, K., et al. (2010) Evidence for an association between KIBRA and late-onset Alzheimer's disease. Neurobiol. Aging 31, 901–909 10.1016/j.neurobiolaging.2008.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tracy, T. E., Sohn, P. D., Minami, S. S., Wang, C., Min, S.-W., Li, Y., Zhou, Y., Le, D., Lo, I., Ponnusamy, R., Cong, X., Schilling, B., Ellerby, L. M., Huganir, R. L., and Gan, L. (2016) Acetylated tau obstructs KIBRA-mediated signaling in synaptic plasticity and promotes tauopathy-related memory loss. Neuron 90, 245–260 10.1016/j.neuron.2016.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ling, J., Huang, Y., Zhang, L., Wei, D., and Cheng, W. (2018) Association of KIBRA polymorphism with risk of Alzheimer's disease: evidence based on 20 case-control studies. Neurosci. Lett. 662, 77–83 10.1016/j.neulet.2017.08.057 [DOI] [PubMed] [Google Scholar]
- 6. Meliambro, K., Wong, J. S., Ray, J., Calizo, R. C., Towne, S., Cole, B., El Salem, F., Gordon, R. E., Kaufman, L., He, J. C., Azeloglu, E. U., and Campbell, K. N. (2017) The Hippo pathway regulator KIBRA promotes podocyte injury by inhibiting YAP signaling and disrupting actin cytoskeletal dynamics. J. Biol. Chem. 292, 21137–21148 10.1074/jbc.M117.819029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Willsey, A. J., Fernandez, T. V., Yu, D., King, R. A., Dietrich, A., Xing, J., Sanders, S. J., Mandell, J. D., Huang, A. Y., Richer, P., Smith, L., Dong, S., Samocha, K. E., Tourette International Collaborative Genetics (TIC Genetics), Tourette Syndrome Association International Consortium for Genetics (TSAICG), et al. (2017) De novo coding variants are strongly associated with Tourette Disorder. Neuron 94, 486.e9–499.e9 10.1016/j.neuron.2017.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wilson, K. E., Yang, N., Mussell, A. L., and Zhang, J. (2016) The regulatory role of KIBRA and PTPN14 in Hippo signaling and beyond. Genes 7, E23 10.3390/genes7060023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hermann, A., Wennmann, D. O., Gromnitza, S., Edeling, M., Van Marck, V., Sudol, M., Schaefer, L., Duning, K., Weide, T., Pavenstädt, H., and Kremerskothen, J. (2018) WW and C2 domain-containing proteins regulate hepatic cell differentiation and tumorigenesis through the hippo signaling pathway. Hepatology 67, 1546–1559 10.1002/hep.29647 [DOI] [PubMed] [Google Scholar]
- 10. Wennmann, D. O., Schmitz, J., Wehr, M. C., Krahn, M. P., Koschmal, N., Gromnitza, S., Schulze, U., Weide, T., Chekuri, A., Skryabin, B. V., Gerke, V., Pavenstädt, H., Duning, K., and Kremerskothen, J. (2014) Evolutionary and molecular facts link the WWC protein family to Hippo signaling. Mol. Biol. Evol. 31, 1710–1723 10.1093/molbev/msu115 [DOI] [PubMed] [Google Scholar]
- 11. Yoshihama, Y., Hirai, T., Ohtsuka, T., and Chida, K. (2009) KIBRA co-localizes with protein kinase Mζ (PKMζ) in the mouse hippocampus. Biosci. Biotechnol. Biochem. 73, 147–151 10.1271/bbb.80564 [DOI] [PubMed] [Google Scholar]
- 12. Yoshihama, Y., Sasaki, K., Horikoshi, Y., Suzuki, A., Ohtsuka, T., Hakuno, F., Takahashi, S., Ohno, S., and Chida, K. (2011) KIBRA suppresses apical exocytosis through inhibition of aPKC kinase activity in epithelial cells. Curr. Biol. 21, 705–711 10.1016/j.cub.2011.03.029 [DOI] [PubMed] [Google Scholar]
- 13. Duning, K., Schurek, E. M., Schlüter, M., Bayer, M., Reinhardt, H. C., Schwab, A., Schaefer, L., Benzing, T., Schermer, B., Saleem, M. A., Huber, T. B., Bachmann, S., Kremerskothen, J., Weide, T., and Pavenstädt, H. (2008) KIBRA modulates directional migration of podocytes. J. Am. Soc. Nephrol. 19, 1891–1903 10.1681/ASN.2007080916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rosse, C., Formstecher, E., Boeckeler, K., Zhao, Y., Kremerskothen, J., White, M. D., Camonis, J. H., and Parker, P. J. (2009) An aPKC-Exocyst complex controls Paxillin phosphorylation and migration through localised JNK1 activation. PLos Biol. 7, e1000235 10.1371/journal.pbio.1000235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Papassotiropoulos, A., Stephan, D. A., Huentelman, M. J., Hoerndli, F. J., Craig, D. W., Pearson, J. V., Huynh, K. D., Brunner, F., Corneveaux, J., Osborne, D., Wollmer, M. A., Aerni, A., Coluccia, D., Hänggi, J., Mondadori, et al. (2006) Common Kibra alleles are associated with human memory performance. Science 314, 475–478 10.1126/science.1129837 [DOI] [PubMed] [Google Scholar]
- 16. Milnik, A., Heck, A., Vogler, C., Heinze, H.-J., de Quervain, D. J. F., and Papassotiropoulos, A. (2012) Association of KIBRA with episodic and working memory: a meta-analysis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 159B, 958–969 10.1002/ajmg.b.32101 [DOI] [PubMed] [Google Scholar]
- 17. Kawai, E., Shibata, N., Nagata, T., Shinagawa, S., Tagai, K., Ohnuma, T., Shimazaki, H., Toda, A., Kasanuki, K., Takayama, T., Suzuki, A., Nakayama, K., Yamada, H., and Arai, H. (2015) Genetic association between KIBRA polymorphism and Alzheimer's disease within a Japanese population. Neuromolecular Med. 17, 170–177 10.1007/s12017-015-8348-8 [DOI] [PubMed] [Google Scholar]
- 18. Zhang, N., Liu, H., Qin, W., Liu, B., Jiang, T., and Yu, C. (2017) APOE and KIBRA interactions on brain functional connectivity in healthy young adults. Cereb. Cortex 27, 4797–4805 10.1093/cercor/bhw276 [DOI] [PubMed] [Google Scholar]
- 19. Piras, I. S., Krate, J., Schrauwen, I., Corneveaux, J. J., Serrano, G. E., Sue, L., Beach, T. G., and Huentelman, M. J. (2017) Whole transcriptome profiling of the human hippocampus suggests an involvement of the KIBRA rs17070145 polymorphism in differential activation of the MAPK signaling pathway. Hippocampus 27, 784–793 10.1002/hipo.22731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Johannsen, S., Duning, K., Pavenstädt, H., Kremerskothen, J., and Boeckers, T. M. (2008) Temporal-spatial expression and novel biochemical properties of the memory-related protein KIBRA. Neuroscience 155, 1165–1173 10.1016/j.neuroscience.2008.06.054 [DOI] [PubMed] [Google Scholar]
- 21. Schneider, A., Huentelman, M. J., Kremerskothen, J., Duning, K., Spoelgen, R., and Nikolich, K. (2010) KIBRA: a new gateway to learning and memory? Front. Aging Neurosci. 2, 4 10.3389/neuro.24.004.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sacktor, T. C. (2011) How does PKMζ maintain long-term memory? Nat. Rev. Neurosci. 12, 9–15 10.1038/nrn2949 [DOI] [PubMed] [Google Scholar]
- 23. Büther, K., Plaas, C., Barnekow, A., and Kremerskothen, J. (2004) KIBRA is a novel substrate for protein kinase Cζ. Biochem. Biophys. Res. Commun. 317, 703–707 10.1016/j.bbrc.2004.03.107 [DOI] [PubMed] [Google Scholar]
- 24. Makuch, L., Volk, L., Anggono, V., Johnson, R. C., Yu, Y., Duning, K., Kremerskothen, J., Xia, J., Takamiya, K., and Huganir, R. L. (2011) Regulation of AMPA receptor function by the human memory-associated gene KIBRA. Neuron 71, 1022–1029 10.1016/j.neuron.2011.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vogt-Eisele, A., Krüger, C., Duning, K., Weber, D., Spoelgen, R., Pitzer, C., Plaas, C., Eisenhardt, G., Meyer, A., Vogt, G., Krieger, M., Handwerker, E., Wennmann, D. O., Weide, T., Skryabin, B. V., et al. (2014) KIBRA (KIdney/BRAin protein) regulates learning and memory and stabilizes protein kinase Mζ. J. Neurochem. 128, 686–700 10.1111/jnc.12480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kremerskothen, J., Plaas, C., Büther, K., Finger, I., Veltel, S., Matanis, T., Liedtke, T., and Barnekow, A. (2003) Characterization of KIBRA, a novel WW domain-containing protein. Biochem. Biophys. Res. Commun. 300, 862–867 10.1016/S0006-291X(02)02945-5 [DOI] [PubMed] [Google Scholar]
- 27. Kremerskothen, J., Plaas, C., Kindler, S., Frotscher, M., and Barnekow, A. (2005) Synaptopodin, a molecule involved in the formation of the dendritic spine apparatus, is a dual actin/α-actinin binding protein. J. Neurochem. 92, 597–606 10.1111/j.1471-4159.2004.02888.x [DOI] [PubMed] [Google Scholar]
- 28. Tracy, T. E., and Gan, L. (2017) Acetylated tau in Alzheimer's disease: an instigator of synaptic dysfunction underlying memory loss: increased levels of acetylated tau blocks the postsynaptic signaling required for plasticity and promotes memory deficits associated with tauopathy. BioEssays 39 10.1002/bies.201600224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Heitz, F. D., Farinelli, M., Mohanna, S., Kahn, M., Duning, K., Frey, M. C., Pavenstädt, H., and Mansuy, I. M. (2016) The memory gene KIBRA is a bidirectional regulator of synaptic and structural plasticity in the adult brain. Neurobiol. Learn. Mem. 135, 100–114 10.1016/j.nlm.2016.07.028 [DOI] [PubMed] [Google Scholar]
- 30. Duning, K., Wennmann, D. O., Bokemeyer, A., Reissner, C., Wersching, H., Thomas, C., Buschert, J., Guske, K., Franzke, V., Flöel, A., Lohmann, H., Knecht, S., Brand, S.-M., Pöter, M., Rescher, U., et al. (2013) Common exonic missense variants in the C2 domain of the human KIBRA protein modify lipid binding and cognitive performance. Transl. Psychiatry 3, e272 10.1038/tp.2013.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Corbalan-García, S., and Gómez-Fernández, J. C. (2014) Signaling through C2 domains: more than one lipid target. Biochim. Biophys. Acta 1838, 1536–1547 10.1016/j.bbamem.2014.01.008 [DOI] [PubMed] [Google Scholar]
- 32. Lemmon, M. A. (2008) Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 9, 99–111 10.1038/nrm2328 [DOI] [PubMed] [Google Scholar]
- 33. Shen, Y., and Bax, A. (2010) SPARTA+: a modest improvement in empirical NMR chemical shift prediction by means of an artificial neural network. J. Biomol. NMR 48, 13–22 10.1007/s10858-010-9433-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shen, Y., Delaglio, F., Cornilescu, G., and Bax, A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 10.1007/s10858-009-9333-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yamamoto, E., Kalli, A. C., Yasuoka, K., and Sansom, M. S. P. (2016) Interactions of pleckstrin homology domains with membranes: adding back the bilayer via high-throughput molecular dynamics. Structure 24, 1421–1431 10.1016/j.str.2016.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kalli, A. C., and Sansom, M. S. (2014) Interactions of peripheral proteins with model membranes as viewed by molecular dynamics simulations. Biochem. Soc. Trans. 42, 1418–1424 10.1042/BST20140144 [DOI] [PubMed] [Google Scholar]
- 37. Khanal, P., Jia, Z., and Yang, X. (2018) Cysteine residues are essential for dimerization of Hippo pathway components YAP2L and TAZ. Sci. Rep. 8, 3485 10.1038/s41598-018-21828-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cumming, R. C., Andon, N. L., Haynes, P. A., Park, M., Fischer, W. H., and Schubert, D. (2004) Protein disulfide bond formation in the cytoplasm during oxidative stress. J. Biol. Chem. 279, 21749–21758 10.1074/jbc.M312267200 [DOI] [PubMed] [Google Scholar]
- 39. Ashraf, A., and Pervaiz, S. (2015) Hippo circuitry and the redox modulation of hippo components in cancer cell fate decisions. Int. J. Biochem. Cell Biol. 69, 20–28 10.1016/j.biocel.2015.10.001 [DOI] [PubMed] [Google Scholar]
- 40. Cheignon, C., Tomas, M., Bonnefont-Rousselot, D., Faller, P., Hureau, C., and Collin, F. (2018) Oxidative stress and the amyloid beta peptide in Alzheimer's disease. Redox Biol. 14, 450–464 10.1016/j.redox.2017.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ubach, J., Lao, Y., Fernandez, I., Arac, D., Südhof, T. C., and Rizo, J. (2001) The C2B domain of synaptotagmin I is a Ca2+-binding module. Biochemistry 40, 5854–5860 10.1021/bi010340c [DOI] [PubMed] [Google Scholar]
- 42. Voskoboinik, I., Thia, M.-C., Fletcher, J., Ciccone, A., Browne, K., Smyth, M. J., and Trapani, J. A. (2005) Calcium-dependent plasma membrane binding and cell lysis by perforin are mediated through its C2 domain. J. Biol. Chem. 280, 8426–8434 10.1074/jbc.M413303200 [DOI] [PubMed] [Google Scholar]
- 43. Ubach, J., García, J., Nittler, M. P., Südhof, T. C., and Rizo, J. (1999) Structure of the Janus-faced C2B domain of rabphilin. Nat. Cell Biol. 1, 106–112 10.1038/10076 [DOI] [PubMed] [Google Scholar]
- 44. Montaville, P., Schlicker, C., Leonov, A., Zweckstetter, M., Sheldrick, G. M., and Becker, S. (2007) The C2A-C2B linker defines the high affinity Ca2+ binding mode of rabphilin-3A. J. Biol. Chem. 282, 5015–5025 10.1074/jbc.M606746200 [DOI] [PubMed] [Google Scholar]
- 45. Dai, H., Shin, O.-H., Machius, M., Tomchick, D. R., Südhof, T. C., and Rizo, J. (2004) Structural basis for the evolutionary inactivation of Ca2+ binding to synaptotagmin 4. Nat. Struct. Mol. Biol. 11, 844–849 10.1038/nsmb817 [DOI] [PubMed] [Google Scholar]
- 46. Verdaguer, N., Corbalan-García, S., Ochoa, W. F., Fita, I., and Gómez-Fernández, J. C. (1999) Ca2+ bridges the C2 membrane-binding domain of protein kinase Cα directly to phosphatidylserine. EMBO J. 18, 6329–6338 10.1093/emboj/18.22.6329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guerrero-Valero, M., Ferrer-Orta, C., Querol-Audí, J., Marin-Vicente, C., Fita, I., Gómez-Fernández, J. C., Verdaguer, N., and Corbalán-García, S. (2009) Structural and mechanistic insights into the association of PKC-C2 domain to PtdIns(4,5)P2. Proc. Natl. Acad. Sci. U.S.A. 106, 6603–6607 10.1073/pnas.0813099106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ochoa, W. F., Corbalán-Garcia, S., Eritja, R., Rodríguez-Alfaro, J. A., Gómez-Fernández, J. C., Fita, I., and Verdaguer, N. (2002) Additional binding sites for anionic phospholipids and calcium ions in the crystal structures of complexes of the C2 domain of protein kinase Cα. J. Mol. Biol. 320, 277–291 10.1016/S0022-2836(02)00464-3 [DOI] [PubMed] [Google Scholar]
- 49. Ferrer-Orta, C., Pérez-Sánchez, M. D., Coronado-Parra, T., Silva, C., López-Martínez, D., Baltanás-Copado, J., Gómez-Fernández, J. C., Corbalán-García, S., and Verdaguer, N. (2017) Structural characterization of the Rabphilin-3A-SNAP25 interaction. Proc. Natl. Acad. Sci. U.S.A. 114, E5343–E5351 10.1073/pnas.1702542114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Coudevylle, N., Montaville, P., Leonov, A., Zweckstetter, M., and Becker, S. (2008) Structural determinants for Ca2+ and phosphatidylinositol 4,5-bisphosphate binding by the C2A domain of rabphilin-3A. J. Biol. Chem. 283, 35918–35928 10.1074/jbc.M804094200 [DOI] [PubMed] [Google Scholar]
- 51. Montaville, P., Coudevylle, N., Radhakrishnan, A., Leonov, A., Zweckstetter, M., and Becker, S. (2008) The PIP2 binding mode of the C2 domains of rabphilin-3A. Protein Sci. 17, 1025–1034 10.1110/ps.073326608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Picas, L., Viaud, J., Schauer, K., Vanni, S., Hnia, K., Fraisier, V., Roux, A. E. L., Bassereau, P., Gaits-Iacovoni, F., Payrastre, B., Laporte, J., Manneville, J.-B., and Goud, B. (2014) BIN1/M-Amphiphysin2 induces clustering of phosphoinositides to recruit its downstream partner dynamin. Nat. Commun. 5, 5647 10.1038/ncomms6647 [DOI] [PubMed] [Google Scholar]
- 53. Posner, M. G., Upadhyay, A., Bagby, S., Hough, D. W., and Danson, M. J. (2009) A unique lipoylation system in the Archaea. FEBS J. 276, 4012–4022 10.1111/j.1742-4658.2009.07110.x [DOI] [PubMed] [Google Scholar]
- 54. Delaglio, F., Grzesiek, S., Vuister, G. W., Zhu, G., Pfeifer, J., and Bax, A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 55. Vranken, W. F., Boucher, W., Stevens, T. J., Fogh, R. H., Pajon, A., Llinas, M., Ulrich, E. L., Markley, J. L., Ionides, J., and Laue, E. D. (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696 10.1002/prot.20449 [DOI] [PubMed] [Google Scholar]
- 56. Williamson, M. P. (2013) Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 73, 1–16 10.1016/j.pnmrs.2013.02.001 [DOI] [PubMed] [Google Scholar]
- 57. Schuck, P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 10.1016/S0006-3495(00)76713-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Otwinowski, Z., and Minor, W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 59. Long, F., Vagin, A. A., Young, P., and Murshudov, G. N. (2008) BALBES: a molecular-replacement pipeline. Acta Crystallogr. D Biol. Crystallogr. 64, 125–132 10.1107/S0907444907050172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Emsley, P., Lohkamp, B., Scott, W. G., and Cowtan, K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Adams, P. D., Afonine, P. V., Bunkóczi, G., Chen, V. B., Davis, I. W., Echols, N., Headd, J. J., Hung, L. W., Kapral, G. J., Grosse-Kunstleve, R. W., McCoy, A. J., Moriarty, N. W., Oeffner, R., Read, R. J., Richardson, D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Davis, I. W., Leaver-Fay, A., Chen, V. B., Block, J. N., Kapral, G. J., Wang, X., Murray, L. W., Arendall, W. B., 3rd, Snoeyink, J., Richardson, J. S., and Richardson, D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 10.1093/nar/gkm216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S., and Richardson, D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Marrink, S. J., Risselada, H. J., Yefimov, S., Tieleman, D. P., and de Vries, A. H. (2007) The MARTINI force field: coarse grained model for biomolecular simulations. J. Phys. Chem. B 111, 7812–7824 10.1021/jp071097f [DOI] [PubMed] [Google Scholar]
- 65. Monticelli, L., Kandasamy, S. K., Periole, X., Larson, R. G., Tieleman, D. P., and Marrink, S.-J. (2008) The MARTINI coarse-grained force field: extension to proteins. J. Chem. Theory Comput. 4, 819–834 10.1021/ct700324x [DOI] [PubMed] [Google Scholar]
- 66. Hess, B., Kutzner, C., van der Spoel, D., and Lindahl, E. (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 10.1021/ct700301q [DOI] [PubMed] [Google Scholar]
- 67. Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., DiNola, A., and Haak, J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 10.1063/1.448118 [DOI] [Google Scholar]
- 68. Atilgan, A. R., Durell, S. R., Jernigan, R. L., Demirel, M. C., Keskin, O., and Bahar, I. (2001) Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 80, 505–515 10.1016/S0006-3495(01)76033-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hess, B., Bekker, H., Berendsen, H. J. C., Fraaije, J. G. E. M. (1997) LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 [DOI] [Google Scholar]
- 70. Scott, W. R. P., Hünenberger, P. H., Tironi, I. G., Mark, A. E., Billeter, S. R., Fennen, J., Torda, A. E., Huber, T., Krüger, P., and van Gunsteren, W. F. (1999) The GROMOS biomolecular simulation program package. J. Phys. Chem. A 103, 3596–3607 10.1021/jp984217f [DOI] [Google Scholar]
- 71. Bussi, G., Donadio, D., and Parrinello, M. (2007) Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101–014108 10.1063/1.2408420 [DOI] [PubMed] [Google Scholar]
- 72. Parrinello, M., and Rahman, A. (1981) Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 10.1063/1.328693 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.