

Abstract
Aims
Everolimus is a drug from the class of mammalian target of rapamycin inhibitors used for both immunosuppressant and oncological indications. We postulate that there is room for improvement of dosing, as the optimal immunosuppressive dose in calcineurin‐free regimens is unknown and since the once daily dosing regimen for oncological indications is often associated with treatment‐limiting toxicity.
Methods
We developed a mechanistic population pharmacokinetic model for everolimus in cancer and transplant patients and explored alternative dosing regimens.
Results
We found that formulation did not influence bioavailability and that use of >20 mg prednisolone daily increased everolimus clearance. In transplant patients, the approved dose of 0.75–1 mg twice daily (BID) results in subtherapeutic trough levels (<6 μg l–1) and that a higher starting dose of 2.25–3 mg BID is required.
Conclusion
For oncological indications, our results encourage the investigation of dosing everolimus 3.75 mg BID in terms of superiority in safety and noninferiority in efficacy.
Keywords: cancer, dose individualization, everolimus, nonmem, population pharmacokinetics, transplant
What is Already Known about this Subject
Everolimus is an effective drug used for prophylaxis of allograft rejection and treatment of various types of cancer, although in different formulations.
Treatment of cancer with everolimus is associated with severe toxicity.
In the transplant setting, it is unknown what the right dose is in calcineurin‐free regimens.
What this Study Adds
The interchangeability of everolimus formulations allows further dose optimization.
In cancer patients, everolimus dose reduction and splitting may result in efficacious treatment with associated with lower and, potentially, less toxic exposure.
Higher doses than approved may be required in calcineurin‐free regimens in transplant patients for adequate immunosuppressive treatment early in therapy.
Introduction
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5889 is an orally administered immunosuppressive and antiproliferative drug that inhibits mammalian target of rapamycin (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2109) signalling. The immunosuppressive effect of everolimus is based on the inhibition of T‐ and B‐cell proliferation, differentiation and antibody production. Everolimus is used as an immunosuppressive drug to prevent allograft rejection. mTOR‐inhibitors are increasingly used in immunosuppressive regimens after organ transplantation due to the unfavourable nephrotoxicity profile of another class of immunosuppressive drugs, the calcineurin inhibitors 1, 2, 3, 4, 5, 6, 7. For prophylaxis of allograft rejection with everolimus in a calcineurin‐free regimen, the consensus therapeutic drug monitoring (TDM) target is a trough level of 6–10 μg l–1 8. It should be noted that, although several studies have proven the benefits of everolimus in calcineurin‐free regimens, it remains unknown which dose is required to reach the target of 6–10 μg l–1 and that frequent dose titration is required to reach this target 8. Furthermore, everolimus in calcineurin‐free regimens is often coadministered with relatively high dose steroids (≥20 mg), which induce the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1337 (CYP3A4), the main enzyme responsible for metabolism of everolimus 9 putting pharmacokinetic (PK) target attainment further at risk.
As an oncolytic drug, everolimus is used in the treatment of several cancers such as advanced renal cell carcinoma, neuroendocrine tumours, advanced breast cancer and tuberous sclerosis complex 10, 11. Everolimus causes, through binding to the mTOR receptor, inactivation of the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1525 (S6 K1). In its turn, S6 K1 stimulates protein synthesis and cell‐cycle progression. Deregulation of the mTOR pathway occurs in many types of cancer and inhibition of this pathway with an mTOR‐inhibitor results in tumour growth inhibition 12. Everolimus inhibits mTORC1 but also blocks http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=285 activation via inhibition of mTORC2 13. Although everolimus is an effective agent in oncology, its use is seriously hampered by its outspoken toxicity profile, often requiring dose reductions or treatment discontinuation 14. A potential explanation for this difference may be the result of the different dosing regimens used. Also, while TDM of everolimus is routinely applied in the immunosuppressive setting, it is still uncommon in oncological indications, despite a clear relationship between drug exposure and treatment outcome 8, 15, 16, 17.
There is increasing evidence that the everolimus continuous adequate exposure is required for efficacy, pointing to a prominent role of trough concentrations as a predictor for efficacy. First, during clinical development of everolimus in cancer patients, it was found that daily dosing of everolimus resulted in continuous high mTOR inhibition and better pharmacodynamic (PD) response, measured as in vivo mTOR inhibition, as well as better clinical response objectified with Response Evaluation Criteria in Solid Tumors (RECIST) criteria, when compared to a weekly schedule, albeit in the same total dose of 70 mg during 1 week 18, 19, 20, 21. Second, in mouse xenograft models of renal and breast carcinoma, it was recently shown that continuous low exposure above the free unbound concentration associated with 50% inhibition (IC50) of proliferation, obtained with subcutaneous infusion of everolimus, resulted in similar efficacy as with standard intermittent oral dosing. The continuous regimen was as effective but was associated with a lower total dose and area under the concentration time curve (AUC) 22 when compared to intermittent dosing. These findings show that continuous adequate exposure to the mTOR‐inhibitor during a dosing interval is a prerequisite for therapeutic success and that, therefore, trough levels are likely to be good PK endpoints to predict efficacy when compared to AUC, a metric that does not necessarily predict the minimum concentration. Also, these findings indicate that dose splitting (e.g. a twice daily schedule instead of once daily) may be useful to decrease the required total daily dose, while maintaining the same trough levels and, thus, a durable mTOR inhibition. Dose splitting will further result in a lower AUC and peak concentrations, which may be useful to reduce the toxicity associated with everolimus, as also previously shown for sirolimus, a chemically and pharmacologically similar drug 23. It is, however, unclear which twice daily everolimus dose is required to maintain a durable mTOR inhibition as achieved with the once daily schedule.
For dose individualization, different tablet sizes may allow dose individualization without the necessity of inaccurate tablet splitting and administration of a formulation independent of indication. Everolimus is available as Afinitor for treatment of malignancies as 2.5 mg, 5 mg, 7.5 mg and 10 mg tablets and available under the trade name Certican (Novartis Pharma AG, Basel, Switzerland) for the prophylaxis of organ rejection as 0.25 mg and 0.75 mg tablets. It remains unknown whether these different formulations can be exchanged from a PK point of view 24.
The aim of our current study was, therefore, two‐fold. First, we aimed to describe the PK of everolimus in transplant and oncology patients and identify covariates for its PK. Second, we aimed to develop alternative dosing regimens to improve treatment of transplant and oncology patients in silico. For transplant patients, we aimed to develop a dosing regimen to achieve a target trough level of 6–10 μg l–1, as required in calcineurin‐free regimens. For oncology patients, we aimed to develop a twice‐daily regimen resulting in comparable trough levels and mTOR inhibition as with the once‐daily regimen, but with a lower total daily dose and, thus, exposure.
Methods
Study population and PK sampling
The data in our PK study were from patients receiving everolimus (Afinitor) 10 once daily for treatment of metastatic thyroid or breast cancer (n = 71) as a part of two clinical studies (http://clinicaltrials.gov identifiers NCT01118065 and NCT01948960) as well as from transplant patients from two studies (Dutch Trial Register: NTR567, NTR1615 and http://clinicaltrials.gov identifier NCT02387151), where everolimus (Certican) was used for prophylaxis of renal allograft rejection (n = 55). All studies were approved by the medical ethics committee and all participants provided informed consent. The study populations and specific characteristics were previously published elsewhere 2, 17, 25. Rich PK sampling was performed with approximately eight (or more) sampling occasions for each individual during a dosing interval to allow adequate estimation of all relevant PK parameters.
Bioanalysis
Everolimus EDTA whole blood concentrations were quantified with liquid chromatography coupled with tandem mass spectrometry assays, validated according to the European Medicines Agency guidelines on bioanalytical method validation 26, as described previously 27. The assay could quantify everolimus concentrations over the range of 2 to 160 μg l–1, with within‐run and between‐run accuracy and precision of 90–110% across validated range. The performance of the assay was tested in each analytical run by incorporating external quality controls of Recipe or Chromsystems, which met the acceptance criteria (<15% bias) for all analyses.
PK analysis
Statistics
All whole blood concentration data were log‐transformed before analysis. Population PK modelling was performed with the non‐linear mixed effects modelling software package NONMEM v7.3, using the Stochastic Approximation Expectation Maximization estimation method followed by Importance Sampling (SAEM‐IMP) to obtain the objective function. The residual error was modelled with an additive error on the log scale, thus approximating a proportional error. The inter‐ and intraindividual variability was modelled using an exponential error model. The 95% confidence intervals (95% CI) of the parameter estimates were calculated with the sampling importance resampling method as recently proposed by Dosne et al. 28 and as implemented in the software package Perl Speaks Nonmem 4.7.0 29. Model development was guided by physiological plausibility, standard goodness‐of‐fit plots, visual predictive checks and the Akaike Information Criterion (AIC) 30. Prediction‐corrected visual predictive checks were constructed as described by Bergstrand et al., based on 1000 simulations of the final model 31.
Structural model development
A previously developed mechanistic population PK model for everolimus in cancer patients was used as a starting point for our analysis. This 2‐compartment distribution model accounts for the physiologically plausible relationship between central and first‐pass metabolism with a well‐stirred liver model 25.
Everolimus PK are routinely assessed in whole blood. As everolimus extensively accumulates in erythrocytes 24, haematocrit (Ht) is a confounder for the whole blood PK of everolimus. Although, ideally, plasma concentrations of everolimus are measured, this is impossible for everolimus as slight haemolysis during blood collection will already result in spuriously high plasma concentrations. In the previously developed model, we calculated plasma PK from the paired observations of whole blood concentrations and Ht, assuming a fixed erythrocyte‐to‐plasma accumulation ratio of 85:15 25, as reported by the manufacturer for the concentration range usually observed in clinical practice 24. However, because of known non‐linear erythrocyte binding of everolimus at higher concentrations, we aimed to extend our model to also capture nonlinear binding as well. Therefore, specific and nonspecific everolimus erythrocyte binding constants were determined from in vitro blood distribution data from the manufacturer 24. Nonlinear erythrocyte binding was captured from a graph showing distribution of [3H]everolimus between erythrocytes and plasma vs. everolimus concentration over a concentration range of 5–5000 ng ml–1 in whole blood using Webplot digitizer 3.11 32. For the derivation of the nonlinear erythrocyte binding of everolimus, we assumed a Ht of 45% (v/v) in the source data of healthy volunteers and we fitted the data to equation (1) by means of least squares regression:
| (1) |
In this equation, Crb is the everolimus erythrocyte concentration, Bmax is the maximal everolimus concentration specifically bound to erythrocytes, Kd is the predicted dissociation constant, Kns is the constant expressing non‐specific binding of everolimus to erythrocytes and Cp is the plasma concentration 33. In the population PK analysis, we calculated the plasma concentrations from the paired observations of whole blood concentrations and Ht using the derived values for Bmax, Kd and Kns. All parameter estimates of the PK model are, therefore, shown as the respective plasma PK parameters.
Delay in drug absorption after ingestion was modelled with a chain of four transition compartments. The mean absorption time (MAT) was estimated and the rate constant (ktr) for these transition compartments was calculated using equation (2):
| (2) |
In this equation, n is the number of transition compartments (n = 4).
The well‐stirred liver model was implemented as described previously 25. We estimated the apparent intrinsic clearance (CLint) and assumed a liver blood flow (QH) of 90 l h–1 and calculated liver volume (VL) as a function of body size and age, with the equation proposed by Small et al. 34. The liver plasma flow (QHP) was calculated from liver blood flow and Ht with equation (3):
| (3) |
Hepatic extraction (EH) was defined calculated with equation (4).
| (4) |
In this equation fu is the known concentration independent unbound fraction of 0.27 with an interindividual variability of 3% 35.
The apparent hepatic plasma clearance (CLH) was calculated with equation (5).
| (5) |
To account for impact of body size as a confounder for PK a priori, all flow parameters (hepatic blood flow QH of 90 l h–1 and intercompartmental clearance Q) as well as the volume parameters central volume (VC) and peripheral volume (Vp) were allometrically scaled to body size. We compared the performance of measured total body weight, as well as the calculated normal fat mass and fat‐free mass as size descriptors for this purpose, as recently described by Holford et al. 36 during base model development.
The rate constants describing the PK model are summarized below:
k12 = k23 = k34 = k45 = k56 = 5/MAT
k60 = CLH / VL
k67 = (QHP×(1‐EH))/VL
k76 = QHP / VC
k78 = Q / VC
k87 = Q / VP
A schematic depiction of the model can be found in Figure 1. The NONMEM control stream for reference is provided in supplemental material S2.
Figure 1.
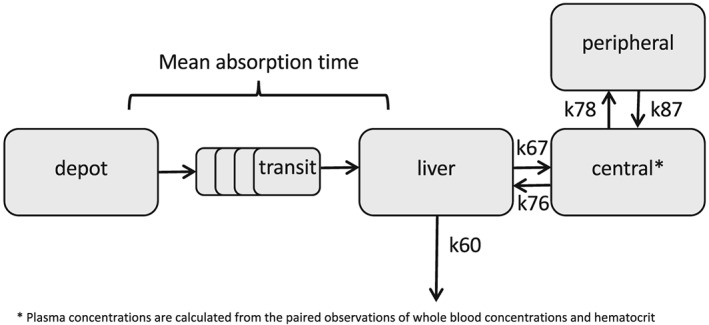
Schematic depiction of the pharmacokinetic model
Covariate analysis
After development of the base model, we investigated the impact of drug formulation (Certican vs. Afinitor) on relative bioavailability as a binary covariate. Furthermore, we investigated the use of high daily dose prednisolone (≥20 mg) as a binary covariate on intrinsic clearance, as it is a known inducer of cytochrome p450 isoenzyme 3A4 (CYP3A4) isoenzyme, the main responsible enzyme for the metabolism of everolimus 37. A covariate was included in the model when it was physiologically plausible, significantly improved model fit (P < 0.05) and if the effect was considered clinically relevant, with a cut‐off for relevancy of 25% change in a PK parameter. The P value was calculated from the reduction in objective function. A reduction of 3.84 corresponds to a P < 0.05 (from chi squared distribution with one degree of freedom). A 25% change in PK was considered a clinically relevant change, based on the fact that smaller dose units are required for dose individualization for oncological indications, and that for this indication everolimus is not considered a drug with a narrow therapeutic window by the European Medicines Agency everolimus product‐specific bioequivalence guidance 38.
As our PK data were not specifically collected for a covariate analysis, we performed an a posteriori power analysis to assure that our data were adequate for the proposed covariate analysis, as proposed earlier 39, 40. For this purpose, we simulated for each covariate 500 virtual studies with a covariate effect (25% change in PK) and performed a re‐estimation to calculate the power at a significance level of 5%. This power calculation was implemented in the Stochastic Simulation and Estimation option in Perl Speaks Nonmem 41.
Investigation of improved dosing regimens
For prophylaxis of allograft rejection in a calcineurin‐free regimen, the consensus PK target is a whole blood trough level of 6–10 μg l–1 at steady state 8 to assure adequate immunosuppression with limited toxicity during twice daily dosing of everolimus. This is underlined by the fact that in most randomized clinical trials with a regimen of everolimus in absence of calcineurin inhibitors; however, the average whole blood everolimus trough level at the end of the study was found to be 7 μg l–1 2, 3, 42. For exploration of an improved dosing regimen for this indication, we simulated the typical steady state PK curve for everolimus in whole blood in the approved dose of 0.75 mg and 1 mg twice daily. If these dosing regimens did not result in target trough levels of 6–10 μg l–1, we explored other twice daily dosing regimens to reach this target.
For oncological indications, it has been reported that everolimus PK correlate with efficacy and toxicity 16. However, there is currently no specific PK target known to aim for during treatment. However, as the approved dosing regimen of 10 mg is known to result in an effective treatment we, therefore, simulated the typical whole blood trough level associated with everolimus dosed 10 mg once daily to determine the trough level associated with clinical efficacy. Also, as inhibition of S6 kinase 1 (S6 K1), a downstream effector of mTOR and a well‐established PD biomarker of everolimus 43, directly relates with everolimus plasma PK, we calculated the corresponding S6 K1 inhibition in tumour tissue as described previously 25. Since increasing the dosing frequency will allow maintaining similar trough levels with a lower total daily dose, we simulated various twice daily regimens to obtain the twice daily dose associated with trough concentrations and tumour S6 K1 inhibition comparable to the once daily schedule. All simulations were performed assuming PK steady state in a typical individual of age 40 years, total body weight 70 kg, height 1.8 m and Ht 45%.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 44, and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 45.
Results
Study population
A summary of the population characteristics is shown in Table 1. In short, these populations consisted of patients on everolimus for treatment of cancer or prophylaxis of renal allograft rejection. In these populations, everolimus was used in line with the drug label and the use of strong inhibitors or inducers of CYP3A4 or p‐glycoprotein were prohibited. Tacrolimus was used as comedication besides everolimus and prednisolone in 22 renal transplant recipients who received alemtuzumab induction therapy, the other renal transplant recipients were patients on everolimus and prednisolone combination therapy. Other comedication that was used in the renal transplant population were statins (atorvastatin, pravastatin, simvastatin, rosuvastatin), sulfamethoxazole/trimethoprim, calcium antagonists (without CYP3A inhibition) and proton pump inhibitors (pantoprazole, omeprazole and esomeprazole). All data were selected without prior intervention with TDM. This allowed unbiased estimation of the PK parameters 46.
Table 1.
Baseline characteristics of included patients
| Number | Mean | Median | Standard deviation | Minimum | Maximum | |
|---|---|---|---|---|---|---|
| Renal transplant recipients | 55 | |||||
| Male | 38 | |||||
| Female | 17 | |||||
| Age (years) | 53 | 55 | 14.0 | 19 | 74 | |
| Length (cm) | 175 | 175 | 9.3 | 153 | 191 | |
| Weight (kg) | 81 | 79 | 13.7 | 52 | 110.3 | |
| Ht (fraction) | 0.35 | 0.36 | 0.04 | 0.28 | 0.45 | |
| Everolimus Dose (twice daily) | 2.4 | 3 | 0.74 | 1.5 | 3 | |
| Prednisolone dose (total daily dose) | 14.5 | 10 | 6 | 7.5 | 40 | |
| Total number of PK samples | 347 | |||||
| Number of PK samples per patient | 6.3 | 7 | 1.2 | 1 | 7 | |
| Metastatic thyroid or breast cancer patients | 71 | |||||
| Male | 20 | |||||
| Female | 51 | |||||
| Age (years) | 61 | 62 | 10.0 | 37 | 80 | |
| Length (cm) | 170 | 168 | 8.9 | 152 | 189 | |
| Weight (kg) | 73.1 | 73 | 12.6 | 45 | 105 | |
| Ht (fraction) | 0.38 | 0.38 | 0.05 | 0.25 | 0.50 | |
| Everolimus Dose (once daily) | 10 | 10 | 10 | 0 | 10 | 10 |
| Prednisolone dose (total daily dose) | 0 | 0 | 0 | 0 | 0 | |
| Total number of PK samples | 893 | |||||
| Number of PK samples per patient | 12.6 | 12 | 5.8 | 3 | 21 |
Base model development
The everolimus blood binding constants Bmax, Kd and Kns were estimated to be 0.964, 0.0920 and 0.153 mg l–1, respectively. The predicted relationship between erythrocyte and plasma concentrations, together with the observed plasma and erythrocyte concentrations is shown in supplemental material S1. In total, we had 1239 PK observations available in 126 individuals. Everolimus PK were well‐described with the previously developed two‐compartment disposition model 25. We observed considerable intra‐individual variability in absorption. No inter‐individual variability could be estimated for VP and Q. Furthermore, estimated fat‐free mass best described the relationship of PK with body size. Therefore, fat‐free mass was used for allometric scaling of flow and volume parameters in the base model. The parameter estimates of the base model are shown in Table 2.
Table 2.
Parameter estimates for the base and final model
| Base model | Final model | |||
|---|---|---|---|---|
| Estimate | 95% CI | Estimate | 95% CI | |
| MAT (h) | 0.451 | 0.393–0.508 | 0.404 | 0.347–0.457 |
| CLint (l h–1) | 364 | 344–386 | 340 | 319–362 |
| Increase in CLint due high dose prednisolone (%) | ‐ | ‐ | 31 | 9.10–55.8 |
| VC (L) | 176 | 158–193 | 175 | 157–193 |
| VP (L) | 577 | 546–613 | 577 | 542–609 |
| Q (l h–1) | 86.4 | 80.3–92.0 | 85.7 | 80.3–91.5 |
| QH (l h–1) | 90 (FIX) | ‐ | 90 (FIX) | ‐ |
| Interindividual variability CLint (%) | 35.2 | 30.4–39.4 | 33.9 | 29.4–38.2 |
| Interindividual variability VC (%) | 40.0 | 39.4–48.4 | 40.6 | 29.3–50.0 |
| Intraindividual variability MAT (%) | 103 | 86.6–121 | 110 | 90.9–127 |
| Residual variability (%) | 17.5 | 16.6–18.2 | 17.9 | 17.0–18.7 |
All pharmacokinetic parameters are shown as the respective plasma pharmacokinetic parameters. All volume and flow parameters are allometrically scaled to a fat‐free mass of 57.2 kg, corresponding with the fat‐free mass of a man with a total body weight of 70 kg and a length of 1.80 m and should be interpreted as apparent volumes and flows, since the absolute bioavailability of everolimus is unknown. FIX means this parameter was not estimated, but fixed.
Power and covariate analysis
In our population, we had 82% and 64% power, respectively, to detect a 25% change in bioavailability as a result of formulation or in apparent intrinsic clearance as a result of use of high‐dose prednisolone. In the covariate analysis, only concomitant use of high‐dose (≥20 mg) prednisolone was identified as a significant covariate and it increased apparent intrinsic clearance with 31% (95% CI 9–51%). Formulation did not explain variability in relative bioavailability or absorption rate. Therefore, only high‐dose prednisolone was included as covariate for intrinsic clearance in the final model.
Final model evaluation
The final model parameter estimates are shown in Table 2. All parameter estimates could be reliably estimated, as shown in the narrow 95% CI of the parameter estimates. Shrinkage for the variability parameters as well as the residual error was <30%, indicating that the data were informative for description of variability. Standard goodness‐of‐fit plots of the final model are shown in Figure 2. As seen in the prediction‐corrected visual predictive check (Figure 2A,B), the simulated data corresponded well the observed data, because the observed 10th, 50th and 90th percentile of the data matched with the predicted percentiles for both oncology and transplant patients. Furthermore, the plots showing the observed vs. (individually) predicted whole blood concentrations (Figure 2F,G) show that the data are narrowly scattered around the line of unity. Figure 2E, showing the conditional weighted residuals vs. Ht, shows an even distribution of the residuals, demonstrating an unbiased prediction of whole blood concentrations over a wide range of Ht. The NONMEM control stream for the analysis has been included in supplementary material S2.
Figure 2.
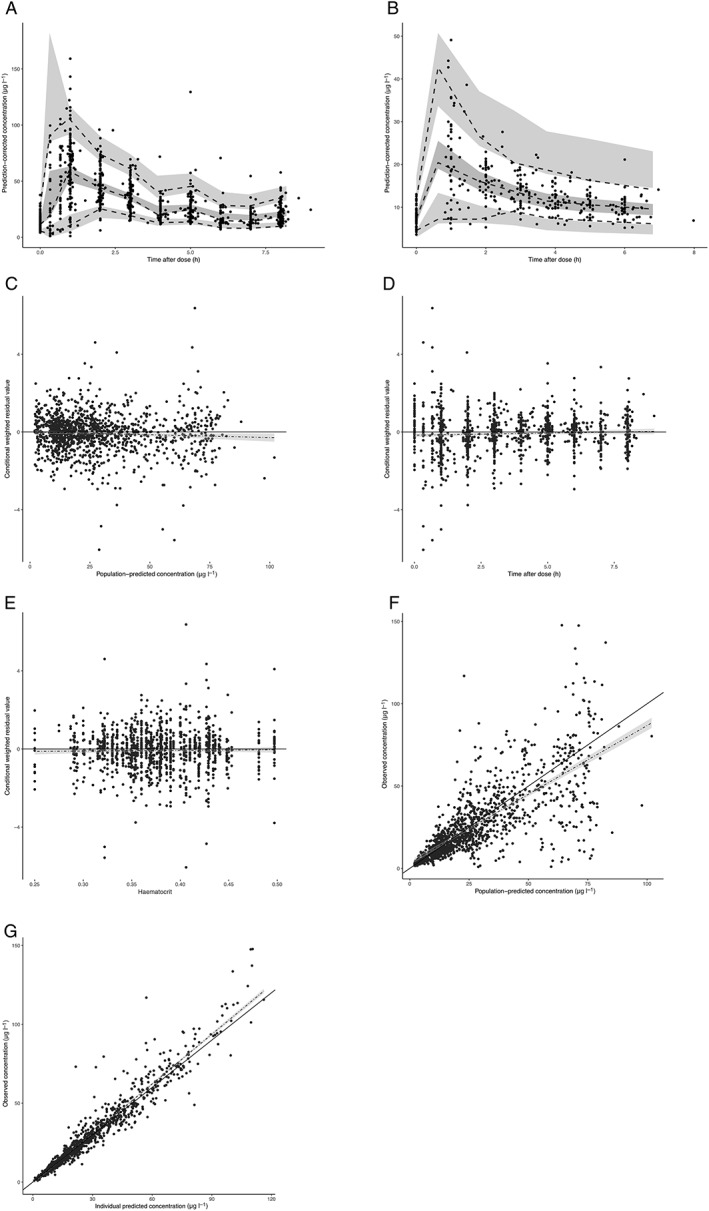
Goodness of fit plots of the final everolimus model. (A) prediction‐corrected visual predictive check for everolimus in cancer patients. The black dots are the individual observed data points. The dashed lines connect the observed 5th, 50th and 95th percentiles per bin. The grey areas are the 95% confidence interval of the simulated percentiles. (B) Prediction‐corrected visual predictive check for everolimus in renal transplant patients. The black dots are the individual observed data points. The dashed lines connect the observed 5th, 50th and 95th percentiles per bin. The grey areas are the 95% confidence interval of the simulated percentiles. (C) Conditional weighted residuals vs. population predicted whole blood concentration. (D) Conditional weighted residuals vs. time after dose. (E) Conditional weighted residuals vs. Ht. (F) Observed concentration vs. population predicted concentrations. (G) Observed concentration vs. individual predicted concentrations
Alternative dosing regimens
For use of everolimus in the transplantation setting with a calcineurin‐free regimen, our model predicted that the approved doses of 0.75 mg and 1 mg twice daily resulted in steady state whole blood trough levels of 2.37 and 3.16 μg l–1, respectively. Notably, these trough levels are well‐below the target trough level of 6–10 μg l–1 required for optimal treatment response. In combination with high dose (≥20 mg) prednisolone these trough levels were predicted to be even lower at 1.75 and 2.33 μg l–1. To obtain an everolimus whole blood trough level of approximately 7 μg l–1 without concomitant use of high dose (≥20 mg) prednisolone, the dose had to be increased to 2.25 mg twice daily (resulting in a trough level of 6.70 μg l–1) and to 3 mg twice daily in combination with high dose (≥20 mg) prednisolone to reach the PK target (resulting in a trough level of 7.01 μg l–1). The typical steady state PK curves in whole blood, with or without high dose (≥20 mg) prednisolone, for the 0.75, 1, 2.25 and 3 mg twice daily regimens are shown in Figure 3.
Figure 3.
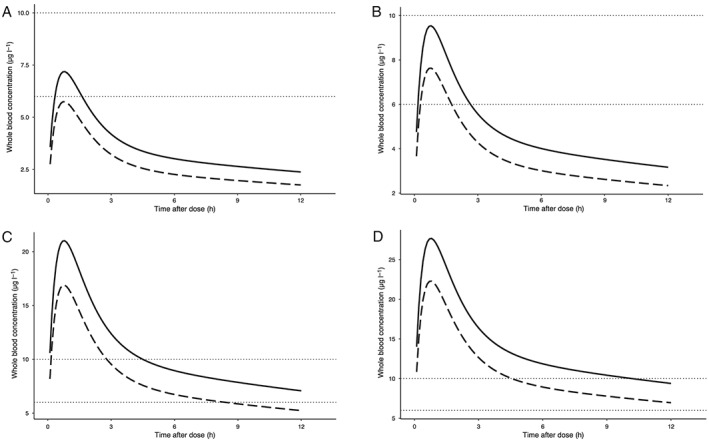
Typical steady‐state pharmacokinetic curves of everolimus in whole blood. The dotted horizontal lines indicate pharmacokinetic target, a trough level of 6–10 μg l–1. The dashed line indicates the typical pharmacokinetic curve when co‐administered with high dose (≥20 mg) prednisolone, the solid line indicates the typical pharmacokinetic curve when administered without prednisolone. (A) Dose 0.75 mg twice daily. (B) Dose 1 mg twice daily. (C) Dose 2.25 mg twice daily. (D) Dose 3 mg twice daily
For oncological indications, the predicted whole blood trough level and corresponding tumour S6 K1 inhibition for the approved 10 mg once daily dose were 12.3 μg l–1 and 89.9%, respectively. The peak concentration was 68.8 μg l–1 associated with a maximum S6 K1 inhibition of 96%. When the daily dose of 10 mg was reduced to 7.5 mg and divided in two separate administrations (3.75 mg twice daily), we predicted a whole blood trough concentration of 11.7 μg l–1, with a corresponding S6 K1 inhibition of 89.6%. The peak concentration was reduced almost a twofold in this twice daily dosing regimen at 34.2 μg l–1, but with only a limited decrease in S6 K1 inhibition of 94%. This shows that mTOR inhibition is at a plateau level in these concentrations. These results indicate that with a 25% lower total dose, administered twice daily, a similar mTOR inhibition can be maintained as in the approved dose of 10 mg once daily. To allow comparison, the predicted PK and PD profiles during a dosing interval in oncology patients for the approved and alternative dosing regimen are shown in Figure 4.
Figure 4.
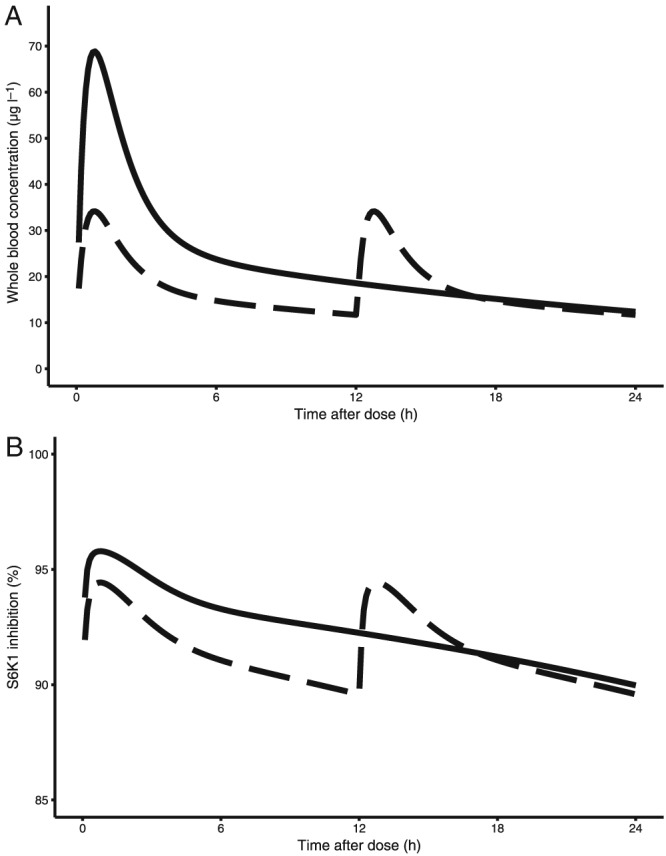
Typical steady‐state pharmacokinetic curves of everolimus in whole blood and corresponding S6 K1 inhibition during a 24‐h interval. The dashed lines show the predictions for the 3.75 mg twice daily regimen, the solid lines show the predictions for the approved 10 mg once daily regimen. (A) Whole blood pharmacokinetics. (B) S6 K1 inhibition
Discussion
For the first time, we describe the population PK of everolimus independent of its indication. In our analysis, we did not find any impact of formulation on bioavailability. Although absence of evidence is not evidence of absence, we showed that we had a high power to detect a clinically relevant difference in relative bioavailability. A clinical implication of our findings may therefore be that both the Certican (available as 0.25 and 0.75 mg tablets) and Afinitor (available as 2.5, 5 and 10 mg tablets) formulations may be used interchangeably to obtain an optimal individualized dose without the necessity of inaccurate tablet splitting. Secondly, we found that the approved twice daily dose of 0.75 mg or 1 mg is inadequate to reach the PK target (a whole blood trough level of 6–10 μg l–1) for calcineurin‐free regimens in solid organ transplant patients. To prevent subtherapeutic exposure early in immunosuppressive therapy, we suggest increasing the initial dose to 2.25 mg twice daily when administered without or 3 mg twice daily when dosed concomitantly with high dose (≥20 mg) prednisolone. Although TDM is routinely applied in transplant patients, resulting in dose titration after treatment, giving the proposed dose a priori will result in faster PK target attainment after start of treatment.
For oncological indications, we found that similar whole blood trough levels and durable S6 K1 inhibition could be maintained as with the approved once daily 10 mg dose with a 25% lower daily dose when administered as 3.75 mg every 12 h. This dosing regimen was also associated with an approximate twofold reduction in maximum concentration. Although once daily dosing is often preferred over twice daily dosing for reasons of treatment adherence 47, we think that it has been shown for the transplant setting that twice daily dosing of everolimus is feasible and that, therefore, this is an attractive regimen to potentially decrease toxicity and treatment costs, without compromising efficacy, also for oncological indications.
Although our predictions show that, on a population level, everolimus dosing can be improved, it does not overcome the necessity of dose individualization by means of clinical and TDM, considering the high variability in PK and treatment response in both oncology and transplant patients 8, 48. In clinical practice, it is most likely that PK and PD variability will be further reduced, due to dose tailoring during treatment guided by TDM or clinical response.
We recently demonstrated that variability in Ht causes variability in whole blood PK of everolimus, without affecting plasma concentrations or S6 K1 inhibition 25. Therefore, when performing TDM by measurement of whole blood concentrations, measured whole blood concentrations should be corrected for Ht, to allow comparison with reference values. This is especially relevant considering the high variability in Ht in patients treated with everolimus 49, as well as the fact that everolimus itself may cause anaemia 50. We advise to calculate the plasma concentrations from the paired observations of whole blood concentrations and Ht using our derived relationship between plasma and erythrocyte concentrations, since it is more representative of the pharmacologically active (unbound) plasma concentrations. The target trough level of 6–10 μg l–1 for adequate immunosuppression in a whole blood sample with a Ht of 45% would correspond to a plasma concentration of 1.1–1.9 μg l–1. The typical whole blood trough concentration of 11.7 μg l–1 associated with the once daily 10 mg dose in cancer patients would correspond with a plasma concentration of approximately 2.3 μg l–1.
Our study may have several limitations. First, in our model, we assume similar erythrocyte binding for all individuals. It is known, however, that some drugs may be displaced from erythrocytes by other drugs 51. Although there is currently no evidence that displacement of everolimus from erythrocytes may occur, altered erythrocyte binding kinetics could result in biased predictions of everolimus plasma concentrations. In our analysis we assume that potential PK differences are a result of different drug formulation for the different indications. In our mechanistic PK analysis, we corrected for the potential confounders body size by allometric scaling as well as for differences in Ht. To rule out any effect of population or indication on the PK of everolimus, a prospective PK cross‐over study is required to investigate the impact of formulation on everolimus PK. Our study consisted of a heterogeneous population. One might argue that this a shortcoming of our study. Nonetheless, we think that a PK study in a real‐world heterogeneous setting is of added value as it may be better representative of the clinical situation. We have focused our analysis on everolimus and not on its metabolites. Although everolimus is not known to have active metabolites 24, several metabolites have been identified 52 and their exact role in predicting efficacy or side effects has not yet been established. The use of strong CYP3A inhibitors or inducers was prohibited in our population. With regard to the other comedications used [tacrolimus, statins (atorvastatin, pravastatin, simvastatin, rosuvastin), sulfamethoxazole/trimethoprim, calcium antagonists (without CYP3A4 inhibition), proton pump inhibitors (pantoprazole, omeprazole and esomeprazole)] several publications have shown no significant influence everolimus PK 8, 9. Prednisolone, by contrast, is known to induce CYP3A activity in the gut and liver and was, therefore, specifically tested for effect on everolimus PK.
The role of PK/PD modelling during early drug development of everolimus has been pivotal for its development for oncological indications. Preclinical PK/PD modelling was performed to link mTOR‐inhibition to PK and this PK/PD‐model was then validated in humans and used to guide dosing in early clinical trials in cancer patients 18, 43. In our analysis we have developed a semi‐mechanistic PK model and linked the previously established relationships between exposure and outcome to devise alternative dosing regimens. Although application of our model for use of Bayesian dose tailoring by means of TDM was not a purpose of our analysis, the developed model could be used for this purpose. The predictive performance of the model for the desired PK target (e.g. trough levels) should then be prospectively assessed before implementation in the clinic.
Together with the recent study by Verheijen et al. 53, who showed that dose splitting everolimus 10 mg once daily in 5 mg twice daily resulted in lower maximum concentrations, our findings encourage a prospective study in cancer patients investigating everolimus 10 mg once daily vs. 3.75 mg twice daily in terms of noninferiority for efficacy and superiority for toxicity. Furthermore, the use of 2.25 mg everolimus [or 3 mg in combination with high dose (≥20 mg) prednisolone] twice daily in the transplant setting should be prospectively investigated in terms of PK target attainment after treatment initiation as well as safety and efficacy.
Competing Interests
The authors have received funding from Novartis pharma to perform the clinical studies where the PK data were obtained.
Supporting information
Figure S1 Predicted and observed relationship between plasma and red blood cell everolimus concentrations
Data S2 Example control stream
ter Heine, R. , van Erp, N. P. , Guchelaar, H. J. , de Fijter, J. W. , Reinders, M. E. J. , van Herpen, C. M. , Burger, D. M. , and Moes, D. J. A. R. (2018) A pharmacological rationale for improved everolimus dosing in oncology and transplant patients. Br J Clin Pharmacol, 84: 1575–1586. doi: 10.1111/bcp.13591.
References
- 1. Pape L, Offner G, Kreuzer M, Froede K, Drube J, Kanzelmeyer N, et al De novo therapy with everolimus, low‐dose ciclosporine A, basiliximab and steroid elimination in pediatric kidney transplantation. Am J Transplant Off J Am Soc Transplant Am Soc Transpl Surg 2010; 10: 2349–2354. [DOI] [PubMed] [Google Scholar]
- 2. Bemelman FJ, de Fijter JW, Kers J, Meyer C, Peters‐Sengers H, de Maar EF, et al Early conversion to prednisolone/everolimus as an alternative weaning regimen associates with beneficial renal transplant histology and function: the randomized‐controlled MECANO trial. Am J Transplant 2017; 17: 1020–1030. [DOI] [PubMed] [Google Scholar]
- 3. Budde K, Becker T, Arns W, Sommerer C, Reinke P, Eisenberger U, et al Everolimus‐based, calcineurin‐inhibitor‐free regimen in recipients of de‐novo kidney transplants: an open‐label, randomised, controlled trial. Lancet 2011; 377: 837–847. [DOI] [PubMed] [Google Scholar]
- 4. Kacar S, Gurkan A, Karaca C, Varılsuha C, Tilif S. Low‐dose calcineurin inhibitor regimen combined with mammalian target of rapamycin inhibitors preserves kidney functions in renal transplant recipients without allograft nephropathy. Transplant Proc 2010; 42: 3513–3516. [DOI] [PubMed] [Google Scholar]
- 5. Ferraresso M, Belingheri M, Ginevri F, Murer L, Dello Strologo L, Cardillo M, et al Three‐yr safety and efficacy of everolimus and low‐dose cyclosporine in de novo pediatric kidney transplant patients. Pediatr Transplant 2014; 18: 350–356. [DOI] [PubMed] [Google Scholar]
- 6. Lin M, Mittal S, Sahebjam F, Rana A, Sood GK. Everolimus with early withdrawal or reduced‐dose calcineurin inhibitors improves renal function in liver transplant recipients: a systematic review and meta‐analysis. Clin Transplant 2017; 31: e12872. [DOI] [PubMed] [Google Scholar]
- 7. de Fijter JW. Cancer and mTOR inhibitors in transplant recipients. Transplantation 2017; 101: 45–55. [DOI] [PubMed] [Google Scholar]
- 8. Shipkova M, Hesselink DA, Holt DW, Billaud EM, van Gelder T, Kunicki PK, et al Therapeutic drug monitoring of everolimus: a consensus report. Ther Drug Monit 2016; 38: 143–169. [DOI] [PubMed] [Google Scholar]
- 9. Moes DJAR, Swen JJ, den Hartigh J, van der Straaten T, van der Heide JJH, Sanders JS, et al Effect of CYP3A4*22, CYP3A5*3, and CYP3A combined genotypes on cyclosporine, everolimus, and tacrolimus pharmacokinetics in renal transplantation. CPT Pharmacometrics Syst Pharmacol 2014; 3: e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Santoni M, Pantano F, Amantini C, Nabissi M, Conti A, Burattini L, et al Emerging strategies to overcome the resistance to current mTOR inhibitors in renal cell carcinoma. Biochim Biophys Acta ‐ Rev Cancer 2014; 1845: 221–231. [DOI] [PubMed] [Google Scholar]
- 11. Novartis Pharma . Afinitor prescribing information. 2016.
- 12. Pópulo H, Lopes JM, Soares P. The mTOR signalling pathway in human cancer. Int J Mol Sci 2012; 13: 1886–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zeng Z, Sarbassov DD, Samudio IJ, Yee KWL, Munsell MF, Ellen Jackson C, et al Rapamycin derivatives reduce mTORC2 signaling and inhibit AKT activation in AML. Blood 2007; 109: 3509–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rugo HS, Pritchard KI, Gnant M, Noguchi S, Piccart M, Hortobagyi G, et al Incidence and time course of everolimus‐related adverse events in postmenopausal women with hormone receptor‐positive advanced breast cancer: insights from BOLERO‐2. Ann Oncol 2014; 25: 808–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pharma N. Summary of Product Charactaristics Votubia. 2015.
- 16. Deppenweiler M, Falkowski S, Saint‐Marcoux F, Monchaud C, Picard N, Laroche ML, et al Towards therapeutic drug monitoring of everolimus in cancer? Results of an exploratory study of exposure‐effect relationship. Pharmacol Res 2017; 121: 138–144. [DOI] [PubMed] [Google Scholar]
- 17. de Wit D, Schneider TC, Moes DJAR, Roozen CFM, den Hartigh J, Gelderblom H, et al Everolimus pharmacokinetics and its exposure‐toxicity relationship in patients with thyroid cancer. Cancer Chemother Pharmacol 2016; 78: 63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tanaka C, O'Reilly T, Kovarik JM, Shand N, Hazell K, Judson I, et al Identifying optimal biologic doses of everolimus (RAD001) in patients with cancer based on the modeling of preclinical and clinical pharmacokinetic and pharmacodynamic data. J Clin Oncol 2008; 26: 1596–1602. [DOI] [PubMed] [Google Scholar]
- 19. Tabernero J, Rojo F, Calvo E, Burris H, Judson I, Hazell K, et al Dose‐ and schedule‐dependent inhibition of the mammalian target of rapamycin pathway with everolimus: a phase I tumor pharmacodynamic study in patients with advanced solid tumors. J Clin Oncol 2008; 26 (10): 1603. [DOI] [PubMed] [Google Scholar]
- 20. O'Donnell A, Faivre S, Burris HA, Rea D, Papadimitrakopoulou V, Shand N, et al Phase I pharmacokinetic and pharmacodynamic study of the oral mammalian target of rapamycin inhibitor everolimus in patients with advanced solid tumors. J Clin Oncol 2008; 26: 1588–1595. [DOI] [PubMed] [Google Scholar]
- 21. Ellard SL, Clemons M, Gelmon KA, Norris B, Kennecke H, Chia S, et al Randomized phase II study comparing two schedules of everolimus in patients with recurrent/metastatic breast cancer: NCIC clinical trials group IND.163. J Clin Oncol 2009; 27: 4536–4541. [DOI] [PubMed] [Google Scholar]
- 22. Laborde L, Oz F, Ristov M, Guthy D, Sterker D, McSheehy P. Continuous low plasma concentrations of everolimus provides equivalent efficacy to oral daily dosing in mouse xenograft models of human cancer. Cancer Chemother Pharmacol. 2017; 80: 869–878. [DOI] [PubMed] [Google Scholar]
- 23. Meier‐Kriesche HU, Kaplan B. Toxicity and efficacy of sirolimus: relationship to whole‐blood concentrations. Clin Ther 2000; 22: B93–B100. [DOI] [PubMed] [Google Scholar]
- 24. Novartis Pharma . Clinical Pharmacology and Biopharmaceutics review Afinitor, 2008.
- 25. van Erp NP, van Herpen CM, de Wit D, Willemsen A, Burger DM, Huitema ADR, et al A semi‐physiological population model to quantify the effect of hematocrit on everolimus pharmacokinetics and pharmacodynamics in cancer patients. Clin Pharmacokinet 2016; 55: 1447–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. European Medicines Agency . Guideline Bioanalytical Method Validation. 2011. [DOI] [PubMed]
- 27. Moes DJAR, Press RR, de Fijter JW, Guchelaar HJ, den Hartigh J. Liquid chromatography‐tandem mass spectrometry outperforms fluorescence polarization immunoassay in monitoring everolimus therapy in renal transplantation. Ther Drug Monit 2010; 32: 413–419. [DOI] [PubMed] [Google Scholar]
- 28. Dosne A‐G, Bergstrand M, Harling K, Karlsson MO. Improving the estimation of parameter uncertainty distributions in nonlinear mixed effects models using sampling importance resampling. J Pharmacokinet Pharmacodyn 2016; 43: 583–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lindbom L, Ribbing J, Jonsson EN. Perl‐speaks‐NONMEM (PsN) – a Perl module for NONMEM related programming. Comput Methods Programs Biomed 2004; 75: 85–94. [DOI] [PubMed] [Google Scholar]
- 30. Olofsen E, Dahan A. Using Akaike's information theoretic criterion in mixed‐effects modeling of pharmacokinetic data: a simulation study. F1000Research, 2014. [DOI] [PMC free article] [PubMed]
- 31. Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J 2011; 13: 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rohatgi Ankit. WebPlotDigitizer. Austin, Texas, USA; 2017.
- 33. Snoeck E, Piotrovskij V, Jacqmin P, Van Peer A, Danhof M, Ver Donck K, et al Population analysis of the non linear red blood cell partitioning and the concentration‐effect relationship of draflazine following various infusion rates. Br J Clin Pharmacol 1997; 43: 603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Small BG, Wendt B, Jamei M, Johnson TN. Prediction of liver volume – a population‐based approach to meta‐analysis of paediatric, adult and geriatric populations – an update. Biopharm Drug Dispos 2017; 38: 290–300. [DOI] [PubMed] [Google Scholar]
- 35. Kovarik JM, Sabia HD, Figueiredo J, Zimmermann H, Reynolds C, Dilzer SC, et al. Influence of hepatic impairment on everolimus pharmacokinetics: implications for dose adjustment. Clin Pharmacol Ther 2001; 70: 425–430. [PubMed] [Google Scholar]
- 36. Holford NHG, Anderson BJ. Allometric size: the scientific theory and extension to normal fat mass. Eur J Pharm Sci 2017; 109: S59–S64. [DOI] [PubMed] [Google Scholar]
- 37. Kovarik JM, Beyer D, Bizot MN, Jiang Q, Shenouda M, Schmouder RL. Blood concentrations of everolimus are markedly increased by ketoconazole. J Clin Pharmacol 2005; 45: 514–528. [DOI] [PubMed] [Google Scholar]
- 38. Everolimus tablets 0.25 mg, 0.5 mg, 0.75 mg and 1 mg; 2.5 mg, 5 mg and 10 mg, dispersible tablets 0.1 mg and 0.25 mg; 2 mg, 3 mg and 5 mg product‐specific bioequivalence guidance [Internet]. 2016. [cited 2018 Feb 13]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2017/01/WC500219286.pdf
- 39. Yang S, Beerahee M. Power estimation using a population pharmacokinetics model with optimal design by clinical trial simulations: application in pharmacokinetic drug‐drug interaction studies. Eur J Clin Pharmacol 2011; 67: 225–233. [DOI] [PubMed] [Google Scholar]
- 40. Ueckert S, Karlsson MO, Hooker AC. Accelerating Monte Carlo power studies through parametric power estimation. J Pharmacokinet Pharmacodyn 2016; 43: 223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Keizer RJ, Karlsson MO, Hooker A. Modeling and simulation workbench for NONMEM: tutorial on Pirana, PsN, and Xpose. CPT Pharmacometrics Syst Pharmacol. 2013; 2: e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. de Fijter JW, Holdaas H, Øyen O, Sanders JS, Sundar S, Bemelman FJ, et al Early conversion from calcineurin inhibitor‐ to everolimus‐based therapy following kidney transplantation: results of the randomized ELEVATE trial. Am J Transplant 2017; 17: 1853–1867. [DOI] [PubMed] [Google Scholar]
- 43. O'Reilly T, McSheehy PM. Biomarker development for the clinical activity of the mTOR inhibitor everolimus (RAD001): processes, limitations, and further proposals. Transl Oncol 2010; 3: 65–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, et al The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 2018; 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E, et al The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 2017; 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ahn JE, Birnbaum AK, Brundage RC. Inherent correlation between dose and clearance in therapeutic drug monitoring settings: possible misinterpretation in population pharmacokinetic analyses. J Pharmacokinet Pharmacodyn 2005; 32: 703–718. [DOI] [PubMed] [Google Scholar]
- 47. Saini SD, Schoenfeld P, Kaulback K, Dubinsky MC. Effect of medication dosing frequency on adherence in chronic diseases. Am J Manag Care 2009; 15: e22–e33. [PubMed] [Google Scholar]
- 48. Davies M, Saxena A, Kingswood JC. Management of everolimus‐associated adverse events in patients with tuberous sclerosis complex: a practical guide. Orphanet J Rare Dis 2017; 12: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Joist H, Brennan DC, Coyne DW. Anemia in the kidney‐transplant patient. Adv Chronic Kidney Dis 2006; 13: 4–10. [DOI] [PubMed] [Google Scholar]
- 50. Xu J, Tian D. Hematologic toxicities associated with mTOR inhibitors temsirolimus and everolimus in cancer patients: a systematic review and meta‐analysis. Curr Med Res Opin 2014; 30: 67–74. [DOI] [PubMed] [Google Scholar]
- 51. Lettieri JT, Portelli ST. Effects of competitive red blood cell binding and reduced hematocrit on the blood and plasma levels of [14C]Indapamide in the rat. J Pharmacol Exp Ther 1983; 224: 269–272. [PubMed] [Google Scholar]
- 52. Strom T, Haschke M, Zhang YL, Bendrick‐Peart J, Boyd J, Roberts M, et al Identification of everolimus metabolite patterns in trough blood samples of kidney transplant patients. Ther Drug Monit 2007; 29: 592–599. [DOI] [PubMed] [Google Scholar]
- 53. Verheijen RB, Atrafi F, Schellens JHM, Beijnen JH, Huitema ADR, Mathijssen RHJ, et al Pharmacokinetic optimization of everolimus dosing in oncology: a randomized crossover trial. Clin Pharmacokinet 2018; 57: 637–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Predicted and observed relationship between plasma and red blood cell everolimus concentrations
Data S2 Example control stream