

Abstract
Aims
The aim of this study is to develop a population pharmacokinetic (PopPK) model for intravenous busulfan in children that incorporates variants of GSTA1, gene coding for the main enzyme in busulfan metabolism.
Methods
Busulfan concentration–time data was collected from 112 children and adolescents (median 5.4 years old, range: 0.1–20) who received intravenous busulfan during the conditioning regimen prior to stem cell transplantation. Weight, sex, baseline disease (malignant vs. non‐malignant), age, conditioning regimen and GSTA1 diplotypes were evaluated as covariates of pharmacokinetic parameters by using nonlinear mixed effects analysis. The ability to achieve the target AUC24h (3600–6000 μM min−1) was assessed by estimating the first dose based on the present PopPK model and by comparing the results with other available models in children.
Results
A one‐compartment model with first‐order elimination best described the data. Allometric scaling of weight and a factor of busulfan metabolism maturation were included in the base model. GSTA1 diplotypes were found to be a significant covariate of busulfan clearance, which was 7% faster in rapid metabolizers and 12% slower in poor metabolizers, in comparison with normal ones. Busulfan doses calculated using the parameters of the proposed PopPK model were estimated to achieve the target AUC in 85.2% of the cases (95% CI 78.7–91.7%).
Conclusion
This is the first PopPK for busulfan that successfully incorporated GSTA1 genotype in a paediatric population. Its use may contribute to better prediction of busulfan exposure in children and adolescents since the first dose, by tailoring the dose according to the individual metabolic capacity.
Keywords: busulfan, children, GSTA1, polymorphisms, population pharmacokinetics, stem cell transplantation
What is Already Known about this Subject
In children, busulfan (Bu) has been associated with high pharmacokinetic variability, in spite of the use of IV formulation.
Although genetic variations have been associated with pharmacokinetic variability, Bu dosing in children and adolescents are still sustained by models built with anthropometric data such as weight and/or age.
GSTA1 genetic variations have been consistently associated with Bu pharmacokinetics (PK) and were recently successfully introduced into a population pharmacokinetic (PopPK) model in adults.
What this Study Adds
This study provides the first PopPK model for IV Bu in children and adolescents that incorporates a genetic background (GSTA1 genetic variations) to better predict the PK parameters. This information can be used to tailor treatment according to the genotype, intending to minimize the occurrence of treatment‐related toxicities.
Introduction
The bi‐functional alkylating agent busulfan (Bu) is a key component of several conditioning regimens used before stem cell transplantation (SCT). In children, it has been used as an alternative to total body irradiation (TBI) with comparable event‐free survival (EFS) in patients with acute myeloblastic leukaemia (AML) 1 and less developmental and cognitive impacts in long‐term survivors 2, 3. Bu is also included in conditioning regimens before allogeneic SCT for acute lymphoblastic leukaemia 4 and non‐malignant diseases 5, 6 as well as in autologous transplantation regimens for high‐risk neuroblastoma 7, 8, 9, 10 and relapsed/refractory Hodgkin lymphoma 11.
Despite its wide utilization, Bu presents a narrow therapeutic window. Higher rates of relapse and rejection occur among patients with a low Bu exposure, whereas over‐exposure correlates with transplant‐related toxicities such as acute graft versus host disease (aGVHD), sinusoidal obstruction syndrome (SOS) and death 12, 13, 14, 15. Pharmacokinetic (PK) studies have demonstrated a high intra‐ and inter‐patient variability despite equivalent doses of Bu 16, 17, 18, 19, 20, 21, 22. Dosing recommendations in children are supported by population PK (PopPK) studies, which describe concentration–time data according to weight and/or age 22, 23, 24, 25, 26, 27, 28, 29, 30, 31.
Recently, single nucleotide variations (SNV) of the gene coding for glutathione S‐transferase alpha‐1 (GSTA1), implicated in the main metabolic pathway of Bu conjugation to glutathione, was successfully introduced into a PopPK model in adults, suggesting that patients heterozygous for GSTA1 g.‐69C>T variant (haplotype *B) should have their doses decreased 32.
Although another study has failed to include the same GSTA1 variants into a PopPK model in children 33, data from Choi et al. 32 agrees with previous findings from our group, whereby paediatric patients homozygous for haplotype *B, combined with those heterozygous for haplotype *B1b (g.‐69C>T, g.‐513A>G, g.‐1142C>G), presented lower Bu clearance and had higher risk of treatment‐related toxicities 34. Moreover, as reported in a recent publication, three distinct groups of patients were identified based on GSTA1 haplotypes combinations (diplotypes), which were associated with different gene expression and consequently with Bu metabolizing capacity 13.
Based on a wider analysis of SNVs occurring in GSTA1 promoter region and previously described occurring haplotypes, this current study aims to include that genetic information in a PopPK model to improve individualization of Bu dosing and consequently optimize Bu PK parameters in paediatric patients.
Methods
Patients
After approval by the local Institutional Review Board, 247 medical charts of paediatric patients who received IV Bu as part of the conditioning regimen in preparation for autologous or allogeneic SCT between April 2002 and August 2016 at CHU Sainte‐Justine (261 transplantations) were reviewed. Signed informed consent was available for 173 patients. The regimens that include other chemotherapeutic drugs before or during the days of Bu infusion were not considered in the analysis. Out of the remaining 137 patients, five were further excluded because no PK data was available, ten were excluded due to absence of GSTA1 genetic information and ten due to missing crucial clinical data. The present analysis is part of an ongoing study registered at http://clinicaltrials.gov site (NCT01257854) and a subset of patients was reported in previous studies 13, 34, 35, 36.
Treatment regimen
Busulfan (Busulfex, Otsuka Pharmaceuticals, Saint‐Laurent, Montreal, QC, Canada) was administered intravenously (IV) for four consecutive days, according to two different dosing schedules: from April 2002 to April 2012, a 2‐h infusion every 6 h (Bu6) was used. Starting May 2012, the previous schedule was replaced by a 3‐h infusion every 24 h (Bu24). Bu6 initial doses were prescribed as follows: 16 mg m−2 in infants ≤3 months of age; 0.8 mg kg−1 in children >3 months and <1 year or ≥4 years old; and 1.0 mg kg−1 in those ≥1 year and <4 years old 35. Busulfan once‐daily doses (Bu24) were obtained by multiplying Bu6 doses by four. PK‐guided dose adjustments were possible from the 2nd (Bu24) or the 5th (Bu6) dose and so on, aiming for a total cumulative AUC of 18 000 μM min−1 every 4 days. Oral lorazepam (q6h) or levetiracetam (q12h) were used during the days of Bu infusion as seizure prophylaxis, starting in the morning of the first Bu dose, up to 24 h after the last infusion. Other drugs presenting a potential drug–drug interaction with Bu, such as the azole antifungals, were temporarily withheld or replaced during the days of Bu infusion, when possible.
PK analysis and genotyping
Samples were obtained from a central venous line (not used to administer Bu) immediately before and at 120, 135, 150, 180, 240, 300 and 360 min after the start of infusion of Bu6 doses and 180, 195, 240, 300, 360 and 480 min for Bu24 doses. Plasma concentrations of Bu were determined using a high‐performance liquid chromatographic (HPLC) assay with ultraviolet detection 37. PK parameters were estimated using non‐compartmental analysis (NCA) in WinNonlin (Pharsight, version 3.1). GSTA1 genotyping was performed as previously described 38 from peripheral mononuclear cells or saliva samples, prior to the first Bu infusion. Haplotypes were determined based on single nucleotide variants of GSTA1 occurring at the following loci: ‐69 (rs3957357); ‐513 (rs11964968); ‐631 (rs4715333) and ‐1142 (rs58912740), as detailed in Supplemental Material, Table S1. Those haplotypes were previously reported as being associated with differences in promoter activity 13, as shown in Supplemental Material, Figure S1. Hence, considering both alleles (diplotypes), patients were classified into three distinct groups, based on the potential gene expression as rapid (G1), normal (G2) and poor metabolizers (G3), according to Supplemental Material, Figure S2.
Population pharmacokinetic analysis
Bu IV administration was assumed to be constant with an infusion duration of 120 min for Bu6 and 180 min for Bu24 doses. Concentration–time data was analysed using Phoenix – NLME 6.4 (Certara USA Inc., Princeton, NJ, USA), using the first‐order conditional estimation (FOCE) and the interaction option. Nonlinear mixed effects models were used to describe typical PK compartmental models (e.g. one‐ or two‐compartmental models with linear or nonlinear elimination) that better fit the Bu concentration–time data. Between‐subject variability (BSV) and inter‐occasion variability (IOV) in PK parameters were modelled as exponential random‐effect models in order to constrain the individual parameter values positively, which were thus assumed to follow a log‐normal distribution.
Model evaluation and selection were based on pertinent graphical representations of goodness‐of‐fit and on the minimization of −2 log (likelihood), which was presented as the objective function value (OFV). A decrease in the OFV of 3.84 and 5.99 (P = 0.05), respectively for 1 and 2 degrees of freedom were considered as statistically significant.
Anthropometric data descriptors
Body weight, body surface area (BSA) and body composition were applied to the base model. For weight, theoretical and empirical allometric scaling were tested. Another allometric scaled size factor was also tested in the model, which is composed by free fat mass 39 added to the fraction of fat mass that interferes on Bu PK. As proposed by McCune et al. 24, that fraction is 51% for the Bu CL and 20% for the V. Bu metabolism maturation on CL was evaluated as a maturation function (Fmat), which was derived from patients' post‐menstrual age (PMA), obtained by adding a gestational age of 40 weeks to post‐natal age, as also described by McCune et al. 24. All respective equations used for calculations are available in the supplemental material (Supplemental Material, Table S2, Equations A, B, C, D and E).
Covariate analysis and sources of variability
Covariate analysis was carried out using visual inspection followed by a formal evaluation in Phoenix. The latter consisted of a stepwise forward additive approach (P = 0.05) followed by backward elimination (P = 0.01). Potential covariates included: sex, baseline disease (malignant vs. non‐malignant) and GSTA1‐based group (G1, G2 or G3), PMA, post‐natal age and conditioning regimen (BuCy vs. others).
During forward selection, covariates were tested in univariate analysis and deemed significant if the OFV decreased by at least 3.84 (P ≤ 0.05, DF = 1) followed by their inclusion in the model. During backward elimination, significance of the covariates was confirmed by removing one at a time from the full model and required an increase in the OFV of at least 6.63 (P ≤ 0.01, DF = 1) to remain in the model.
Model evaluation
The performance of the final PopPK model was evaluated with diagnostic plots and prediction‐corrected visual predictive check (pcVPC). In pcVPC, both the observations and the model predictions were normalized to population prediction in each bin of the independent variable (i.e., time after last dose). Based on the estimates of the final model, time profiles of concentrations were simulated using 1000 replicates. Within each bin, nonparametric 95% percentile intervals of the 2.5th, 50th and 97.5th percentiles of predicted‐corrected concentrations were computed and compared to the 2.5th, 50th and 97.5th percentiles of observed‐corrected concentrations. In order to assess whether the final model could be used to estimate individual PK parameters based on population means and sparse PK data, changes in the estimates of BSV, residual variability and shrinkage in individual random effects were evaluated 39. Shrinkage values of ≤20% indicate good individual estimates of a parameter of interest, while larger shrinkage values show that individual Bayesian estimates ‘shrunk’ towards the population mean values.
In addition, the stability and precision of the model were evaluated using a nonparametric bootstrap within Perl‐Speaks‐NONMEM (PsN V3.4.2) 40. The bootstrap technique involves resampling from the original data, with each individual subject considered as a sampling unit. One thousand replicates of the data were generated by bootstrap to obtain the median and 95% percentile of PK parameters and the fixed‐ and random‐effect parameters. The bias of each parameter was calculated by computing the difference between the median value derived from the bootstrap and the final parameter estimate.
Comparison with other available PopPK models
A simple comparison was performed with other available paediatric PopPK models for Bu, which do not include GSTA1 as a covariate. First doses were calculated according to the estimated individualized Bu CL from our model as well as according to four other models 23, 24, 25, 27, recently reported as the best performing ones 36. Two additional models were included, upon which FDA 29 and EMA 31 have built their dosing recommendations for Bu in children (see Supplemental Material, Table S3). For the models that allow a flexible target Css 24, 25, 27, a value of 0.77 mg l−1 was used (equivalent to AUC24h of 4500 μM min−1 or AUC6h of 1125 μM min−1). By assuming a linear relationship between the administered dose and the measured AUC, a predicted AUC was calculated as follows: AUCpredicted = (AUCobserved/Doseadministrated) * Doseproposed. Individual predicted AUCs for each model were then classified as below, within or above the therapeutic window, set as 3600–6000 μM min−1 for Bu24 and 900–1500 μM min−1 for Bu6 doses. McNemar's test for related samples was used for comparison between the GSTA1‐based and the other PopPK models and P values were considered statistically significant if <0.05.
Results
A total of 199 PK profiles were obtained from 112 paediatric patients, 0.1–20 years of age (median 5.4 years old), of which 115 received a Bu‐containing conditioning regimen before an SCT. Patients' clinical and demographic characteristics are summarized in Table 1. A hundred and fourteen PK profiles (57%) were obtained from 31 conditioning regimens using Bu24 doses (mean of 3.7 PK profiles per regimen). In contrast, PK profiles of Bu6 doses were almost exclusively derived after the first dose (99%). A total of 1735 blood samples were available for the PopPK analysis (median 9, range 7–18 samples from patients receiving Bu6; median 28, range 14–28 samples from patients receiving Bu24). Observed concentration–time profiles of Bu are plotted in Supplemental Material, Figure S3.
Table 1.
Patients' demographic and clinical characteristics
| Demographic characteristics | Overall (n = 112) | Once daily (n = 31) | Four times daily (n = 81) | |||
|---|---|---|---|---|---|---|
| n | (%) | n | (%) | n | (%) | |
| Gender | ||||||
| Male | 53 | (47.3) | 14 | (45.2) | 39 | (48.1) |
| Female | 59 | (52.7) | 17 | (54.8) | 42 | (51.9) |
| Diagnosis | ||||||
| Malignancies | 74 | (66.1) | 22 | (71.0) | 52 | (64.2) |
| AML | 34 | (30.4) | 7 | (22.6) | 27 | (33.3) |
| MDS | 19 | (17.0) | 1 | (3.2) | 18 | (22.2) |
| ALL | 8 | (7.1) | 2 | (6.5) | 6 | (7.4) |
| Neuroblastoma | 12 | (10.7) | 12 | (38.7) | 0 | (0.0) |
| MPS | 1 | (0.9) | 0 | (0.0) | 1 | (1.2) |
| Non‐Malignancies | 38 | (33.9) | 9 | (29.0) | 29 | (35.8) |
| Immunodeficiencies | 10 | (8.9) | 0 | (0.0) | 10 | (12.3) |
| Hemoglobinopathy | 16 | (14.3) | 7 | (22.6) | 9 | (11.1) |
| Metabolic diseases | 7 | (6.3) | 2 | (6.5) | 5 | (6.2) |
| Hemophagocytic syndrome | 5 | (4.5) | 0 | (0.0) | 5 | (6.2) |
| Conditioning regimen | ||||||
| BuCy | 91 | (81.3) | 17 | (54.8) | 74 | (91.4) |
| BuCyVP16 | 8 | (7.1) | 0 | (0.0) | 8 | (9.9) |
| BuMel | 14 | (12.5) | 12 | (38.7) | 2 | (2.5) |
| BuCyMel | 1 | (0.9) | 1 | (3.2) | 0 | (0.0) |
| BuMelAraC | 1 | (0.9) | 1 | (3.2) | 0 | (0.0) |
| Number of HSCT | ||||||
| 2 | 7 | (6.3) | 0 | (0.0) | 7 | (8.6) |
| 3 or more | 1 | (0.9) | 0 | (0.0) | 1 | (1.2) |
| Ethnic groups | ||||||
| Caucasian | 94 | (83.9) | 65 | (80.2) | 29 | (93.5) |
| African | 12 | (10.7) | 10 | (12.3) | 2 | (6.5) |
| Other | 6 | (5.4) | 6 | (7.4) | 0 | (0.0) |
| Weight adequacy a | ||||||
| Overweight | 14 | (12.5) | 1 | (4.3) | 13 | (23.2) |
| Obesity | 7 | (6.3) | 1 | (4.3) | 6 | (10.7) |
| GSTA1 diplotype group | ||||||
| Group 1 | 16 | (14.3) | 6 | (19.4) | 10 | (12.3) |
| Group 2 | 80 | (71.4) | 20 | (64.5) | 60 | (74.1) |
| Group 3 | 16 | (14.3) | 5 | (16.1) | 11 | (13.6) |
| Age (years) | ||||||
| Median | 5.4 | 4.8 | 5.6 | |||
| Minimum | 0.1 | 0.4 | 0.1 | |||
| Maximum | 20 | 16 | 20 | |||
AML, acute myeloid leukemia; MDS, myelodisplastic syndrome; ALL, acute lymphoblastic leukemia; MPS, myeloproliferative disease; Cy, cyclophosphamide; VP16, etoposide; Mel, melphalan; AraC, cytarabine.
Evaluated according to Barlow et al. 45 only in children older than 2 years old
Base model
Bu concentration–time data was best described by a one‐compartment model with a first‐order elimination. The maturation factor of Bu metabolism (Fmat, see Supplemental Material, Table S2, equation D) and theoretical allometric scaling of the actual body weight (ABW) were included in the base model. During covariate analysis, PMA (∆OFV = −26.7, P = 2.3 × 10−7) and GSTA1 diplotypes groups (∆OFV = −11.7, P = 0.003) were retained as significant covariates on V and CL, respectively. Compared to normal metabolizers (G2), rapid metabolizers had 7% faster CL rates (G1) while poor metabolizers (G3) had 12% slower CL rates (see Supplemental Material, Figure S4). The PopPK parameter estimates of the final model for a patient with a median body weight of 70 kg as well as the results from the bootstrapping are presented in Table 2. Goodness‐of‐fit plots in Figure 1 show that the model predictions were in reasonable agreement with the observed plasma concentrations. The pcVPC plots shown in Figure 2 suggest that the model adequately describes Bu levels by accounting for differences in body size, maturation of CL and GSTA1 diplotypes groups. The robustness of the final model was shown by the bootstrap results (Table 2). The population estimates for the final model were similar to the mean of the bootstrap replicates and within the 95% confidence interval obtained from the bootstrap analysis. The individualized clearance was predicted by the following equation:
where FGSTA1 = 1.07 for G1, 1 for G2 and 0.88 for G3 patients.
Table 2.
Final PopPK model: pharmacokinetic parameter estimates and bootstrap results
| Base model | Final model | Bootstrap (1000 runs) | ||||||
|---|---|---|---|---|---|---|---|---|
| Estimate | RSE (%) | Shrinkage (%) | Estimate | RSE (%) | Shrinkage (%) | Median value | 95% CI | |
| PK parameter | ||||||||
| CL 70 kg (l h −1 ) | 13.58 | 1.97 | 13.70 | 2.43 | 13.69 | 13.12–14.16 | ||
| V 70 kg (l) | 50.05 | 1.21 | 49.57 | 1.15 | 49.57 | 49.72–50.76 | ||
| GSTA1 ‐group on CL (Reference G2) | ||||||||
| G3 | 0.88 | 0.40 | 0.89 | 0.82–0.96 | ||||
| G1 | 1.07 | 4.40 | 1.07 | 1.05–1.21 | ||||
| PMA on V | −0.06 | 23.40 | −0.06 | −0.18–0.09 | ||||
| Between‐subject variability (BSV) | ||||||||
| BSV CL (%) | 18.30 | 9.00 | 6.20 | 13.30 | 8.80 | 6.00 | 18.80 | 15.00–21.20 |
| BSV V (%) | 11.50 | 15.40 | 22.30 | 7.00 | 19.20 | 24.60 | 7.48 | 3.64–9.95 |
| Between‐occasion variability (BOV) | ||||||||
| BOV CL (%) | 7.40 | 8.00 | 46.10 | 7.30 | 8.10 | 46.60 | 7.44 | 6.12–8.28 |
| BOV V (%) | 10.00 | 18.60 | 21.50 | 9.60 | 16.40 | 9.30 | 9.57 | 4.94–12.40 |
| Random residual variability | ||||||||
| σ [Proportional error (%)] | 7.40 | 7.90 | 7.40 | 7.90 | 27.20 | 25.00–29.00 | ||
Figure 1.
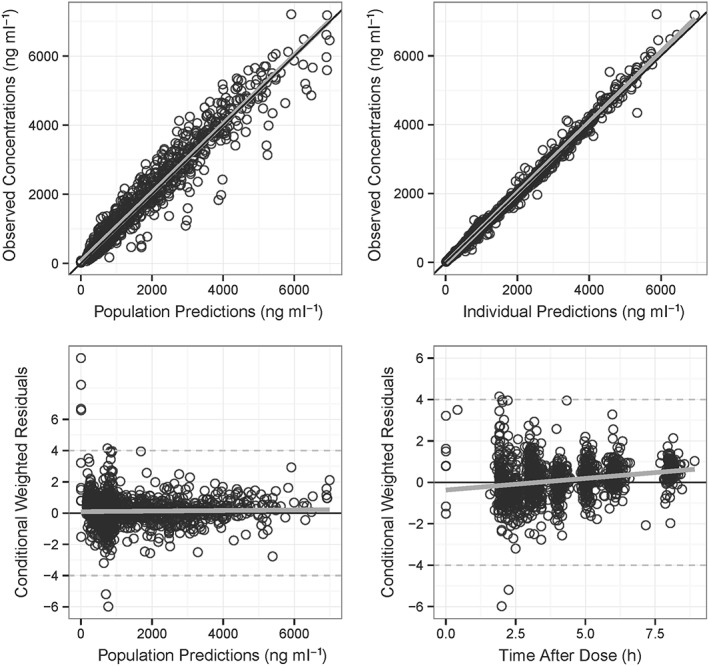
Goodness‐of‐fit of the final model. (A) Observed individual concentrations of Bu vs. population predicted concentrations; (B) observed individual concentration of Bu vs. individual predicted concentration; (C) conditional weighted residuals vs. population predicted concentrations; (D) conditional weighted residuals vs. time after the first dose
Figure 2.
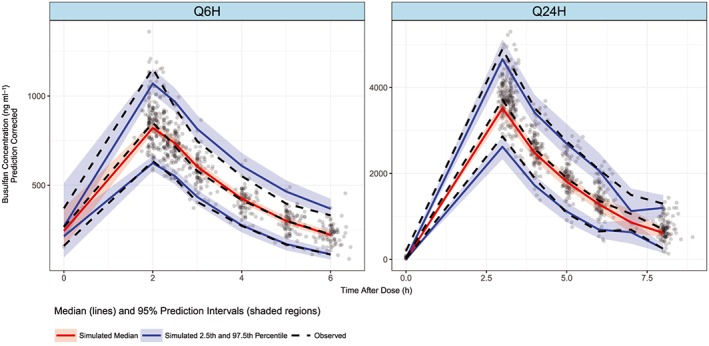
Prediction‐corrected visual prediction check (pcVPC) of final model. Q6H for doses every 6 h and Q24H for doses every 24 h. The dots represent observed concentrations and dashed lines represent 2.5th, 50th and 97.5th percentiles of observed‐corrected concentrations; solid lines represent 2.5th, 50th and 97.5th simulated percentiles within each bin; shaded areas represent 95% confidence interval of the 2.5th, 50th and 97.5th prediction intervals
Comparison with other PopPK models
Doses obtained through the parameters of the GSTA1‐based PopPK model achieved the target AUC in 85.2% of cases (95% CI 78.7–91.7%), which was significantly higher in comparison with other tested models, as detailed in Table 3. The performance of the different models, across the age groups, is shown in Table 3. Among 16 patients classified as poor metabolizers (G3), three patients had a predicted AUC in the toxic range using GSTA1‐based doses. A similar number was observed when doses were calculated using McCune's or Bartelink's model. On the other hand, GSTA1‐based doses resulted in no G1 patients outside the target AUC, contrarily to the doses calculated by the models of Bartelink (18.8%, P = 0.25), McCune (43.8%, P = 0.02), Long‐Boyle (37.5%, P = 0.03), Paci and Nguyen (68.8%, P < 0.001) and Booth (81.3%, P < 0.001) (Supplemental Material, Figure S5).
Table 3.
Frequency of achievement of target AUC6h of 900–1500 M min−1 or AUC24h 3600–6000 M min−1 according to different models stratified by age
| Age group (years) | N | GSTA1‐based | Bartelink | McCune | Long‐Boyle | Paci | Nguyen | Booth |
|---|---|---|---|---|---|---|---|---|
| 0–1 | 20 | 70.0 | 70.0 | 65.0 | 65.0 | 60.0 | 30.0 | 50.0 |
| 1–2 | 12 | 83.3 | 83.3 | 66.7 | 58.3 | 50.0 | 50.0 | 41.7a |
| 2–4 | 16 | 93.8 | 87.5 | 81.3 | 43.8a | 56.3 | 62.5 | 12.5a |
| 4–6 | 11 | 100.0 | 90.9 | 81.8 | 45.5a | 45.5a | 45.5a | 0a |
| 6–8 | 7 | 100.0 | 85.7 | 85.7 | 57.1 | 28.6 | 42.9 | 0a |
| 8–10 | 11 | 90.9 | 90.9 | 63.6 | 54.5 | 27.3a | 27.3a | 9.1a |
| 10–12 | 15 | 86.7 | 66.7 | 73.3 | 86.7 | 33.3a | 40.0a | 26.7a |
| >12 | 23 | 78.3 | 47.8 | 73.9 | 56.5 | 52.2 | 43.5 | 43.5 |
| Total | 115 | 85.2 | 73.9 a | 73.0 a | 59.1 a | 47.0 a | 42.6 a | 37.4 a |
P value < 0.05 for pairwise comparison between other models with the GSTA1‐based model, by using McNemar's related‐samples test.
Discussion
This study has provided the first PopPK model for Bu in paediatrics that successfully incorporated a genetic factor as a covariate into the PK parameter estimation. Although consistent data exist on GSTA1 genotype as an influencing factor of Bu PK (studies are summarized in Supplemental Material, Table S4), it was only recently introduced in a PopPK model in adults. Choi et al. 32 reported that patients heterozygous for GSTA1 haplotype *B have 15% lower CL compared to homozygous haplotype (*A), consequently requiring a dose reduction. Due to the rarity of haplotype *B in the Asian population 41, no homozygous patients were included in that model, revealing a limitation in its applicability to other ethnic groups. In Caucasians, for example, for whom the prevalence of *B homozygosis has been reported as higher than 50%, Zwaveling et al. failed to incorporate the GST genetic variants (GSTA1, GSTM1, GSTT1 and GSTP1 genes) into a PopPK for Bu in children and adolescents 33. However, for GSTA1, that study accounted only for the ‐69C/T locus, which does not seem to be the best marker to assess the potential activity of the gene promoter. The latter fact could possibly explain the negative finding in that study.
Although ‐69C/T can adequately identify *B homozygous, it does not distinguish the *B1b haplotype, which was associated with a reduced promoter activity, even in heterozygosis. Moreover, haplotypes *A2 and *A3, in homozygosis or compound‐heterozygosis, stand out as related to a higher promoter activity compared to *A1 carriers, and, again, those cannot be distinguished from *A1 haplotype by assessing only the ‐69 locus 13. However, in the present study, a more reliable classification was used, based on GSTA1 haplotypes and diplotypes, grouped according to the available in‐vitro and clinical studies. That way, three distinct groups were identified and successfully incorporated into a PopPK model, which resulted in a better performance when compared with other available models that do not account for the GSTA1 genotypic variability.
The structural model that best fitted our data was a one‐compartment model with first‐order elimination, which is comparable to most of the available Bu PopPK models developed in paediatrics. As detailed in Supplemental Material, Table S5, several PopPK models are available in paediatrics. Invariably, the source of variability of the PK parameters has been attributed to anthropometric characteristics of the patients, with no successful incorporation into those models of external factors, such as the drug combinations used during conditioning regimens, or other intrinsic factors, such as genetic markers. However, concerning the external factors, some publications suggested a decrease of Bu CL of 10–30% after 3–4 days of fludarabine (Flu), a chemotherapeutic drug frequently used along with Bu before SCT 42, 43, 44. It is worth noting that that association, BuFlu, has been included in the development cohort of some of those PopPK models, for whom the presence of a covariate analysis including Flu is not clear, as detailed in Supplemental Material, Table S6. A recent publication from our group also suggested that Bu CL would be lower in BuFlu associations, even though a biological rationale is still unknown 36. In the present model, in an attempt to avoid that bias, only conditioning regimens including Bu as the first drug, administered with no other chemotherapeutic drug during the 4 days of the infusion, were considered in the model development.
Besides the genetic background as a covariate, the proposed model is based on the maturation of the metabolism of Bu in children, as proposed by McCune et al. 24. The authors suggested an E max sigmoid curve to explain the Bu maturation, which would reach 50% of the adult metabolic rate at 46 weeks of PMA. The PMA is calculated from the first day of the last menstruation period of the mother prior to the birth, which is set to 40 weeks due to missing information. Based on McCune et al.'s finding, this maturation factor (Fmat) was tested in our model and was a more significant covariate on clearance than post‐natal age. As proposed by McCune et al., the E max sigmoid curve of Fmat could guarantee the reliability of the model in patients aged in months through to young adulthood. Other models such as those described by Bartelink et al. 23 and Paci et al. 27 have employed a weight‐dependent empirical allometric scaling on CL, which also seemed to improve the predictability of their models with ageing. Contrarily, in the model proposed by Long‐Boyle et al. 25, two different age‐dependent linear corrections for CL were used, establishing 12 years old as a cut‐off. That method, in comparison with the present model, resulted more frequently in toxic AUC, especially in patients over 10 years old. The current dosing recommendations for the use of Bu in children and adolescents are based on the recommendations of the regulatory agencies such as the European Medicines Agency (EMA) and the Food and Drug Administration (FDA). Nguyen et al. 31, who used a log‐linear function of the weight and Booth et al. 29 who used a fixed empirical allometric scaling of the weight on CL, described the two models that support EMA and FDA recommendations, respectively. Both models included only 24 children and adolescents in their development cohort. Compared to our model, the results of those models revealed that the rate of Bu metabolism is, at least in children, only partially explained by the weight and the size.
The body composition was also evaluated by McCune et al. in the prediction of PK parameters. Initially, the authors described that a fraction of the fat mass (51% on CL and 20% on V) in addition to the free fat mass (FFM) would describe better the Bu PK parameters than absolute body weight (ABW) 24. The resulting normal fat mass (NFM), as it was called by the authors, along with theoretical allometric scaling, were tested in the present data with no advantage in comparison to ABW. On the other hand, it is worth mentioning that only a few obese patients were included in the current study, equivalent to 6% of the available concentration–time data.
The current model proposes that GSTA1 diplotypes may be a useful tool to identify patients with significant differences in Bu PK and confirms the previously reported classification of poor (G3), normal (G2) and rapid (G1) metabolizers 13, 36. However, as much as 13% and 7% of the variability between subjects (BSV) on CL and V, respectively, remained unexplained. Other genetic markers not included in the present analysis, such as GSTM1‐null, also found in association with PK in older children 13, could partially explain that variability, as well as other genetic and non‐genetic factors. Especially the G2 patients, considered normal metabolizers and comprising 71% of our cohort, presented a high variability, and certainly consist of a group to be better described in terms of PK influencing factors and potentially sub‐grouped, establishing a more reliable classification. Meanwhile, the incorporation of the GSTA1 diplotypes seemed to reduce the overall BSV of CL by 27% (from 18.3% to 13.3%), by addressing the extremes of the individual metabolic potential of Bu.
Among G1 individuals, GSTA1‐based doses resulted in a better achievement of AUC target. Those patients were previously reported as having higher EFS even with lower first‐dose AUCs and less treatment‐related toxicities 13. In spite of the improvement of the prediction, it is still not clear whether GSTA1‐adjusted doses may impact clinical outcomes in those patients. On the other hand, among G3 patients, who were recognized as having higher risk of toxicities, the dose prediction based on the model's parameters resulted in three patients in the toxic range, which was similar to other models that did not account for genetic markers, as presented in Supplemental Material, Figure S4. As only 16 G3 patients were included in the development cohort, comparisons between the models must be done with caution. To assess those questions, a larger, multicentre prospective study is needed to better evaluate the performance of the present model.
Conclusion
The present maturation‐ and pharmacogenetic‐based PopPK model for Bu is the first to be described in a paediatric population. Based on the identification of three groups of diplotypes of GSTA1 associated with distinct gene activity, this model may contribute considerably to better predict Bu exposure in children and adolescents, by providing a reliable tool for dose tailoring according to the individual metabolic capacity. A larger study, preferably a prospective trial, is needed to confirm our results and to assess the safety and the feasibility of that personalized approach in Bu prescription in children and adolescents.
Competing Interests
There are no competing interests to declare.
We warmly thank the patients and their parents for consenting to participate in this HSCT study. We also thank R. Lo Piccolo, S. Mezziani, M‐F .Vachon and M. Cortier for the help in this study as well as the Fondation Charles‐Bruneau and the CANSEARCH Foundation for being the sponsors of this study. This study was supported partly by the Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung (Grant number153389).
Supporting information
Table S1 Haplotype composition based on single nucleotide variants of GSTA1 [13]
Table S2 Equations used in McCune's models [25] ABW: actual body weight (kg); Css: concentration of steady‐state; FFatCL and FFatV: fraction of fat mass respectively implicated in clearance (CL) and volume of distribution (V); FFM: free fat mass (kg); Fmat: maturation function; Fsize: size function; HT: height (m); NFM: normal fat mass (kg); PMA: post‐menstrual age; TM50: PMA when Bu metabolism reaches 50% of adult levels; t: dose frequency; WHS50: ABW value when FFM is half of WHSmax; WHSmax: maximum FFM for a given height
Table S3 Nomograms and equations used to obtain IV Bu doses according to each method AUCtarget: target area under the curve (μM min−1 l−1); BSA: body surface area (m2); BW: body weight (kg) a0.0041 is the conversion factor to convert the AUC units from μM min−1 l−1 to mg h−1 l−1. b4.72 mg h−1 l−1 = 1150 μM min−1. McCune et al.’s predicted doses were obtained from the web‐based calculator available at http://www.nextdose.org [24]
Table S4 Evidence of the GSTs genetic variations as associated factor with clinical and/or PK outcomes aGVHD: acute graft versus host disease; AUC: area under the curve; CL: clearance; CL/F: apparent clearance; C max: maximal concentration; Css: concentration of steady‐state; Cy2: cyclophosphamide 60 mg kg−1 day−1 for 2 consecutive days; Cy4: cyclophosphamide 50 mg kg−1 day−1 for 4 consecutive days; Flu: fludarabine; NA: not available; NS: not significant; SOS: sinusoidal obstructive syndrome
Table S5 Available PopPK models in paediatric population ABW: actual body weight; BSA: body surface area; CL: clearance; Fmat: factor of maturation of Bu metabolism; Fsize: Size factor; Kmat: maturation coefficient; Magmat: magnitude of maturation of Bu metabolism; Q: inter‐compartmental clearance; V and V1: central volume of distribution; V2: volume of distribution in the peripheral compartment
Table S6 Characteristics of the models used in performance comparison ABW: absolute body weight; AUC: area under the curve; Css: concentration of steady‐state; Cy: cyclophosphamide; Flu: fludarabine; IV: intravenous; NA: not available; PMA: post‐menstrual age; PO: oral. aAuthors stated that conditioning regimens including Flu were used, but the proportion was not reported
Figure S1 GSTA1 haplotype and reporter gene assay of GSTA1 promoter Luciferase activities of the proximal promoters of GSTA1 variants (GSTA1*A1, GSTA1*A2, GSTA1*A3, GSTA1*B1a, GSTA1*B2, GSTA1*B1b) in transient transfection in HepG2 cells. Error bars represent the standard deviations. Pairwise comparisons by analysis of variance (ANOVA) between data for the GSTA1*A1 vs. any other haplotype, after Bonferroni correction. *** P < 0.001; **** P < 0.0001, ***** P < 0.000001
Figure S2 Composition of groups based on GSTA1 diplotypes [36]
Figure S3 Blood Bu concentration vs. time after first dose. Q6H for doses every 6 h and Q24H for doses every 24 h
Figure S4 Box plot of random effects (ETA) on clearance (CL) and volume of distribution (V) from structural model according to GSTA1‐based groups before their inclusion as covariates
Figure S5 Performance of models in G1 and G3 patients. Predicted first dose AUCs in the toxic range among G3 patients (A) and sub‐therapeutic first dose predicted AUCs among G1 patients (B) aSignificant differences between respective models and GSTA1‐based model (P < 0.05)
Nava, T. , Kassir, N. , Rezgui, M. A. , Uppugunduri, C. R. S. , Huezo‐Diaz Curtis, P. , Duval, M. , Théoret, Y. , Daudt, L. E. , Litalien, C. , Ansari, M. , Krajinovic, M. , and Bittencourt, H. (2018) Incorporation of GSTA1 genetic variations into a population pharmacokinetic model for IV busulfan in paediatric hematopoietic stem cell transplantation. Br J Clin Pharmacol, 84: 1494–1504. doi: 10.1111/bcp.13566.
References
- 1. Sisler IY, Koehler E, Koyama T, Domm JA, Ryan R, Levine JE, et al Impact of conditioning regimen in allogeneic hematopoetic stem cell transplantation for children with acute myelogenous leukemia beyond first complete remission: a pediatric blood and marrow transplant consortium (PBMTC) study. Biol Blood Marrow Transplant 2009; 15: 1620–1627. [DOI] [PubMed] [Google Scholar]
- 2. Xu S‐X, Tang X‐H, Xu H‐Q, Feng B, Tang X‐F. Total body irradiation plus cyclophosphamide versus busulphan with cyclophosphamide as conditioning regimen for patients with leukemia undergoing allogeneic stem cell transplantation: a meta‐analysis. Leuk Lymphoma 2010; 51: 50–60. [DOI] [PubMed] [Google Scholar]
- 3. Bernard F, Auquier P, Herrmann I, Contet A, Poiree M, Demeocq F, et al Health status of childhood leukemia survivors who received hematopoietic cell transplantation after BU or TBI: an LEA study. Bone Marrow Transplant 2014; 49: 709–716. [DOI] [PubMed] [Google Scholar]
- 4. Bartelink IH, van Reij EM, Gerhardt CE, van Maarseveen EM, de Wildt A, Versluys B, et al Fludarabine and exposure‐targeted busulfan compares favorably with busulfan/cyclophosphamide‐based regimens in pediatric hematopoietic cell transplantation: maintaining efficacy with less toxicity. Biol Blood Marrow Transplant 2014; 20: 345–353. [DOI] [PubMed] [Google Scholar]
- 5. Gungor T, Teira P, Slatter M, Stussi G, Stepensky P, Moshous D, et al Reduced‐intensity conditioning and HLA‐matched haemopoietic stem‐cell transplantation in patients with chronic granulomatous disease: a prospective multicentre study. Lancet 2014; 383: 436–448. [DOI] [PubMed] [Google Scholar]
- 6. Pai SY, Logan BR, Griffith LM, Buckley RH, Parrott RE, Dvorak CC, et al Transplantation outcomes for severe combined immunodeficiency, 2000–2009. N Engl J Med 2014; 371: 434–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Proust‐Houdemont S, Pasqualini C, Blanchard P, Dufour C, Benhamou E, Goma G, et al Busulfan‐melphalan in high‐risk neuroblastoma: the 30‐year experience of a single institution. Bone Marrow Transplant 2016; 51: 1076–1081. [DOI] [PubMed] [Google Scholar]
- 8. Molina B, Alonso L, Gonzalez‐Vicent M, Andion M, Hernandez C, Lassaletta A, et al High‐dose busulfan and melphalan as conditioning regimen for autologous peripheral blood progenitor cell transplantation in high‐risk neuroblastoma patients. Pediatr Hematol Oncol 2011; 28: 115–123. [DOI] [PubMed] [Google Scholar]
- 9. De Ioris MA, Contoli B, Jenkner A, De Pasquale MD, Serra A, De Sio L, et al Comparison of two different conditioning regimens before autologous transplantation for children with high‐risk neuroblastoma. Anticancer Res 2012; 32: 5527–5533. [PubMed] [Google Scholar]
- 10. Soni S, Pai V, Gross TG, Ranalli M. Busulfan and melphalan as consolidation therapy with autologous peripheral blood stem cell transplantation following Children's Oncology Group (COG) induction platform for high‐risk neuroblastoma: early results from a single institution. Pediatr Transplant 2014; 18: 217–220. [DOI] [PubMed] [Google Scholar]
- 11. Nieto Y, Thall P, Valdez B, Andersson B, Popat U, Anderlini P, et al High‐dose infusional gemcitabine combined with busulfan and melphalan with autologous stem‐cell transplantation in patients with refractory lymphoid malignancies. Biol Blood Marrow Transplant 2012; 18: 1677–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bolinger AM, Zangwill AB, Slattery JT, Risler LJ, Sultan DH, Glidden DV, et al Target dose adjustment of busulfan in pediatric patients undergoing bone marrow transplantation. Bone Marrow Transplant 2001; 28: 1013–1018. [DOI] [PubMed] [Google Scholar]
- 13. Ansari M, Huezo‐Diaz Curtis P, Uppugunduri CRS, Nava T, Rezgui MA, Mlakar V, et al GSTA1 diplotypes affect busulfan clearance and toxicity in children undergoing allogenic HSCT: a multicenter study. Oncotarget 2017; 8: 90852–90867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McCune JS, Gibbs JP, Slattery JT. Plasma concentration monitoring of busulfan: does it improve clinical outcome? Clin Pharmacokinet 2000; 39: 155–165. [DOI] [PubMed] [Google Scholar]
- 15. Bartelink IH, Bredius RG, Belitser SV, Suttorp MM, Bierings M, Knibbe CA, et al Association between busulfan exposure and outcome in children receiving intravenous busulfan before hematologic stem cell transplantation. Biol Blood Marrow Transplant 2009; 15: 231–241. [DOI] [PubMed] [Google Scholar]
- 16. McCune JS, Holmberg LA. Busulfan in hematopoietic stem cell transplant setting. Expert Opin Drug Metab Toxicol 2009; 5: 957–969. [DOI] [PubMed] [Google Scholar]
- 17. Vassal G, Fischer A, Challine D, Boland I, Ledheist F, Lemerle S, et al Busulfan disposition below the age of three: alteration in children with lysosomal storage disease. Blood 1993; 82: 1030–1034. [PubMed] [Google Scholar]
- 18. Gibbs JP, Murray G, Risler L, Chien JY, Dev R, Slattery JT. Age‐dependent tetrahydrothiophenium ion formation in young children and adults receiving high‐dose busulfan. Cancer Res 1997; 57: 5509–5516. [PubMed] [Google Scholar]
- 19. Gibbs JP, Liacouras CA, Baldassano RN, Slattery JT. Up‐regulation of glutathione S‐transferase activity in enterocytes of young children. Drug Metab Dispos 1999; 27: 1466–1469. [PubMed] [Google Scholar]
- 20. Veal GJ, Nguyen L, Paci A, Riggi M, Amiel M, Valteau‐Couanet D, et al Busulfan pharmacokinetics following intravenous and oral dosing regimens in children receiving high‐dose myeloablative chemotherapy for high‐risk neuroblastoma as part of the HR‐NBL‐1/SIOPEN trial. Eur J Cancer 2012; 48: 3063–3072. [DOI] [PubMed] [Google Scholar]
- 21. Lee JW, Kang HJ, Lee SH, Yu KS, Kim NH, Yuk YJ, et al Highly variable pharmacokinetics of once‐daily intravenous busulfan when combined with fludarabine in pediatric patients: phase I clinical study for determination of optimal once‐daily busulfan dose using pharmacokinetic modeling. Biol Blood Marrow Transplant 2012; 18: 944–950. [DOI] [PubMed] [Google Scholar]
- 22. Wall DA, Chan KW, Nieder ML, Hayashi RJ, Yeager AM, Kadota R, et al Safety, efficacy, and pharmacokinetics of intravenous busulfan in children undergoing allogeneic hematopoietic stem cell transplantation. Pediatr Blood Cancer 2010; 54: 291–298. [DOI] [PubMed] [Google Scholar]
- 23. Bartelink IH, Boelens JJ, Bredius RG, Egberts AC, Wang C, Bierings MB, et al Body weight‐dependent pharmacokinetics of busulfan in paediatric haematopoietic stem cell transplantation patients: towards individualized dosing. Clin Pharmacokinet 2012; 51: 331–345. [DOI] [PubMed] [Google Scholar]
- 24. McCune JS, Bemer MJ, Barrett JS, Scott Baker K, Gamis AS, Holford NH. Busulfan in infant to adult hematopoietic cell transplant recipients: a population pharmacokinetic model for initial and Bayesian dose personalization. Clin Cancer Res 2014; 20: 754–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Long‐Boyle JR, Savic R, Yan S, Bartelink I, Musick L, French D, et al Population pharmacokinetics of busulfan in pediatric and young adult patients undergoing hematopoietic cell transplant: a model‐based dosing algorithm for personalized therapy and implementation into routine clinical use. Ther Drug Monit 2015; 37: 236–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Savic RM, Cowan MJ, Dvorak CC, Pai SY, Pereira L, Bartelink IH, et al Effect of weight and maturation on busulfan clearance in infants and small children undergoing hematopoietic cell transplantation. Biol Blood Marrow Transplant 2013; 19: 1608–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Paci A, Vassal G, Moshous D, Dalle JH, Bleyzac N, Neven B, et al Pharmacokinetic behavior and appraisal of intravenous busulfan dosing in infants and older children: the results of a population pharmacokinetic study from a large pediatric cohort undergoing hematopoietic stem‐cell transplantation. Ther Drug Monit 2012; 34: 198–208. [DOI] [PubMed] [Google Scholar]
- 28. Buffery PJ, Allen KM, Chin PK, Moore GA, Barclay ML, Begg EJ. Thirteen years' experience of pharmacokinetic monitoring and dosing of busulfan: can the strategy be improved? Ther Drug Monit 2014; 36: 86–92. [DOI] [PubMed] [Google Scholar]
- 29. Booth BP, Rahman A, Dagher R, Griebel D, Lennon S, Fuller D, et al Population pharmacokinetic‐based dosing of intravenous busulfan in pediatric patients. J Clin Pharmacol 2007; 47: 101–111. [DOI] [PubMed] [Google Scholar]
- 30. Trame MN, Bergstrand M, Karlsson MO, Boos J, Hempel G. Population pharmacokinetics of busulfan in children: increased evidence for body surface area and allometric body weight dosing of busulfan in children. Clin Cancer Res 2011; 17: 6867–6877. [DOI] [PubMed] [Google Scholar]
- 31. Nguyen L, Fuller D, Lennon S, Leger F, Puozzo C. IV busulfan in pediatrics: a novel dosing to improve safety/efficacy for hematopoietic progenitor cell transplantation recipients. Bone Marrow Transplant 2004; 33: 979–987. [DOI] [PubMed] [Google Scholar]
- 32. Choi B, Kim MG, Han N, Kim T, Ji E, Park S, et al Population pharmacokinetics and pharmacodynamics of busulfan with GSTA1 polymorphisms in patients undergoing allogeneic hematopoietic stem cell transplantation. Pharmacogenomics 2015; 16: 1585–1594. [DOI] [PubMed] [Google Scholar]
- 33. Zwaveling J, Press RR, Bredius RG, van Derstraaten TR, den Hartigh J, Bartelink IH, et al Glutathione S‐transferase polymorphisms are not associated with population pharmacokinetic parameters of busulfan in pediatric patients. Ther Drug Monit 2008; 30: 504–510. [DOI] [PubMed] [Google Scholar]
- 34. Ansari M, Rezgui MA, Theoret Y, Uppugunduri CR, Mezziani S, Vachon MF, et al Glutathione S‐transferase gene variations influence BU pharmacokinetics and outcome of hematopoietic SCT in pediatric patients. Bone Marrow Transplant 2013; 48: 939–946. [DOI] [PubMed] [Google Scholar]
- 35. Ansari M, Theoret Y, Rezgui MA, Peters C, Mezziani S, Desjean C, et al Association between busulfan exposure and outcome in children receiving intravenous busulfan before hematopoietic stem cell transplantation. Ther Drug Monit 2014; 36: 93–99. [DOI] [PubMed] [Google Scholar]
- 36. Nava T, Rezgui MA, Uppugunduri CRS, Huezo‐Diaz Curtis P, Theoret Y, Duval M, et al GSTA1 genetic variants and conditioning regimen: missing key factors in dosing guidelines of busulfan in pediatric hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2017; 23: 1918–1924. [DOI] [PubMed] [Google Scholar]
- 37. Rifai N, Sakamoto M, Lafi M, Guinan E. Measurement of plasma busulfan concentration by high‐performance liquid chromatography with ultraviolet detection. Ther Drug Monit 1997; 19: 169–174. [DOI] [PubMed] [Google Scholar]
- 38. Ansari M, Lauzon‐Joset JF, Vachon MF, Duval M, Theoret Y, Champagne MA, et al Influence of GST gene polymorphisms on busulfan pharmacokinetics in children. Bone Marrow Transplant 2010; 45: 261–267. [DOI] [PubMed] [Google Scholar]
- 39. Karlsson MO, Savic RM. Diagnosing model diagnostics. Clin Pharmacol Ther 2007; 82: 17–20. [DOI] [PubMed] [Google Scholar]
- 40. Henderson AR. The bootstrap: a technique for data‐driven statistics. Using computer‐intensive analyses to explore experimental data. Clinica Chimica Acta 2005; 359: 1–26. [DOI] [PubMed] [Google Scholar]
- 41. Yin J, Xiao Y, Zheng H, Zhang YC. Once‐daily iv BU‐based conditioning regimen before allogeneic hematopoietic SCT: a study of influence of GST gene polymorphisms on BU pharmacokinetics and clinical outcomes in Chinese patients. Bone Marrow Transplant 2015; 50: 696–705. [DOI] [PubMed] [Google Scholar]
- 42. de Castro FA, Lanchote VL, Voltarelli JC, Colturato VA, Simoes BP. Influence of fludarabine on the pharmacokinetics of oral busulfan during pretransplant conditioning for hematopoietic stem cell transplantation. J Clin Pharmacol 2013; 53: 1205–1211. [DOI] [PubMed] [Google Scholar]
- 43. Perkins JB, Kim J, Anasetti C, Fernandez HF, Perez LE, Ayala E, et al Maximally tolerated busulfan systemic exposure in combination with fludarabine as conditioning before allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant 2012; 18: 1099–1107. [DOI] [PubMed] [Google Scholar]
- 44. Yeh RF, Pawlikowski MA, Blough DK, McDonald GB, O'Donnell PV, Rezvani A, et al Accurate targeting of daily intravenous busulfan with 8‐hour blood sampling in hospitalized adult hematopoietic cell transplant recipients. Biol Blood Marrow Transplant 2012; 18: 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Barlow SE. Expert committee recommendations regarding the prevention, assessment, and treatment of child and adolescent overweight and obesity: summary report. Pediatrics 2007; 120 (Suppl 4): S164–S192. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Haplotype composition based on single nucleotide variants of GSTA1 [13]
Table S2 Equations used in McCune's models [25] ABW: actual body weight (kg); Css: concentration of steady‐state; FFatCL and FFatV: fraction of fat mass respectively implicated in clearance (CL) and volume of distribution (V); FFM: free fat mass (kg); Fmat: maturation function; Fsize: size function; HT: height (m); NFM: normal fat mass (kg); PMA: post‐menstrual age; TM50: PMA when Bu metabolism reaches 50% of adult levels; t: dose frequency; WHS50: ABW value when FFM is half of WHSmax; WHSmax: maximum FFM for a given height
Table S3 Nomograms and equations used to obtain IV Bu doses according to each method AUCtarget: target area under the curve (μM min−1 l−1); BSA: body surface area (m2); BW: body weight (kg) a0.0041 is the conversion factor to convert the AUC units from μM min−1 l−1 to mg h−1 l−1. b4.72 mg h−1 l−1 = 1150 μM min−1. McCune et al.’s predicted doses were obtained from the web‐based calculator available at http://www.nextdose.org [24]
Table S4 Evidence of the GSTs genetic variations as associated factor with clinical and/or PK outcomes aGVHD: acute graft versus host disease; AUC: area under the curve; CL: clearance; CL/F: apparent clearance; C max: maximal concentration; Css: concentration of steady‐state; Cy2: cyclophosphamide 60 mg kg−1 day−1 for 2 consecutive days; Cy4: cyclophosphamide 50 mg kg−1 day−1 for 4 consecutive days; Flu: fludarabine; NA: not available; NS: not significant; SOS: sinusoidal obstructive syndrome
Table S5 Available PopPK models in paediatric population ABW: actual body weight; BSA: body surface area; CL: clearance; Fmat: factor of maturation of Bu metabolism; Fsize: Size factor; Kmat: maturation coefficient; Magmat: magnitude of maturation of Bu metabolism; Q: inter‐compartmental clearance; V and V1: central volume of distribution; V2: volume of distribution in the peripheral compartment
Table S6 Characteristics of the models used in performance comparison ABW: absolute body weight; AUC: area under the curve; Css: concentration of steady‐state; Cy: cyclophosphamide; Flu: fludarabine; IV: intravenous; NA: not available; PMA: post‐menstrual age; PO: oral. aAuthors stated that conditioning regimens including Flu were used, but the proportion was not reported
Figure S1 GSTA1 haplotype and reporter gene assay of GSTA1 promoter Luciferase activities of the proximal promoters of GSTA1 variants (GSTA1*A1, GSTA1*A2, GSTA1*A3, GSTA1*B1a, GSTA1*B2, GSTA1*B1b) in transient transfection in HepG2 cells. Error bars represent the standard deviations. Pairwise comparisons by analysis of variance (ANOVA) between data for the GSTA1*A1 vs. any other haplotype, after Bonferroni correction. *** P < 0.001; **** P < 0.0001, ***** P < 0.000001
Figure S2 Composition of groups based on GSTA1 diplotypes [36]
Figure S3 Blood Bu concentration vs. time after first dose. Q6H for doses every 6 h and Q24H for doses every 24 h
Figure S4 Box plot of random effects (ETA) on clearance (CL) and volume of distribution (V) from structural model according to GSTA1‐based groups before their inclusion as covariates
Figure S5 Performance of models in G1 and G3 patients. Predicted first dose AUCs in the toxic range among G3 patients (A) and sub‐therapeutic first dose predicted AUCs among G1 patients (B) aSignificant differences between respective models and GSTA1‐based model (P < 0.05)