

Abstract
Background
The influenza virus is reportedly associated with 3‐5 million cases of severe illness and 250 000‐500 000 deaths annually worldwide.
Objectives
We investigated the variation of influenza A virus in Korea and examined the association with death.
Methods
A total of 13 620 cases were enrolled in the Hospital‐based Influenza Morbidity & Mortality surveillance system in Korea during 2011‐2016. Among these cases, a total of 4725 were diagnosed with influenza using RT‐PCR (influenza A; n = 3696, influenza B; n = 928, co‐infection; n = 101). We used 254 viral sequences from the 3696 influenza A cases for phylogenetic analysis using the BioEdit and MEGA 6.06 programs.
Results
We found that the sequences of A/H3N2 in the 2011‐2012 season belong to subgroup 3C.1, whereas the sequences in the 2012‐2013 season pertain to subgroup 3C.2. The sequences in the 2013‐2014 and 2014‐2015 seasons involve subgroups 3C.3a and 3C.2a. The A/H1N1pdm09 subtype belongs to subgroup 6 and contains two clusters. In addition, sequence analysis confirmed the several substitutions of internal genes and gene substitutions associated with drug resistance (I222V in NA and S31N in M2) in the fatal cases. While statistical analysis found no significant associations between genetic differences in the viruses and mortality, mortality was associated with certain host factors, such as chronic lung disease.
Conclusions
In conclusion, influenza A virus clade changes occurred in Korea during the 2011‐2016 seasons. These data, along with antigenic analysis, can aid in selecting effective vaccine strains. We confirmed that fatality in influenza A cases was related to underlying patient diseases, such as chronic lung disease, and further studies are needed to confirm associations between mortality and viral genetic substitutions.
Keywords: hospital‐based influenza morbidity and mortality (HIMM), influenza A virus, substitution
1. INTRODUCTION
Influenza A viruses are classified under the Orthomyxoviridae family and consist of several subtypes, based on their hemagglutinin (HA) and neuraminidase (NA) composition. In some cases, reassortment of the 8 viral RNA segments may occur if different influenza viruses infect the same host, thereby generating new viral strains (antigenic shift). Mutations in the genes of influenza A viruses are enhanced by the absence of RNA proofreading enzymes; in particular, substitutions in the HA protein alter the viral antigenic epitopes sufficiently to avoid the host immune response (antigenic drift). These antigenic changes can trigger the generation of new strains and subgroups, leading to pandemics or new epidemics worldwide.1, 2, 3
Influenza A virus subtypes H3N2 (A/H3N2) and H1N1 (A/H1N1), and influenza B virus have been circulating in the human population every winter, and account for 3 to 5 million cases of severe illness and 250 000 to 500 000 deaths annually, mostly caused by secondary bacterial pneumonia in young children and the elderly.4, 5, 6, 7 To reduce the public health burden of influenza, the World Health Organization (WHO) recommends annual influenza vaccination and recommends the four strains to be included in vaccine composition. Seasonal vaccine strains are recommended twice a year because of the differences in the duration of winter in the Southern Hemisphere and Northern Hemisphere. Vaccine strain recommendations for effective influenza vaccination are based on antigenic analysis and viral genome data worldwide in addition to global surveillance systems of the circulating viruses. In addition, genome sequencing and analysis of circulating influenza viruses have provided comprehensive understanding of evolutionary models as well as epidemiologic insight based on analysis of antigenic determinants, drug resistance, and a variety of sequence‐based bioinformatics methods.2, 8, 9
Here, we analyzed the HA and NA genes of influenza A viruses prevalent in Korea during the 2011‐2016 seasons. These viruses were identified through the Hospital‐based Influenza Morbidity & Mortality (HIMM) surveillance system.10 In addition, we investigated the effect of mutations in the 8 segmented genes of influenza A viruses on risk of mortality for infected patients.
2. MATERIALS AND METHODS
2.1. Sample collection and viral isolation
Through the HIMM system, clinical and virological surveillance was conducted for patients visiting the emergency departments or being hospitalized due to influenza‐like illness at 10 university hospitals in Korea from 2011 to 2016 (n = 13 620). We obtained nasopharyngeal swab samples during the 2011‐2016 season and identified influenza viruses using RT‐PCR. Influenza A virus‐positive samples were identified as either influenza A/H3N2 or 2009 pandemic A/H1N1 (A(H1N1) pdm09) and subjected to sequencing for the determination of HA and NA genes. A total of 254 sequences were used in the phylogenetic study. Using influenza A virus‐positive samples, Madin‐Darby canine kidney (MDCK) cells were inoculated with severe acute respiratory infection (SARI) samples and cultured for 48‐72 h. The hemagglutination assay was performed to detect the influenza virus growth.11 Unfortunately, samples were not obtained for the 2011‐2013 seasons. Therefore, analysis of fatal cases was performed only for the 2013‐2016 seasons.
2.2. Viral RNA extraction and sequencing
Viral RNA was extracted using the QIAamp Viral RNA mini kit (Qiagen, Hilden, Germany) and then reverse‐transcribed using influenza A virus universal primers (Uni 12, 5′‐AGCAAAAGCAGG‐3′) with the Primescript 1st strand cDNA synthesis kit (Takara, Shiga, Japan). PCR amplification was carried out using primers specific for the viral RNA segments coding for HA (forward, 5′‐ AGCAAAAGCAGGGG‐3′; reverse, 5′‐AGTAGA AAC AAGGGTGTTTT‐3′), NA (forward, 5′‐AGCRAAAGCAGGRGTTTAAAA‐3′; reverse, 5′‐AGTAGAAACAAGGAGTTTTTT‐3′), nucleoprotein (NP; forward, 5′‐AGCRAAAGCAG GGTARATAAT‐3′; reverse, 5′‐AGTAGAAACAAGGGTATTTTT‐3′), nonstructural protein (NS; forward, 5′‐AGCRAAAGCAGGGTGACAAA‐3′; reverse, 5′‐AGTAGAAACAAGGGTGTTTTTTAT‐3′), matrix protein (M; forward, 5′‐AGCRAAAGCAGGTAGATATT‐3′; reverse, 5′‐AGTAGAAACAAGGTAGTT TTT‐3′), and RNA polymerase subunits PA (forward, 5′‐AGCRAAAGCAGGTACTGATYCGAAATG‐3′; reverse, 5′‐AGTAGAAACAAGGTACTTTTTTGGACA‐3′), PB1 (forward, 5′‐AGCRAAAGCAGGCA A ACCATTTGAATG‐3′; reverse, 5′‐AGTAGAAACAAGGCATTTTTTCATGAA‐3′), and PB2 forward, 5′‐AGCRAAAGCAGGTCAATTATATTCA‐3′; reverse, 5′‐AGTAGAAACAAGGTCGTTTTTAAACTA‐3′). After amplification, the sequence readouts of the PCR products were analyzed. The sequences obtained from this study were deposited in GenBank under accession no. KY063619 through KY063705, KY509553‐KY509793.
2.3. Sequence analysis
For the phylogenetic study, the sequences were compared with the NCBI‐registered full‐length nucleotide sequences of influenza A/H3N2 and A(H1N1) pdm09 viruses and vaccine strains (H1N1: A/California/07/2009; H3N2: A/Perth/16/2009, A/Victoria/361/2011, A/Texas/50/2012, and A/Switzerland/9715293/2013). The sequences were aligned using the BioEdit program. MEGA 6.06 was used to build the phylogenetic tree, using the maximum‐likelihood method by obtaining the initial tree for the heuristic search based on the neighbor‐joining method through a matrix of pairwise distances evaluated by the maximum composite likelihood approach.12 The bootstrap scores were set to 1000 (bootstrap values over 50 are shown above the tree branches).
2.4. Hemagglutination inhibition (HAI) assay
HAI titers were determined using standard procedures. In brief, antisera were pre‐treated overnight with receptor‐destroying enzyme (RDE) at 37°C and heat inactivated at 56°C for 30 min. Twofold serial dilutions of RDE‐treated antisera (50 μL) starting at a 1:10 dilution were incubated with 4 HA unit/25 μL of each virus and incubated at RT for 1 h. Next, 50 μL of 0.75% guinea pig red blood cells (gRBCs) was added, followed by 1‐h incubation at RT. We confirmed the coagulation of gRBCs and interpreted HAI results.
2.5. Neuraminidase inhibitor (NAI) assay
The NA‐FluorTM assay kit (Thermo Fisher Scientific, Waltham, MA, USA) was used according to the manufacturer′s protocol. Neuraminidase inhibitor was prepared in 10‐fold serial dilutions at 4× the final concentration in assay buffer (16.65 mm MES, 2 mm CaCl2, pH 6.5), and 25 μL was added to wells of a black flat‐bottom 96‐well microplate. Viral samples were diluted in assay buffer, and 25 μL was added to the NAI serial dilutions, and the samples were then incubated for 30 min at 37°C. NA‐Fluor Substrate (50 μL) was added to create a final assay concentration of 100 μm 4‐Methylumbelliferone sodium salt, and the samples incubated at 37°C for 1 h. No‐virus controls were included on each assay plate. Assay was terminated by addition of 100 μL of Stop Solution, and plates were read on a Victor 3 plate reader using Ex 355 nm/Em 460 nm settings. For data analysis, the relative fluorescence unit values of the no‐virus control wells were subtracted from the virus‐containing well values and data were processed using Graphpad® Prism software.
2.6. Statistics analysis
To elucidate the factors associated with the death of influenza cases, fatal and non‐fatal cases were selected (1:3 ratio) on the collection date, sex, and age in the 2013‐2016 seasonal isolates. The A(H1N1)pdm09 fatal case sample was excluded because one fatal case was not appropriate for statistical analysis. The 2011‐2013 fatal case samples were excluded due to lack of appropriate non‐fatal case samples. Therefore, we analyzed the data pertaining to a total of 24 samples including 6 fatal and 18 non‐fatal cases in the 2013‐2016 season using Fisher's exact test. A P‐value <.05 was considered statistically significant.
3. RESULTS
3.1. HA and NA diversity of seasonal influenza A virus in Korea, 2011‐2016
A total of 13 620 patients were enrolled through the HIMM surveillance system during the 2011‐2016 seasons. These include 4725 patients positive for influenza A or B viruses with RT‐PCR, and 3696 (78.2%) of them were confirmed with influenza A virus, including subtype A(H1N1)pdm09 and H3N2 (Table 1). During this period, A/H3N2 was the predominant epidemic strain with the exception of 2015‐2016 season during which A/H1N1 co‐circulated. Because of this dominant pattern, we collected 43 sequences of A(H1N1)pdm09 in the 2012‐2014 and 2015‐2016 seasons, and 211 sequences of A/H3N2 in the 2011‐2015 seasons. A phylogenetic tree was constructed to confirm the substitution patterns of HA and NA genes in the influenza A virus.
Table 1.
Influenza types in Korea during the 2011‐2016 seasons based on RT‐PCR
| Seasons | Number of cases enrolled in HIMM | RT‐PCR | Influenza type | Number of samples analyzed | |||||
|---|---|---|---|---|---|---|---|---|---|
| Positive | Negative | Non‐sample | FluA | FluB | Co‐infection | A/H1N1 | A/H3N2 | ||
| 2011‐2012 | 2252 | 927 (41.1%) | 1243 (55.2%) | 82 (3.6%) | 731 (78.9%) | 182 (19.6%) | 14 (1.5%) | ‐ | 2 |
| 2012‐2013 | 1667 | 681 (40.9%) | 882 (52.9%) | 104 (6.2%) | 676 (99.3%) | 4 (0.6%) | 1 (0.1%) | 1 | 93 |
| 2013‐2014 | 3082 | 1295 (42.0%) | 1328 (43.1%) | 459 (14.9%) | 978 (75.5%) | 307 (23.7%) | 10 (0.8%) | 22 | 46 |
| 2014‐2015 | 3901 | 1298 (33.3%) | 2002 (51.3%) | 601 (15.4%) | 962 (74.1%) | 273 (21.0%) | 63 (4.9%) | ‐ | 70 |
| 2015‐2016 | 2718 | 524 (19.3%) | 1543 (56.8%) | 651 (24.0%) | 349 (66.6%) | 162 (30.9%) | 13 (2.5%) | 20 | ‐ |
| Total | 13 620 | 4725 (34.7%) | 6998 (51.4%) | 1897 (13.9%) | 3696 (78.2%) | 928 (19.6%) | 101 (2.1%) | 43 | 211 |
The HA phylogenetic tree was developed using 43 HA sequences of A(H1N1)pdm09, 211 HA sequences of A/H3N2 and the recommended vaccine strains (A/Perth/16/2009, A/Victoria/361/2011, A/Texas/50/2012, A/Switzerland/9715293/2013, and A/California/07/2009). In Figure 1, most of the A/H1N1 sequences clustered in clade 6B with the exception of A/Cheongju/G792/2013. The clade 6B was differentiated into two groups by the amino acid substitutions V152T and V173I in group 1 and S84N, S162N, and I216T in group 2. The phylogenetic tree of A(H1N1)pdm09 NA genes has shown that the two groups varied in terms of amino acid substitutions V67I, T381I in group 1 and V13I, I34V in group 2. These groups were generally congruent in the A(H1N1)pdm09 HA and NA phylogenetic tree. In the HA phylogenetic tree of A/H3N2, the viruses could be categorized into clades 3C.2 and 3C.3. Specifically, viruses from the 2012‐2013 and 2013‐2014 seasons clustered in clades 3C.2 and 3C.3a, respectively. Viruses from the 2014‐2015 season could be split into two clades based on the amino acid substitutions A138S, F159S, and N225D for clade 3C.3a and L3I, N144S, F159Y, and N225D for clade 3C.2a. The antigenic characterization was performed to determine whether the HA sequence substitution of A/H3N2, which indicates clade classification, is indicative of antigenic variation. We also confirmed significant antigenic changes in influenza A virus as a result of genetic variation in influenza A/H3N2 viruses (Table 2).
Figure 1.
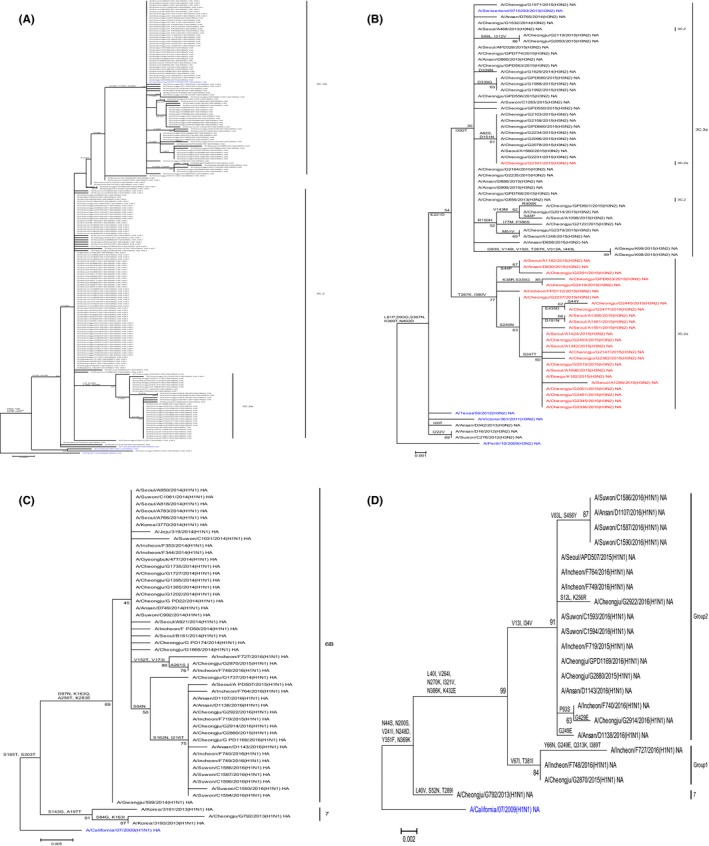
Phylogenetic tree of HA and NA genes of influenza A virus in Korea during the 2011‐2016 seasons. The (A) HA and (B) NA sequences of A/H3N2 isolate in Korea showed clustering in the phylogenetic tree. The sequences of clade 3C.2a in HA tree are shown in red font (B) NA of A/H3N2. The phylogenetic tree of A/H1N1 isolate (C) HA and (D) NA sequences reveals distinct groups. The phylogenetic tree was inferred from the recommended H1N1 vaccine strains (A/California/07/2009), H3N2 vaccine (A/Perth/16/2009, A/Victoria/361/2011, A/Texas/50/2012, A/Switzerland/9715293/2013), and clinical isolates identified in Korea (2011‐2016). The font colors represent the vaccine strain (blue)
Table 2.
Antigenic characterization of H3N2 influenza A viruses
| Reference viruses | Post‐infection sheep antisera | Genetic clade | |||
|---|---|---|---|---|---|
| A/Victoria/361/2011 | A/Texas/50/2012 | A/Switzerland/9715293/2013 | |||
| A/H3N2 vaccine strains | >1280 | >1280 | >1280 | ||
| Test viruses | |||||
| 2013‐2014 season isolates | A/Ansan/D765/2014 | 640 | 320 | 1280 | 3C.3a |
| A/Cheongju/G1532/2014 | 320 | 160 | 640 | 3C.3a | |
| A/Cheongju/G1629/2014 | 640 | 320 | 1280 | 3C.3a | |
| A/Ansan/D799/2014 | 320 | 160 | 1280 | 3C.3a | |
| A/Cheongju/G1739/2014 | 640 | 320 | 1280 | 3C.3a | |
| 2014‐2015 season isolates | A/Cheongju/G2156/2015 | >1280 | 1280 | >1280 | 3C.3a |
| A/Suwon/K98/2015 | 160 | 80 | 1280 | 3C.3a | |
| A/Ansan/D858/2015 | 320 | 160 | 1280 | 3C.3a | |
| A/Ansan/D906/2015 | 320 | 160 | 1280 | 3C.3a | |
| A/Cheongju/G1971/2015 | 160 | 80 | >1280 | 3C.3a | |
| A/Cheongju/G2103/2015 | 640 | 320 | 1280 | 3C.3a | |
| A/Seoul/A1284/2015 | 1280 | 1280 | 1280 | 3C.2a | |
| A/Seoul/A1251/2015 | 1280 | 640 | 1280 | 3C.2a | |
| A/Seoul/A1342/2015 | 80 | 40 | 80 | 3C.2a | |
| A/Ansan/D830/2015 | 160 | 80 | 160 | 3C.2a | |
| A/Seoul/A1299/2015 | 160 | 40 | 80 | 3C.2a | |
| A/Cheongju/G2477/2015 | 80 | 40 | 160 | 3C.2a | |
Based on the NA phylogenetic tree, viruses from the 2014‐2015 season fell into two distinct clusters based on amino acid substitution I392T for clade 3C.3a and T267K and I380V for clade 3C.2a. Interestingly, these differences in the NA cluster were consistent with differences in the HA clusters 3C.2a and 3C.3a, except for strains A/Cheongju/G2161/2015(H3N2), A/Seoul/A468/2013(H3N2), and A/Cheongju/G856/2013(H3N2).
3.2. Genetic analysis of 8 segmented genes in the fatal influenza A virus isolates
Viral virulence can be increased by mutating non‐structural proteins (Table S1). Therefore, internal gene sequences from influenza A viruses from fatal cases were used in the genetic analysis. Genetic analysis of 8 segments (HA, NA, M, NP, NS, PA, PB1, and PB2) was performed using sequences of fatal isolates (A(H1N1)pdm09; n = 1, A/H3N2; n = 11) and vaccine strains. Most of the fatal cases had underlying conditions, such as diabetes, chronic respiratory diseases, and chronic medical illnesses (Table 3).
Table 3.
Clinical and laboratory characteristics of the 12 fatal cases of influenza
| Season | Isolated virus | Age/sex | BMI | Body temperature at presentation (°C) | Pre‐existing condition | Cause of death | Microbiologic test | Antiviral drug treatment (days from symptom onset to administration of antiviral) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Sputum culture | Blood culture | Pneumococcal urinary antigen test | ||||||||
| 2011‐2012 | A/Ansan/D16/2012 (H3N2) | 78/M | 27.5 | None | Diabetes, hypertension, chronic cerebrovascular disease | Pneumonia | Streptococcus pneumoniae | Not done | Positive | Oseltamivir 75 mg bid (0) plus |
| Peramivir 300 mg qd (1) | ||||||||||
| A/Suwon/C276/2012 (H3N2) | 89/M | 24.2 | 37.3 | Diabetes, hypertension, chronic respiratory diseases, BPH | Exacerbation of COPD | MRSA | Not done | Negative | Oseltamivir 75 mg bid (1) | |
| 2012‐2013 | A/Seoul/A468/2013 (H3N2) | 89/M | 23.7 | 37.6 | None | Multi‐organ failure | Streptococcus pneumoniae | Not done | Positive | Peramivir 100 mg qd (5) |
| A/Cheongju/G792/2013 (H1N1) | 74/F | 15.2 | 38 | Cancer | Pneumonia | Normal flora | Not done | Positive | Oseltamivir 150 mg bid (1) plus | |
| Peramivir 300 mg qd (3) | ||||||||||
| A/Ansan/D342/2013 (H3N2) | 72/M | 20.7 | 38.6 | Chronic cardiovascular disease, AML | Pneumonia | Pseudomonas aeruginosa | Pseudomonas aeruginosa | Negative | Oseltamivir 75 mg bid (2) plus | |
| Peramivir 250 mg qd (2) | ||||||||||
| A/Cheongju/G856/2013 (H3N2) | 93/F | 20 | Non | Chronic cerebrovascular disease | Pneumonia | No growth | Not done | Negative | Oseltamivir 75 mg bid (1) | |
| 2013‐2014 | A/Ansan/D765/2014 (H3N2) | 80/M | 23.1 | 38.6 | Chronic respiratory diseases, COPD | Pneumonia, sepsis | Normal flora | Not done | Not done | Oseltamivir 75 mg bid (1) |
| A/Cheongju/G1532/2014 (H3N2) | 86/F | 17.8 | 36.8 | Hypertension | Pneumonia, sepsis | Normal flora | Staphylococcus aureus | Negative | Oseltamivir 75 mg bid (1) | |
| A/Cheongju/G1629/2014 (H3N2) | 86/M | 16.5 | 37.5 | COPD | Pneumonia | MRSA | Not done | Negative | Oseltamivir 75 mg bid (7) | |
| 2014‐2015 | A/Seoul/A1284/2015 (H3N2) | 97/F | 20.2 | 37.8 | Diabetes, chronic respiratory diseases, anemia, dementia, osteoporosis | Pneumonia | Pseudomonas fluorescens | Not done | Negative | Peramivir 300 mg qd (1) |
| A/Seoul/A1251/2015 (H3N2) | 92/M | 17.9 | 38.2 | Diabetes, asthma, chronic respiratory diseases, TB, BPH | Pneumonia, sepsis | MRSA, Escherichia coli | Not done | Negative | Peramivir 300 mg qd (2) | |
| A/Cheongju/G2156/2015 (H3N2) | 83/F | 17.3 | None | Hypertension, chronic respiratory diseases | Pneumonia | Not done | Not done | Negative | Oseltamivir 75 mg bid (3) | |
BMI, body mass index; BPH, benign prostatic hyperplasia; COPD, chronic obstructive pulmonary disease; TB, tuberculosis; AML, acute myeloid leukemia; MRSA, methicillin‐resistant Staphylococcus aureus.
Compared with vaccine strain, amino acid mutations were found in the A/Cheongju/G792/2013(H1N1) HA gene: the P83S, S84G, S143G, K163I, G170R, S185T, A197T, S203T, A261T, G262E, H273N, and I321V substitutions were located in the HA1 region, the E47K, S124N, I183V, and V193A substitutions were located in the HA2 region (Table 4).
Table 4.
Amino acid substitutions in the 8 segments of influenza strains isolated in Korea during 2011‐2015 seasons compared to the vaccine strains
| Subtype | Seasons | Sample name | HA | NA | M1 | M2 | NP | NS1 | NS2 | PA | PB1 | PB2 | Genetic clade (HA based) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H3N2 | H3N2 Vaccine | A/Victoria/361/2011 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| 2011‐2012 | A/Suwon/C276/2012 | Q33R, E50K, V186G, D188G, R255T, P273R, N278K, I149T | I222V, K258E, T329N | ‐ | V51I, | S359P, M374I, M440V | D209N, K229E | ‐ | E351D, R356K, M407I | D581N | V606A | 3C.1 | |
| A/Ansan/D16/2012 | Q33R, V186G, N278K | I222V, K258E, T329N | ‐ | G16E, V51I, E97G | V197A, M374I | V22F, D209N, K229E | K39R | E351D, M407I | ‐ | M66T, R389K | 3C.1 | ||
| 2012‐2013 | A/Ansan/D342/2013 | Q33R, N145S, V186G, N278K | I20T, K258E, T329N | ‐ | ‐ | K470M | E26K, K78R, D209N, K229E | ‐ | M407I | R361K | Y55F, R194Q, L384I | 3C.2 | |
| A/Seoul/A468/2013 | Q33R, N145S, V186G, N246Y, N278K, D160N | E221D, K258E, T329N, I392T | ‐ | ‐ | K357R | E26K, I182V, D209N, K229E | ‐ | M407I, V668I, N675K | M317T | ‐ | 3C.2 | ||
| A/Cheongju/G856/2013 | Q33R, N145S, V186G, N278K, D160N | E221D, K258E, T329N, I392T | ‐ | ‐ | ‐ | E26K, I182V, D209N, K229E | ‐ | G240V, M407I, V668I, N675K | ‐ | ‐ | 3C.2 | ||
| H3N2 Vaccine | A/Switzerland/9715293/2013 | Q33R, T128A, A138S, R142G, N145S, F159S, V186G, N225D, N262K, K326R | E221D, K258E, T329N, I392T | ‐ | ‐ | S217G | E26K, M124I, D209N, K229E | ‐ | Q256K, I308V, I554V, K605R, V669I, H713Y | ‐ | ‐ | 3C.3a | |
| 2013‐2014 | A/Cheongju/G1629/2014 | Q33R, T128A, A138S, R142G, N145S, F159S, V186G, N225D, N262K, K326R | E221D, K258E, T329N, D339N, I392T | ‐ | ‐ | S217G, S482N | E26K, M124I, D209N, K229E | ‐ | Q256K, I308V, I554V, K605R, V669I | ‐ | ‐ | 3C.3a | |
| A/Ansan/D765/2014 | Q33R, T128A, A138S, R142G, N145S, F159S, V186G, N225D, N262K, K326R, V73A | Y67F, E221D, K258E, T329N, I392T | ‐ | ‐ | S217G | E26K, M124I, D209N, K229E | ‐ | Q256K, I308V, I554V, K605R, V669I | R52K, L424M | V338I, S709N | 3C.3a | ||
| A/Cheongju/G1532/2014 | Q33R, T128A, A138S, R142G, N145S, F159S, V186G, N225D, N262K, K326R | E221D, K258E, T329N, I392T | ‐ | ‐ | S217G | E26K, M124I, A202T, D209N, K229E | ‐ | Q256K, I308V, A455G, I554V, K605R, V669I | T400K | A559V, S643T | 3C.3a | ||
| 2014‐2015 | A/Cheongju/G2156/2015 | Q33R, T128A, A138S, R142G, N145S, F159S, V186G, A212S, N225D, N262K, K326R, A201V | E221D, K258E, T329N, I392T | ‐ | ‐ | I136L, S217G | E26K, M124I, D209N, K229E | ‐ | Q256K, I308V, D396E, I554V, K605R, V669I | ‐ | R390K, M476L | 3C.3a | |
| A/Seoul/A1284/2015 | L3I, Q33R, N144S, N145S, F159Y, V186G, N225D, N278K, Q311H, D160N | E221D, S245N, S247T, K258E, T267K, T329N, I380V | ‐ | I39M | M481I | E26K, D209N, K229E | ‐ | N272S, D396E, M407I, V668I, N675K | ‐ | V63I, I589T | 3C.2a | ||
| A/Seoul/A1251/2015 | L3I, Q33R, N144S, N145S, F159Y, K160T, V186G, N225D, N278K, Q311H, D160N | D151N, E221D, S245N, S247T, K258E, T267K, T329N, I380V, W383C | ‐ | I39M | M481I | E26K, D209N, K229E | ‐ | N272S, D396E, M407I, V668I, N675K | W580C | V63I, I589T | 3C.2a | ||
| H1N1 | H1N1 Vaccine | A/California/07/2009 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| 2012‐2013 | A/Cheongju/G792/2013 | P83S, S84G, S143G, K163I, G170R, S185T, A197T, S203T, A261T, G262E, H273N, I321V, E47K, S124N, I183V, V193A | L40V, N44S, S52N, N200S, V241I, N248D, T289I, Y351F, N369K | V80I, M192V, K230R | D21G | V100I, L108I | L90I, I111T, V117M, I123V, N205S | T48A | I30T, Y161F, P224S, E252V, N321K, A343T, V407I, R673S | G154D, I397M, I435T, H456Y | D195N, R293K, V344M, I354L, V731I | 7 |
HA2 domain is indicated in bold‐italic font
Compared with the NA gene of the vaccine strain, A/Cheongju/G792/2013(H1N1) carried L40V, N44S, S52N, N200S, V241I, N248D, T289I, Y351F, and N369K substitutions. In addition, several substitutions were identified in the other 6 segments (Table 4).
In the A/H3N2 analysis, we confirmed variations for the cluster differences (3C.2a and 3C.3a) in the HA and NA genes. In addition, the NA sequence of the 2011‐2012 season (n = 2) carried an I222V substitution, an NA inhibitor resistance mutation.13, 14, 15, 16 The NA sequence of several samples carried substitutions: I20T in A/Ansan/D342/2013, D339N in A/Cheongju/G1629/2014, Y67F in A/Ansan/D765/2014, and D151N, W383C in A/Seoul/A1251/2015. The NAI assay was conducted using vaccine strains and viruses isolated in the 2013‐2016 seasons to determine whether NA gene mutations affected virulence due to antiviral resistance. The NAI assay demonstrated the absence of resistant viruses in the fatal influenza cases (Table 5).
Table 5.
Neuraminidase inhibition of fatal case isolates
| Sample | 2013‐2014 season | 2014‐2015 season | A/Switzerland/9715293/2013 | ||||
|---|---|---|---|---|---|---|---|
| D765 | G1532 | G1629 | G2156 | A1284 | A1251 | ||
| IC50 (nm) | 0.275 | 0.283 | 0.279 | 0.245 | 0.155 | 0.210 | 0.218 |
| 95% CI | 0.259‐0.292 | 0.266‐0.300 | 0.267‐0.292 | 0.224‐0.269 | 0.147‐0.163 | 0.192‐0.230 | 0.204‐0.232 |
In the analysis of internal genes, the M sequence of all the isolates investigated harbored the S31N genetic marker for adamantine resistance in M2. By comparing the sequences from each season with the A/H3N2 vaccine strain (A/Victoria/361/2011), we found several amino acid substitutions (Table 4). Interestingly, the PA gene of A/Ansan/D765/2014, A/Cheongju/G1532/2014, A/Cheongju/G1629/2014, A/Cheongju/G2156/2015, and A/Switzerland/ 9715293/2013 carried 5 amino acid mutations: Q256K, I308V, I554V, K605R, and V669I. These PA substitutions were consistent with clade 3C.2a in the HA phylogenetic tree. As shown in Table 4, we confirmed that the substitutions of NA, NS1, and PA coincided with HA‐based genetic clade. In addition, the PB1 and PB2 genes revealed multiple non‐synonymous substitutions (Table S2).
Statistical analysis was performed to study the association between amino acid substitution and death in fatal cases (Table 6). Although some substitutions (I39M of M2 and M481I of NP) showed a P value of .054, they were not significant in the correlation analysis of genetic substitution and mortality. As shown in Table 7, our correlation analysis revealed that chronic lung disease was more frequently associated with fatal than non‐fatal cases.
Table 6.
Amino acid substitutions associated with mortality based on Fisher's exact test
| Substitutions | Fatal cases (n = 6) | Non‐Fatal cases (n = 18) | Fisher's P‐value |
|---|---|---|---|
| M2_I39M | 2 | 0 | .054 |
| NP_I136L | 1 | 4 | 1.000 |
| NP_S217G | 4 | 15 | .568 |
| NP_M481I | 2 | 0 | .054 |
| NP_S482N | 1 | 0 | .250 |
| NS1_M124I | 4 | 15 | .568 |
| NS1_A202T | 1 | 0 | .250 |
| PA_Q256K | 4 | 15 | .568 |
| PA_N272S | 2 | 3 | .568 |
| PA_I308V | 4 | 15 | .568 |
| PA_M407I | 2 | 3 | .568 |
| PA_A455G | 1 | 0 | .250 |
| PA_I554V | 4 | 15 | .568 |
| PA_K605R | 4 | 15 | .568 |
| PA_V668I | 2 | 3 | .568 |
| PA_V669I | 4 | 15 | .568 |
| PA_N675K | 2 | 3 | .568 |
| PB1_R52K | 1 | 0 | .250 |
| PB1_T400K | 1 | 0 | .250 |
| PB1_L424M | 1 | 0 | .250 |
| PB1_W580C | 1 | 0 | .250 |
| PB2_V63I | 2 | 4 | .618 |
| PB2_V338I | 1 | 1 | .446 |
| PB2_R390K | 1 | 4 | 1.000 |
| PB2_M476L | 1 | 4 | 1.000 |
| PB2_A560V | 1 | 0 | .250 |
| PB2_I589T | 2 | 3 | .568 |
| PB2_S644T | 1 | 0 | .250 |
| PB2_S710N | 1 | 0 | .250 |
Table 7.
Demographic characteristics of patients with influenza
| Fatal cases (n = 6) | Non‐fatal cases (n = 18) | P valuea | |
|---|---|---|---|
| Age (year), mean ± SD | 87.3 ± 6.2 | 82.0 ± 7.1 | .12 |
| Sex (male), n (%) | 3 (50.0) | 9 (50.0) | 1 |
| Comorbidity | 4 (66.7) | 14 (77.8) | .62 |
| Diabetes mellitus | 2 (33.3) | 5 (27.8) | 1 |
| Cardiovascular disease | ‐ | 5 (27.8) | .28 |
| Cerebrovascular disease | ‐ | 6 (33.3) | .28 |
| Neuromuscular disease | ‐ | 4 (22.2) | .54 |
| Chronic lung disease | 4 (66.7) | 2 (11.1) | .02 |
| Asthma | 1 (16.7) | 2 (11.1) | 1 |
| Chronic kidney disease | ‐ | 1 (5.6) | 1 |
| Chronic liver disease | ‐ | 3 (16.7) | .55 |
| Malignancy | ‐ | 1 (5.6) | 1 |
Fisher's exact test.
In conclusion, statistical analysis showed no significant association between the viral genetic differences and mortality; however, the mortality was increased by host factors such as chronic lung disease.
4. DISCUSSION
We performed sequencing and phylogenetic analysis of influenza A viruses collected from patients during the 2011‐2016 season in Korea, to verify previously known and novel virulence factors in patients with fatal influenza and the evolution of influenza viruses with time. We confirmed the genetic changes among the influenza A viruses during 2011‐2016 in Korea. The A(H1N1)pdm09 in 2015‐2016 was grouped into clade 6B and separated into two clusters by substitution V152T, V173I in group 1 and S84N, S162N, and I216T in group 2 over time. Interestingly, the differences of HA clusters were consistent with the NA genes. In the A/H3N2, the sequences in the 2013‐2015 season carried substitutions: A138S, F159S, N225D, N241D, and K326R in HA1 or N144S, F159Y, N225D, and Q311H in HA1, and were categorized into clade 3C.3a and 3C.2a. In addition, the NA genes were distinguished into two clusters: I392T in cluster 1 and T267K, I380V in the other cluster. Interestingly, these results were congruent with HA clustering: 3C.3a and 3C.2a. Further, sequences detected in the 2012‐2013 season (HA clade 3C.2) showed I392T substitution in the NA gene. These mutations were generated earlier than the substitution of HA clade 3C.3a. Previous studies recognized several antigenic sites (A–E) and receptor‐binding sites (RBS) in the HA gene of A/H3N2.2, 17, 18, 19, 20 Several researchers reported that antigenic drift was caused by single amino acid substitutions near the RBS of the influenza A virus.2, 20, 21 The HA proteins of 3C.2a and 3C.3a viruses are substituted around the RBS, particularly D225N, 126NWT/AG, and 144NN/SSF in HA1, compared with A/Victoria/361/2011. These mutations in the RBS of HA are associated with changes in the protein surface, electrostatic charge, and N‐linked glycosylation. Based on the results of antigenic characterization, we suggest that the isolates belonging to the 3C.3a clade included the A/Switzerland/297135/2013–like virus, which carried a twofold HAI titer of the A/Switzerland/ virus. In addition, the isolates in the 3C.2a clade differ from the vaccine strain in antigenicity due to the 8‐ to 32‐fold differences from the vaccine strain in the HAI titer.
This difference seems to be due to structural variation in HA from genetic substitution. Our study demonstrated the variation patterns of epidemic influenza viruses in Korea using genetic as well as antigenic analyses. These data can be used to select vaccine strains and to analyze virus substitution patterns over time.
H3N2 seasons become increasingly severe, with higher numbers of hospitalizations and deaths. We studied the molecular genetics of influenza A viruses and factors associated with patient death following infection with influenza A/H3N2 during the 2011‐2016 seasons in Korea. First, we identified genetic substitutions in viruses from fatal cases. The I222V substitution in the NA protein was found in only the two A/H3N2 viruses from the 2011‐2012 season. However, other antidrug mutations, H274Y (N2 numbering) and I119V, were not confirmed. Previous studies have shown that the single I222V/M substitution in the NA protein is associated with marginal levels of resistance to oseltamivir, while synergistically increased drug resistance was associated with E119V and H274Y substitutions.15, 16, 19, 22, 23, 24, 25, 26 The S31N substitution in the M2 protein was frequently detected in the more recent viral sequences and reference sequences.27 In addition, V51I and I39M substitutions were identified in the 2011‐2012 and 2014‐2015 fatal case sequences. Among these two substitutions, V51I may enhance the fitness of M2 protein to increase the frequency of adamantine resistance associated with S31N mutation and the substitution of V51‐affected viral replication.28 The I39 of M2 was located in the transmembrane region, and substitution of the transmembrane region could affect M2 function, aiding in resistance to M2 inhibitors and transport to the cell surface. In the NP sequences, M374I and M481I were identified only in fatal case sequences. These substitutions have been reported to be involved a T‐cell epitope presented by MHC molecules.29, 30 In addition, it was previously reported that other substitutions were identified in the A/Cheongju/G792/2013(H1N1) (T48A in NS2) and in the A/Seoul/A468/2013(H3N2) (K357R in NP). The T48A in NS2 of A(H1N1)pdm09 contributed to the enhanced type I IFN antagonistic property of A/Vietnam/UT3062/04, leading to high virulence in ferrets. Mutations in amino acid 357 of NP, which is in the PB2 binding region of NP protein, have been reported that be involved in a shift in host specificity (Q in avian and K in human).
Statistical analysis was performed to confirm the association between amino acid substitution and death in fatal cases. Unfortunately, statistical analysis was conducted only for the 2013‐2015 season using appropriate controls (non‐fatal case samples). The I39M substitution of M2 and the M481I substitution of NP may be weakly correlated based on the P‐value of .054. Interestingly, the I39M of M2 and M482I of NP were identified in the same isolates (A/Seoul/A1284/2015, A/Seoul/A1251/2015). These internal proteins are involved in a variety of host responses and associated with the infectivity and replication of viruses; therefore, substitutions in these proteins can have a variety of effects.31, 32 A significant association between underlying diseases in patients and mortality revealed a significant correlation with chronic lung disease, confirming a well‐known relationship.
In conclusion, we observed clade changes in influenza A viruses in Korea from 2011 to 2016. Prediction of clade change using bioinformatics analysis of these data, along with antigenic analyses, can help select effective vaccine strains. We confirmed that the severity of influenza A virus infection was related to underlying patient diseases, such as chronic lung disease, and further studies are needed to confirm associations between mortality and genetic substitutions in the viruses.
Supporting information
Lee HS, Noh JY, Song JY, et al. Molecular genetic characteristics of influenza A virus clinically isolated during 2011‐2016 influenza seasons in Korea. Influenza Other Respi Viruses. 2018;12:497–507. https://doi.org/10.1111/irv.12549
Funding information
This work was supported by a grant from TEPIK (Transgovernmental Enterprise for Pandemic Influenza in Korea)‚ which is part of the Korea Healthcare Technology R&D Project funded by the Ministry of Health & Welfare, Republic of Korea (Grant no.: A103001).
REFERENCES
- 1. Kilbourne ED, Johansson BE, Grajower B. Independent and disparate evolution in nature of influenza A virus hemagglutinin and neuraminidase glycoproteins. Proc Natl Acad Sci USA. 1990;87:786‐790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Koel BF, Burke DF, Bestebroer TM, et al. Substitutions near the receptor binding site determine major antigenic change during influenza virus evolution. Science. 2013;342:976‐979. [DOI] [PubMed] [Google Scholar]
- 3. Shao TJ, Li J, Yu XF, Kou Y, Zhou YY, Qian X. Progressive antigenic drift and phylogeny of human influenza A(H3N2) virus over five consecutive seasons (2009‐2013) in Hangzhou, China. Int J Infect Dis. 2014;29:190‐193. [DOI] [PubMed] [Google Scholar]
- 4. Bashir Aamir U, Badar N, Mehmood MR, et al. Molecular epidemiology of influenza A(H1N1)pdm09 viruses from Pakistan in 2009‐2010. PLoS ONE. 2012;7:e41866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Joseph C, Togawa Y, Shindo N. Bacterial and viral infections associated with influenza. Influenza Other Respir Viruses. 2013;7(Suppl 2):105‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shrestha S, Foxman B, Weinberger DM, Steiner C, Viboud C, Rohani P. Identifying the interaction between influenza and pneumococcal pneumonia using incidence data. Sci Transl Med. 2013;5:191ra84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hiller NL, Ahmed A, Powell E, et al. Generation of genic diversity among Streptococcus pneumoniae strains via horizontal gene transfer during a chronic polyclonal pediatric infection. PLoS Pathog. 2010;6:e1001108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lam TT, Chong YL, Shi M, et al. Systematic phylogenetic analysis of influenza A virus reveals many novel mosaic genome segments. Infect Genet Evol. 2013;18:367‐378. [DOI] [PubMed] [Google Scholar]
- 9. Huang P, Liang LJ, Hou NM, et al. Phylogenetic, molecular and drug‐sensitivity analysis of HA and NA genes of human H3N2 influenza A viruses in Guangdong, China, 2007‐2011. Epidemiol Infect. 2013;141:1061‐1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Song JY, Cheong HJ, Choi SH, et al. Hospital‐based influenza surveillance in Korea: hospital‐based influenza morbidity and mortality study group. J Med Virol. 2013;85:910‐917. [DOI] [PubMed] [Google Scholar]
- 11. Balish AL, Katz JM, Klimov AI. Influenza: propagation, quantification, and storage. Current protocols in microbiology. 2013;Chapter 15:Unit 15G 1. [DOI] [PubMed]
- 12. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731‐2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tamura D, Okomo‐Adhiambo M, Mishin VP, et al. Application of a seven‐target pyrosequencing assay to improve the detection of neuraminidase inhibitor‐resistant Influenza A(H3N2) viruses. Antimicrob Agents Chemother. 2015;59:2374‐2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stoner TD, Krauss S, Turner JC, et al. Susceptibility of avian influenza viruses of the N6 subtype to the neuraminidase inhibitor oseltamivir. Antiviral Res. 2012;93:322‐329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Simon P, Holder BP, Bouhy X, Abed Y, Beauchemin CA, Boivin G. The I222V neuraminidase mutation has a compensatory role in replication of an oseltamivir‐resistant influenza virus A/H3N2 E119V mutant. J Clin Microbiol. 2011;49:715‐717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hurt AC, Holien JK, Barr IG. In vitro generation of neuraminidase inhibitor resistance in A(H5N1) influenza viruses. Antimicrob Agents Chemother. 2009;53:4433‐4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang H, Carney PJ, Chang JC, Guo Z, Villanueva JM, Stevens J. Structure and receptor binding preferences of recombinant human A(H3N2) virus hemagglutinins. Virology. 2015;477:18‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pyhala R, Ikonen N, Haanpaa M, Santanen R, Tervahauta R. Phylogenetic and antigenic analysis of influenza A(H3N2) viruses isolated from conscripts receiving influenza vaccine prior to the epidemic season of 1998/9. Epidemiol Infect. 2002;129:347‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dapat C, Suzuki Y, Kon M, et al. Phylogenetic analysis of an off‐seasonal influenza virus A (H3N2) in Niigata, Japan, 2010. Jpn J Infect Dis. 2011;64:237‐241. [PubMed] [Google Scholar]
- 20. Wiley DC, Wilson IA, Skehel JJ. Structural identification of the antibody‐binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature. 1981;289:373‐378. [DOI] [PubMed] [Google Scholar]
- 21. Smith DJ, Lapedes AS, de Jong JC, et al. Mapping the antigenic and genetic evolution of influenza virus. Science. 2004;305:371‐376. [DOI] [PubMed] [Google Scholar]
- 22. Spanakis N, Pitiriga V, Gennimata V, Tsakris A. A review of neuraminidase inhibitor susceptibility in influenza strains. Expert Rev Anti Infect Ther. 2014;12:1325‐1336. [DOI] [PubMed] [Google Scholar]
- 23. Dapat C, Kondo H, Dapat IC, et al. Neuraminidase inhibitor susceptibility profile of pandemic and seasonal influenza viruses during the 2009‐2010 and 2010‐2011 influenza seasons in Japan. Antiviral Res. 2013;99:261‐269. [DOI] [PubMed] [Google Scholar]
- 24. Siddique N, Naeem K, Abbas MA, Ahmed Z, Malik SA. Sequence and phylogenetic analysis of highly pathogenic avian influenza H5N1 viruses isolated during 2006‐2008 outbreaks in Pakistan reveals genetic diversity. Virol J. 2012;9:300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bauer K, Schrader C, Suess J, Wutzler P, Schmidtke M. Neuraminidase inhibitor susceptibility of porcine H3N2 influenza A viruses isolated in Germany between 1982 and 1999. Antiviral Res. 2007;75:219‐226. [DOI] [PubMed] [Google Scholar]
- 26. Baz M, Abed Y, McDonald J, Boivin G. Characterization of multidrug‐resistant influenza A/H3N2 viruses shed during 1 year by an immunocompromised child. Clin Infect Dis. 2006;43:1555‐1561. [DOI] [PubMed] [Google Scholar]
- 27. Wang J, Yibing W, DeGrado WF, et al. Structure and inhibition of the drug‐resistant S31N mutant of the M2 ion channel of influenza A virus. PNAS. 2013;110:1315‐1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stewart SM, Pekosz A. Mutations in the membrane‐proximal region of the influenza A virus M2 protein cytoplasmic tail have modest effects on virus replication. J Virol. 2011;85:12179‐12187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bastin J, Rothbard J, Davey J, Jones I, Townsend A. Use of synthetic peptides of influenza nucleoprotein to define epitopes recognized by class I‐restricted cytotoxic T lymphocytes. J Exp Med. 1987;165:1508‐1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wahla A, Schafera F, Bardet W, et al. HLA class I molecules consistently present internal influenza epitopes. PNAS. 2009;106:540‐545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mayer D, Molawi K, Martinez‐Sobrido L, et al. Identification of cellular interaction partners of the influenza virus ribonucleoprotein complex and polymerase complex using proteomic‐based approaches. J Proteome Res. 2007;6:672‐682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huarte M, Sanz‐Ezquerro JJ, Roncal F, Ortin J, Nieto A. PA subunit from influenza virus polymerase complex interacts with a cellular protein with homology to a family of transcriptional activators. J Virol. 2001;75:8597‐8604. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials