

Abstract
Epstein-Barr virus (EBV) lytic replication proceeds through an ordered cascade of gene expression that integrates lytic DNA amplification and late gene transcription. We and others previously demonstrated that 6 EBV proteins that have orthologs in β- and γ-, but not in α-herpesviruses, mediate late gene transcription in a lytic DNA replication-dependent manner. We proposed a model in which the βγ gene-encoded viral pre-initiation complex (vPIC) mediates transcription from newly replicated viral DNA. While this model explains the dependence of late gene transcription on lytic DNA replication, it does not account for this dependence in α-herpesviruses nor for recent reports that some EBV late genes are transcribed independently of vPIC. To rigorously define which transcription start sites (TSS) are dependent on viral lytic DNA replication or the βγ complex, we performed Cap Analysis of Gene Expression (CAGE)-seq on cells infected with wildtype EBV or EBV mutants defective for DNA replication, βγ function, or lacking an origin of lytic replication (OriLyt). This approach identified 16 true-late, 32 early, and 16 TSS that are active at low levels early and are further upregulated in a DNA replication-dependent manner (leaky late). Almost all late gene transcription is vPIC-dependent, with BCRF1 (vIL10), BDLF2, and BDLF3 transcripts being notable exceptions. We present evidence that leaky late transcription is not due to a distinct mechanism, but results from superimposition of the early and late transcription mechanisms at the same promoter. Our results represent the most comprehensive characterization of EBV lytic gene expression kinetics reported to date and suggest that most, but not all EBV late genes are vPIC-dependent.
Author summary
Herpesvirus lytic replication is characterized by the expression of early genes prior to viral DNA replication followed by late gene expression. Late genes are not only expressed after DNA replication, but cannot be expressed in its absence. We and others have shown that γ-herpesviruses, such as Epstein-Barr virus (EBV), as well as β-herpesviruses accomplish late gene transcription using a viral pre-initiation complex (vPIC). Proteins in this complex are encoded by genes with orthologs found only in β- and γ-herpesviruses (βγ genes) and absent in α-herpesviruses. We used EBV mutants defective for DNA replication or βγ complex/vPIC function to rigorously identify all EBV late genes and determine the extent of their dependence upon vPIC for expression. Because of the extensive bidirectional transcription of the EBV genome during lytic replication, unambiguous quantification of most lytic transcripts is not possible by conventional methods such as RNA-seq and RT-qPCR. We therefore used deep sequencing of non-amplified 5’ ends to uniquely localize and quantify transcription start sites in a strand-specific manner. Our results demonstrate that most, but not all, EBV late transcripts require vPIC for their expression. Furthermore, our results indicate that transcription that appears to be partially dependent on DNA replication (called “leaky” late transcription) results from superimposed early and late transcriptional mechanisms acting on the same promoter.
Introduction
Epstein-Barr virus (EBV) is a γ-herpesvirus that infects more than 95% of the human adult population. If acquired early in life, EBV infection is generally asymptomatic, however infection in adolescence may lead to infectious mononucleosis [1,2]. EBV infection is associated with a wide-spectrum of malignancies, such as Burkitt lymphoma, Hodgkin lymphoma, diffuse large B cell lymphoma, NK/T-cell lymphoma, post-transplant lymphoproliferative disease, nasopharyngeal carcinoma, and gastric carcinoma [3–6]. Although these tumors are characterized by latent EBV infection, an increasing body of evidence indicates that lytic infection is also important for the emergence of EBV-associated malignancies [7–10].
Herpesvirus lytic replication proceeds through a highly ordered cascade of gene expression that integrates viral DNA synthesis with structural protein production, while evading the host immune response. In EBV, this process is initiated by two immediate early viral transcription factors Zta (also called Z, ZEBRA, EB1—encoded by BZLF1) and Rta (also called R–encoded by BRLF1) [11–16], which activate early gene promoters leading to expression of proteins essential for viral DNA synthesis [12,17–19]. Late genes, which mainly encode structural proteins, have long been defined by their strict dependence on viral DNA replication for expression, but the basis for this dependence has been elusive. A key advance in our understanding of late gene transcription was the discovery that β- and γ-herpesviruses transcribe their late genes by a mechanism fundamentally different than the α-herpesviruses. This discovery began with the finding that several genes with orthologs found only in β- and γ-herpesviruses (βγ genes) were essential for late gene expression, but dispensable for DNA replication [20–27]. One of these βγ genes was shown to encode a viral TATA binding protein (vTBP) [28,29]. Studies of the EBV vTBP, encoded by BcRF1, showed that it preferentially bound to an atypical TATA box (TATT) found in many late gene promoters and activated transcription through this element in reporter assays [28]. Subsequently, we and others showed that five other EBV βγ genes are essential for late gene expression and activation of TATT-containing promoters [30–32]. Although the precise roles played by each gene are yet to be defined, they have been demonstrated to form a protein complex with vTBP, termed the viral pre-initiation complex (vPIC) [31]. We demonstrated that vPIC can only mediate late gene expression when the EBV origin of replication (OriLyt) is present in cis and proposed a model in which EBV vPIC mediates late gene expression by recruiting RNA polymerase II to use newly replicated viral DNA as the template for transcription [30].
Although this model provides a mechanistic link between lytic DNA replication and late gene transcription in β- and γ-herpesviruses, it does not explain why this dependence is also found in α-herpesviruses. This exception also raises the question of whether and to what extent EBV late gene expression might be accomplished by vPIC-independent mechanisms. In order to answer this question, it is essential to first distinguish genes that are strictly dependent upon DNA replication (late or true late) from those that are partially dependent (leaky late) as well as from early genes which exhibit no dependence upon DNA replication for their expression. The most comprehensive attempt at such an analysis of EBV lytic gene kinetics published to date was by Yuan et al. [33], which employed a custom oligo array to detect EBV lytic transcripts expressed in Akata cells in response to B cell receptor signaling. While this study was a significant advance in our understanding of late gene expression, it had several important limitations: First, it relied on phosphonoacetic acid (PAA) to block viral DNA replication. This approach identified many late genes, but also impaired expression of many well-characterized early genes such as BMRF1. For genes that were less well-characterized, it was unclear the extent to which partial dependence upon DNA replication was a real phenomenon or attributable to transcript overlap or other artifacts such as PAA toxicity. The EBV DNA replication defective mutants described in our recent study lacking the single-stranded DNA binding protein BALF2, which permits trans-complementation of DNA replication, as well the OriLyt knockout [30], afford the opportunity to study EBV transcript dependence upon DNA replication and OriLyt directly, without the toxicity of DNA polymerase inhibitors. Using the same approach, we can assess transcript dependence on the vPIC complex by trans-complementing EBV mutants deleted for specific βγ genes.
A major impediment to any genome-wide analysis of the EBV lytic transcriptome is that transcripts overlap so extensively that most transcripts cannot be unambiguously quantified by any conventional high-throughput method, including RNA-seq, RT-qPCR, and oligo arrays. Recently, quantitative high throughput methods have been developed to identify and quantify transcription start sites (TSS) which can circumvent the barrier presented by extensively overlapping transcripts. We used one of these methods: non-Amplified non-Tagging Illumina Cap Analysis of Gene Expression (nAnT-iCAGE [34], hereafter referred to as CAGE-seq) to quantify each EBV lytic transcript, its transcription start site (TSS), and its dependence on DNA replication, the presence of an OriLyt, and on the βγ gene-encoded vPIC. This approach not only allowed us to comprehensively define the kinetics of the EBV lytic transcriptome, but also provided insights into the unique features of the early and late transcriptional mechanisms.
Results
Overlapping nature of EBV lytic transcripts confounds quantitative analysis by conventional methods
The EBV transcriptome is organized in clusters, where multiple mRNAs are driven by unique promoters, but use the same polyA signal and are therefore co-terminal. One such cluster is the BFRF0.5-BFRF1-BFRF2-BFRF3 locus shown in Fig 1A. In the absence of an IRES, only the first ORF is efficiently translated, leading to transcripts that are overlapping, but not polycistronic. This organization of transcripts, which is typical, presents technical challenges, limiting the ability to precisely measure the mRNA levels of all, except the longest transcript, by RT-qPCR or RNA-seq. To demonstrate this phenomenon, EBV ΔBALF2 HEK293 cells, in which the gene encoding the single-stranded DNA binding protein BALF2 (required for lytic DNA replication) is disrupted (previously described in [30]]), were either uninduced (U), induced into the lytic phase by transfecting Rta and Zta expression plasmids (I) or induced and trans-complemented by transfecting Rta, Zta, and BALF2 expression plasmids (I + t) to further rescue the DNA replication defect. When RT-qPCR was employed to quantify BFRF3 mRNA levels, a signal was detected in the absence of DNA replication and increased when DNA replication was restored by BALF2 trans-complementation (Fig 1B, I vs I + t). In contrast, the BFRF3 protein product VCAp18 was detectable by western blotting only in the presence of EBV lytic DNA replication (Fig 1C, I + t). This apparent discrepancy arises from the inability of RT-qPCR to distinguish authentic BFRF3 transcripts from overlapping early transcripts in this cluster which contain the BFRF3 sequence in their 3’ UTRs. Thus, the overlapping nature of the EBV lytic transcriptome can confound accurate assessment of EBV lytic mRNA levels.
Fig 1. Nested structure of EBV lytic transcripts confounds quantification of EBV lytic mRNAs.

A) Schematic of the BFRF0.5-BFRF1-BFRF2-BFRF3 nested lytic transcriptional unit, the overlapping structure of which is shared by many EBV lytic transcripts due to frequent use of shared polyA signal sequences. B) Bar plot showing EBV BFRF3 mRNA signal relative to cellular β-actin as measured by RT-qPCR in EBV ΔBALF2 HEK293 cells that were either uninduced (U), induced with transfection of Rta and Zta expression plasmids (I), or induced and trans-complemented with transfection of Rta, Zta, and BALF2 expression plasmids (I + t) at 48 hours post-induction. C) Western blot for BFRF3 protein product (VCAp18) from lysates corresponding to samples in panel B.
Cap Analysis of Gene Expression (CAGE)-seq uniquely quantifies each overlapping transcript
In order to uniquely measure each EBV lytic transcript, we performed CAGE-seq, a technique that sequences only the 5’ portion of each mRNA and provides highly quantitative, strand-specific reads that allow mapping of individual transcription start sites (TSS) [34]. Because CAGE-seq only sequences TSS, it avoids artifacts arising from transcript overlap. For these experiments, we used HEK293 cells infected with wildtype EBV, EBV mutants defective for DNA replication (ΔOriLyt or ΔBALF2), or vPIC (ΔBDLF4) [30]. Cells were either uninduced (U), induced by Rta and Zta (I), or induced and trans-complemented by transfection of a plasmid expressing the missing EBV protein (I + t). Cells were harvested for CAGE-seq at 48 hours post-induction. Resultant CAGE-seq tags were aligned to the human (GRCh38) and EBV genomes. Aligned reads were clustered using Paraclu [35] and each TSS cluster was quantified in tags per million reads mapped (TPM). In uninduced cells, EBV transcripts accounted for approximately 620 per one million mRNAs expressed (Table 1). This number increased to between about 13,000 and 44,000 TPM with induction of EBV replication. We estimate our induction protocol resulted in EBV replication in ~10% of cells, thus actual EBV transcript abundance may be as much as 10-fold higher in cells experiencing the EBV lytic cycle.
Table 1. Summary of EBV CAGE-seq tags detected in each cell line/condition.
| Cell Line | Treatment | TPM |
|---|---|---|
| ΔBALF2 | U | 620 |
| I | 23,461 | |
| I + t | 23,720 | |
| ΔOriLyt | I | 16,986 |
| ΔBDLF4 | I | 28,544 |
| I + t | 44,140 | |
| WT | I | 13,542 |
For each cell line and treatment, the total number of CAGE-seq tags mapping to the EBV genome are shown. Treatment conditions include: uninduced (U), induced by transfection of Rta and Zta (I), and induced by transfection of Rta and Zta and trans-complemented with the missing gene product (I + t).
Examination of well characterized early transcription start sites such as BGLF4 and BGLF5 (Fig 2A) demonstrated that these transcripts were readily detected in the absence of DNA replication (ΔBALF2 I and ΔOriLyt I tracks) and expressed independently of the βγ gene-encoded vPIC complex (ΔBDLF4 I track). In fact, BALF2 trans-complementation appeared to result in a slight decrease in abundance of BGLF5 (301 vs. 225 TPM) and BGLF4 (238 vs. 197), suggesting that early and late transcription may compete for limited cellular resources. This decrease in transcription was observed for most early genes. Although it is formally possible that this observation may be due to differences in efficiency of induction of replication between different conditions, we observed similar total EBV transcripts in the ΔBALF2 I and ΔBALF2 I + t conditions (Table 1). Slight decreases were also observed with BDLF4 trans-complementation for both BGLF5 (220 vs. 200) and BGLF4 (217 vs. 186), once again suggesting that early gene transcription may compete for limiting resources with late transcription mechanisms. This decrease occurred despite the presence of more total EBV transcripts in the ΔBDLF4 I + t condition (44,140 TPM) compared to the ΔBDLF4 I condition (28,544 TPM), as shown in Table 1.
Fig 2. CAGE-seq readily distinguishes late from early gene transcription start sites (TSS) based on their dependence upon lytic DNA replication and the βγ-encoded vPIC.

CAGE-seq tracks from EBV ΔBALF2, ΔOriLyt, ΔBDLF4, and WT infected HEK293 cells showing TSS location and transcript abundance (in tags per million (TPM)) for A) representative early genes BGLF4 and BGLF5 and B) representative late genes BFRF1 and BFRF3. For each track, the treatment condition is indicated: cells were either uninduced (U), induced by transfection of Rta and Zta (I), or induced by transfection of Rta and Zta, and further trans-complemented with either BALF2 or BDLF4 (I + t) in the appropriate mutant cell line (ΔBALF2 or ΔBDLF4, respectively). In each case, cells were harvested for CAGE-seq 48 hours post-transfection. Scale marker in the WT track indicates track heights depicted in all tracks for the same TSS cluster. * indicates TSS signals surpassing the indicated scale. Full tracks are available to view in an interactive viewer (https://go.wisc.edu/58sxkb).
In contrast, transcription of canonical late genes such as BFRF1 or BFRF3 was not detected by CAGE-seq in the absence of DNA replication (Fig 2B, ΔOriLyt I and ΔBALF2 I tracks), but was restored with BALF2 trans-complementation (Fig 2B, ΔBALF2 I + t tracks). These genes were also fully dependent upon the βγ gene-encoded vPIC complex for their expression (Fig 2B, compare ΔBDLF4 I with ΔBDLF4 I + t). It is important to note that the levels and kinetics of expression of the BFRF3 transcripts detected by CAGE-seq, most closely resemble the expression patterns detected by western blotting for the BFRF3 protein product VCAp18 (Fig 1C), in contrast to results obtained by RT-qPCR (Fig 1B). Collectively, these results indicate that CAGE-seq is a reliable method for detection of EBV transcripts on a genome-wide level and is not susceptible to artifacts arising from transcript overlap.
Identification of non-canonical EBV late genes
Although the EBV βγ gene-encoded vPIC complex requires DNA replication to mediate late gene expression, a recent report by McKenzie et al. found that at least two EBV late genes, which by definition require DNA replication, are detectable when BGLF3, an essential vPIC component is knocked down by siRNA [36]. Both of these genes, BCRF1 (vIL-10) and BPLF1 (the largest tegument protein), were found to be expressed at low levels (<40 TPM in all conditions, see Fig 3A) and, consistent with late kinetics, were not detectable in the absence of DNA replication (Fig 3A, ΔOriLyt I and ΔBALF2 I tracks). Our CAGE-seq data suggests that only BCRF1 (vIL10) can be expressed in the absence of vPIC, whereas BPLF1 is a canonical late gene that requires BDLF4 for its expression (Fig 3A, compare ΔBDLF4 I vs ΔBDLF4 I + t tracks). We also identified an additional candidate, non-canonical, late gene: BDLF2 (Fig 3B). BDLF2 exhibits strictly late kinetics and was not detectable in the absence of OriLyt or BALF2 (ΔOriLyt I or ΔBALF2 I tracks, respectively), but was expressed in the absence of BDLF4 (ΔBDLF4 I track). The BDLF2 TSS signal did, however, further increase upon BDLF4 trans-complementation (ΔBDLF4 I + t track) from 120 to 358 TPM, potentially a reflection of higher total EBV lytic transcription in this condition (Table 1). Other genes may be partially transcribed by non-canonical mechanisms. For example low level transcription of BDLF3 was observed in the absence of OriLyt or BALF2 (Fig 3B. ΔOriLyt I and ΔBALF2 I tracks), but its TSS signal increased from 18 to 123 TPM upon BALF2 trans-complementation (Fig 3B. ΔBALF2 I and ΔBALF2 I + t tracks), consistent with leaky late kinetics. A much higher level of transcription was maintained in the absence of BDLF4 (130 TPM, Fig 3B. ΔBDLF4 I track), suggesting that this leaky late expression may be non-canonical. In summary, our CAGE-seq data confirm that some late genes can be expressed independently of vPIC. These include BCRF1 (vIL10), BDLF2, and likely the leaky late gene BDLF3. In contrast, our results suggest that BPLF1 is a canonical late gene, fully dependent on vPIC for its expression.
Fig 3. A minority of EBV late transcripts are expressed independently of the βγ-encoded vPIC.

A) CAGE-seq tracks for two EBV late genes, BCRF1 (vIL10) and BPLF1 previously reported to be expressed independently of the βγ-encoded vPIC. B) CAGE-seq tracks for BDLF2 and BDLF3 showing that BDLF2 is a late gene expressed independently of the βγ-encoded vPIC, whereas BDLF3 exhibits partial dependence upon DNA replication (i.e., “leaky” late kinetics). BDLF2 and BDLF3 transcripts are strongly detected in the absence of BDLF4 (ΔBDLF4 I), but increase upon BDLF4 trans-complementation (ΔBDLF4 (I + t)). Treatment conditions are similar as described for Fig 2. Scale marker in the WT track indicates track heights depicted in all tracks for the same TSS cluster. * indicates TSS signals surpassing the indicated scale. Full tracks are available to view in an interactive viewer (https://go.wisc.edu/58sxkb).
Genome-wide classification of EBV lytic genes based on their dependence on DNA replication and the βγ-encoded vPIC complex for expression
In an effort to systematically organize EBV lytic genes according to their kinetic class, we calculated the ratio of the CAGE-seq signal (from S1 Table) in ΔBALF2 I condition to that observed in the trans-complemented ΔBALF2 I + t condition. As expected, well-established late genes had BALF2 ratios near 0 and early genes had much higher ratios, often greater than 1. As previously stated, it is likely that this increase in early gene transcription in the absence of DNA replication is due to the competition between late gene and early gene transcription. We tentatively classified all genes with BALF2 ratios less than 0.1 as true late genes, those above 0.5 as early, and those with intermediate ratios as “leaky” late genes (Table 2). Based on these criteria, EBV replication is characterized by expression of at least 32 genes with early kinetics, 16 with late, and 16 genes that are expressed with leaky late kinetics. We only identified one TSS that exhibited latent kinetics, the C promoter; however, we did observe some early TSS in the uninduced state, consistent with low level spontaneous replication. As has been reported in other genome-wide analysis of EBV replication [37,38], we observed a large number of TSS that did not correspond to annotated transcripts. While many of these were transcribed at low levels, some “unknown” TSS clusters were expressed at much higher levels (S1 Table). In one case (BFRF0.5), a strong TSS was present just 3’ to the annotated open reading frame, suggesting that the authentic protein product might be initiated from the internal methionine at annotated codon 51. Because this TSS is almost certainly responsible for the early signal observed with the BFRF3 primers (Fig 1B) and because the BFRF0.5 initiator methionine has not be empirically determined, we included this TSS as a possible BFRF0.5 transcript in Table 2. Indeed, during the revision of this manuscript, Bencun et al. published ribosomal profiling experiments that also implicate codon 51 as a likely alternative start site for BFRF0.5 (called BFRF1a in their study) [39]. A similar issue arose with a cluster just upstream of BNLF2a that may result in a 5’ UTR that is too short to allow translation of BNLF2a, but could encode BNLF2b. This TSS was labeled BNLF2a/b to reflect this ambiguity.
Table 2. EBV lytic transcription start sites (TSS) detected by CAGE-seq and their dependence upon DNA replication (BALF2 Ratio) and the βγ gene-encoded vPIC (BDLF4 Ratio).
| Cluster | Strand | Kinetics | BALF2Ratio | BDLF4Ratio | WT TPM | ORF/ promoter | Annotation | ||
|---|---|---|---|---|---|---|---|---|---|
| Start | End | ||||||||
| 1709 | 1729 | + | late | 0.00 | 0.01 | 25 | BNRF1 | major tegument protein | |
| 11330 | 11349 | + | latent | 1.02 | 0.81 | 184 | Cp | latency promoter | |
| 53782 | 53794 | + | early | 0.81 | 1.25 | 758 | BHRF1 | v-Bcl2 | |
| 58113 | 58119 | + | late | 0.00 | 0.00 | 22 | BFRF0.5 | terminase subunit | |
| 58581 | 58593 | + | early | 0.98 | 1.18 | 151 | BFRF0.5? | terminase subunit | |
| 58860 | 58867 | + | late | 0.01 | 0.00 | 104 | BFRF1 | capsid nuclear egress | |
| 61372 | 61381 | + | late | 0.02 | 0.00 | 346 | BFRF3 | capsid—hexon tip | |
| 62231 | 62239 | + | early | 1.18 | 0.88 | 78 | Fp | ||
| 75047 | 75054 | + | leaky | 0.19 | 0.05 | 72 | BORF1 | capsid - 1x triplex | |
| 76198 | 76213 | + | early | 1.05 | 1.30 | 161 | BORF2 | RNR—large subunit | |
| 78831 | 78840 | + | early | 0.93 | 1.26 | 279 | BaRF1 | RNR—small subunit | |
| 79869 | 79874 | + | early | 1.29 | 1.31 | 171 | BMRF1 | processivity factor, DNA polymerase | |
| 80811 | 80863 | + | early | 0.64 | 0.84 | 158 | BMRF2 | virion glycoprotein | |
| 86914 | 86917 | + | early | 0.57 | 0.24 | 30 | BSRF1 | virion protein, palmytoylated | |
| 88540 | 88546 | + | leaky | 0.36 | 0.27 | 66 | BLRF1 | glycoprotein N | |
| 88894 | 88898 | + | leaky | 0.10 | 0.04 | 366 | BLRF2 | tegument protein | |
| 105040 | 105049 | + | early | 0.98 | 1.07 | 200 | BRRF1 | Na | |
| 106271 | 106278 | + | leaky | 0.28 | 0.34 | 42 | BRRF2 | tegument protein | |
| 109934 | 109941 | + | leaky | 0.35 | 0.57 | 22 | BKRF2 | glycoprotein L | |
| 110175 | 110180 | + | early | 1.24 | 1.04 | 300 | BKRF3 | uracil DNA glycosylase | |
| 110924 | 110929 | + | early | 0.95 | 1.18 | 167 | BKRF4 | tegument protein | |
| 113906 | 113915 | + | leaky | 0.24 | 0.14 | 11 | BBRF1 | portal | |
| 115794 | 115797 | + | leaky | 0.47 | 0.40 | 6 | BBRF2 | ||
| 119129 | 119134 | + | leaky | 0.23 | 0.29 | 93 | BBRF3 | glycoprotein M | |
| 137247 | 137251 | + | leaky | 0.23 | 2.20 | 12 | BcRF1 | vTBP; vPIC component | |
| 144610 | 144618 | + | late | 0.01 | 0.00 | 5 | BXRF1 | ||
| 145333 | 145340 | + | early | 0.98 | 0.63 | 9 | BVRF1 | portal "cork" | |
| 147752 | 147758 | + | late | 0.02 | 0.01 | 16 | BVRF2 | scaffold protease | |
| 148650 | 148655 | + | leaky | 0.27 | 0.26 | 52 | BdRF1 | capsid scaffold protein | |
| 165497 | 165499 | + | early | 1.29 | 1.06 | 186 | BARF1 | CSF-1 decoy receptor | |
| 167605 | 167603 | - | early | 0.64 | 2.41 | 6 | BNLF2a | TAP inhibitor | |
| 167499 | 167485 | - | early | 0.66 | 0.21 | 198 | BNLF2a/b | TAP inhibitor/unknown | |
| 165414 | 165410 | - | early | 1.94 | 0.87 | 127 | BALF1 | putative vBcl-2 | |
| 164786 | 164778 | - | early | 1.61 | 0.86 | 44 | BALF2 | ssDNA binding protein | |
| 161637 | 161632 | - | leaky | 0.46 | 0.98 | 9 | BALF3 | terminase subunit, pac binding | |
| 159340 | 159332 | - | leaky | 0.34 | 0.14 | 50 | BALF4 | glycoprotein B | |
| 150544 | 150530 | - | late | 0.00 | 0.00 | 197 | BILF2 | vGPCR | |
| 148156 | 148151 | - | early | 1.08 | 1.41 | 16 | BVLF1 | vPIC component | |
| 145119 | 145100 | - | early | 0.90 | 1.36 | 196 | BXLF1 | thymidine kinase | |
| 143282 | 143257 | - | leaky | 0.13 | 0.11 | 20 | BXLF2 | glycoprotein H | |
| 137683 | 137669 | - | late | 0.00 | 0.02 | 70 | BcLF1 | capsid protein, major | |
| 133323 | 133316 | - | late | 0.00 | 0.00 | 55 | BDLF1 | capsid protein, 2x triplex | |
| 132448 | 132441 | - | late | 0.01 | 0.33 | 80 | BDLF2 | virion glycoprotein | |
| 131078 | 131070 | - | leaky | 0.15 | 0.27 | 89 | BDLF3 | gp150 | |
| 129350 | 129343 | - | early | 1.29 | 1.02 | 38 | BDLF3.5 | vPIC component | |
| 128404 | 128398 | - | late | 0.04 | 0.05 | 3 | BGLF1 | ||
| 126902 | 126893 | - | late | 0.03 | 0.01 | 59 | BGLF2 | virion protein | |
| 125140 | 125083 | - | early | 0.72 | 1.00 | 19 | BGLF3 | vPIC component | |
| 124087 | 124083 | - | early | 0.73 | 0.66 | 34 | BGLF3.5 | vPIC component | |
| 123871 | 123809 | - | early | 1.21 | 1.17 | 94 | BGLF4 | protein kinase | |
| 122429 | 122417 | - | early | 1.34 | 1.10 | 95 | BGLF5 | alkaline exonuclease | |
| 121303 | 121121 | - | leaky | 0.50 | 0.37 | 111 | BBLF1 | virion protein, myristoylated | |
| 119044 | 119040 | - | early | 1.25 | 1.08 | 203 | BBLF2/3 | helicase-primase, acc protein | |
| 114445 | 114361 | - | early | 1.05 | 1.15 | 74 | BBLF4 | helicase | |
| 106186 | 106182 | - | early | 1.07 | 1.96 | 5 | BRLF1 | Rta | |
| 102129 | 102126 | - | late | 0.02 | 0.00 | 210 | BZLF2 | gp42 | |
| 92163 | 92158 | - | late | 0.00 | 0.01 | 171 | BLLF1 | gp350 | |
| 90027 | 90019 | - | early | 1.11 | 1.40 | 133 | BLLF2 | ||
| 88489 | 88480 | - | early | 1.07 | 1.16 | 229 | BLLF3 | dUTPase | |
| 87030 | 87023 | - | early | 1.07 | 1.16 | 14 | BSLF1 | primase | |
| 84330 | 84323 | - | early | 1.05 | 1.08 | 969 | SM | lytic RNA export | |
| 75294 | 75281 | - | late | 0.01 | 0.01 | 6 | BOLF1 | tegument protein, binds BPLF1 | |
| 72163 | 72156 | - | late | 0.00 | 0.01 | 5 | BPLF1 | tegument protein, largest | |
| 58539 | 58534 | - | leaky | 0.19 | 0.14 | 6 | BFLF1 | packaging protein | |
| 57130 | 57048 | - | early | 0.79 | 0.41 | 187 | BFLF2 | capsid nuclear egress | |
| 52821 | 52315 | - | early | 0.51 | 1.80 | 65 | BHLF1 | OriLyt transcript | |
Shown are EBV lytic transcripts identified by CAGE-seq. For each transcription start site (TSS) cluster, the table specifies: the location (B95-8, V01555), strand (+ or -), kinetic class based on BALF2 ratio, BALF2 ratio (ratio of signal in I / I + t for ΔBALF2), BDLF4 ratio (ratio of signal in I / I + t for ΔBDLF4), transcript abundance in tags per million in wildtype EBV induced for replication (WT TPM), and the gene or promoter corresponding to the observed CAGE-seq TSS cluster. Annotation sources include [40,41].
We used a similar approach to estimate the dependence of each transcript on vPIC, by calculating a BDLF4 ratio, corresponding to the CAGE-seq signal in the ΔBDLF4 I condition divided by that observed in the trans-complemented ΔBDLF4 I + t condition (Table 2). Of the 16 true late genes identified here, only BDLF2 had an elevated BDLF4 ratio (0.33), consistent with the hypothesis that the vast majority of genes with late kinetics are dependent on vPIC for their expression. BCRF1 (vIL10) expression levels were too low to be captured by our systematic analysis (see next paragraph). Based on these BDLF4 ratios, we identified two more leaky late genes, BcRF1 and BALF3 that appeared to be less dependent upon vPIC than upon DNA replication. To further evaluate the extent to which dependence upon DNA replication also reflects a dependence upon vPIC for expression, we plotted the BALF2 versus the BDLF4 ratios for each kinetic class of genes (Fig 4). This confirmed that the vast majority of late genes require vPIC for expression and further demonstrated that genes exhibiting leaky late kinetics exhibit a partial dependence on vPIC proportional to their partial dependence on DNA replication.
Fig 4. EBV transcript dependence on the βγ-encoded vPIC is highly correlated with dependence on DNA replication.

Scatter plot of EBV transcripts based on their BALF2 dependence vs. BDLF4 dependence ratios determined by CAGE-seq (from Table 1). Transcripts were classified into 3 groups (early, leaky late, and late) based on their DNA replication dependence (BALF2 ratios).
A small number of well-annotated EBV genes were not detected as significant TSS clusters by our pipeline (details of criteria provided in the materials and methods section) and therefore are not present in Table 2. For completeness, we attempted to manually identify TSS signals in the CAGE-seq data corresponding to their known transcription start sites. This effort is presented separately in Table 3.
Table 3. EBV lytic transcripts not meeting high confidence CAGE-seq analysis criteria.
| Cluster | Strand | Kinetics | BALF2Ratio | BDLF4Ratio | WT TPM | ORF/ promoter | Annotation | |
|---|---|---|---|---|---|---|---|---|
| Start | End | |||||||
| 9660 | 9665 | + | Late | 0.03 | 3.94 | 2 | BCRF1 | vIL10 |
| 59502 | 59508 | + | Early | 0.96 | 1.74 | 3 | BFRF2 | vPIC component |
| 124855 | 124862 | + | Early | 0.85 | 1.08 | 3 | BGRF1-BDRF1 | ATPase subunit of terminase |
| 139541 | 139546 | + | Early | 1.17 | 1.28 | 3 | BTRF1 | |
| 169512 | 169520 | - | Latent | 0.73 | 2.88 | 3 | LMP1 | LMP1 |
| 156873 | 156876 | - | Early | 2.01 | 0.98 | 2 | BALF5 | DNA polymerase |
| 128979 | 128990 | - | Early | 1.18 | 0.08 | 1 | BDLF4 | vPIC component |
Shown are CAGE-seq data for well-annotated transcripts that did not meet detection criteria (see materials and methods). Table format is the same as described for Table 2. Note that for the BDLF4 gene, the BDLF4 ratio (I / I + t) may be artifactually low due to detection of the transfected BDLF4 TSS signal in the I + t condition.
BLRF2, which encodes VCAp23, is a leaky late gene
Although BLRF2 is generally categorized as a true late gene [42–44], we detected its TSS signal in the absence of BALF2, OriLyt, and BDLF4, albeit at reduced levels. These signals dramatically increased upon BALF2 or BDLF4 trans-complementation, suggesting that BLRF2 may in fact be a leaky late gene (S1 Table). Because we calculated a BALF2 ratio of 0.1, which was at the cutoff between leaky late and late genes, we chose to further investigate its kinetics. Because it is not possible to uniquely measure BLRF2 transcript levels by RT-qPCR due to overlap with the BLRF1 transcript, we assessed the BLRF2 protein product VCAp23 by western blotting. As shown in Fig 5, in ΔBALF2 cells induced for replication (I), low level VCAp23 expression is observed and this level dramatically increases upon BALF2 trans-complementation (I + t).
Fig 5. EBV BLRF2 exhibits bona fide leaky late kinetics.
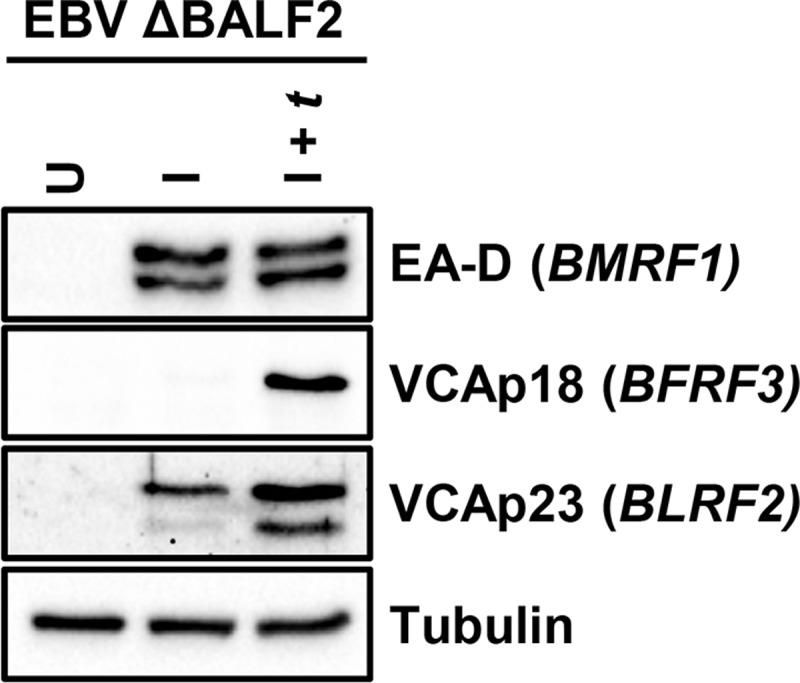
Western blot for VCAp23 (product of EBV BLRF2) in ΔBALF2 HEK293 cells, indicating that in absence of DNA replication (induced only (I)), VCAp23 is still expressed. When DNA replication is restored by trans-complementation with BALF2 (I + t), VCAp23 levels further increase. VCAp18 (product of true late BFRF3 gene), EA-D (product of early BMRF1 gene) and Tubulin blots are shown as controls.
TATTWAA is the only element enriched in late gene promoters
In an effort to determine what promoter elements determine the kinetic class of late and leaky late promoters, we performed Multiple EM for Motif Elicitation analysis (MEME) [45] of elements upstream of observed TSS with these kinetics. For both late (Fig 6A and 6B) and leaky late (Fig 6C and 6D) promoters, we found that the TATT element was enriched, but found no other sequences that were significantly enriched. The sequence was consistently found ~30bp upstream (S1 Fig) of late and most leaky late TSS and conformed to a 7 bp consensus sequence TATTWAA (where W is either A or T). There was no obvious correlation between the strength of late TSS signal and agreement with this sequence. For example, TATTTAA was present upstream of BFRF3 and BGLF1 (Fig 6B) which were transcribed at levels of 346 and 3 TPM (Table 2), respectively. Whereas BILF2 and BOLF1 had the sequence TATTTAG (Fig 6B) and were found to be transcribed at 197 and 6 TPM, respectively (Table 2). There appeared to be more deviation from this consensus in the leaky late genes with some genes such as BRRF2 (TATAAAA) having sequences conforming to the human TBP consensus sequence [46]. TATA boxes are also observed upstream of BDLF3 (Fig 6D) and BCRF1 (TATAAAT) [36], which may be important for their vPIC-independent transcription. It should be noted however, that BDLF2, which is also transcribed independently of vPIC has the sequence (TATTTAA) to which preferential binding of BcRF1 (vTBP) would be expected.
Fig 6. MEME analysis identifies a TATTWAA element in late and leaky late promoters.

A) Consensus motif identified by MEME analysis for 16 late and C) l6 leaky late promoters from clusters in Table 1. Promoter sequences of each gene is further listed for the B) late and D) leaky late classes sorted based on conformity to the identified consensus motif. BcRF1 (vTBP) promoter sequence was excluded by MEME software due to high deviation from the determined consensus motif and is presented separately.
Leaky late kinetics can result from superimposition of early and late transcription mechanisms
For most leaky late TSS clusters, we observed a uniform decrease at each position within the cluster when either lytic DNA replication (ΔOriLyt I and ΔBALF2 I) or vPIC (ΔBDLF4 I) functions were affected. However, close examination of the leaky late BLRF1 TSS cluster (Fig 7) revealed the presence of distinct TSS signals within the cluster that exhibited either early (present in all induced/I tracks) or late (absent in ΔOriLyt I, ΔBALF2 I and ΔBDLF4 I tracks) kinetics. This finding suggests that, at least in the case of BLRF1, the leaky late kinetics result from the production of transcripts initiating at slightly different start sites, but which ultimately encode the same open reading frame. This further suggested, given the apparent importance of vPIC for leaky late kinetics (Fig 4), that in the absence of vPIC activity, leaky late genes would revert to early kinetics. To test this hypothesis, we constructed a double mutant EBV genome in which both the single-stranded DNA binding protein BALF2 and the essential vPIC component BcRF1 (vTBP) are knocked out (i.e. ΔBALF2/ΔBcRF1) and established stable HEK293 cells. Using ΔBALF2/ΔBcRF1 HEK293 cells, we found when induced in absence of lytic DNA replication and a functional vPIC (Fig 8A, I condition) the leaky late VCAp23 protein (product of BLRF2-leaky late expression shown in Fig 8B) was expressed under early conditions. In presence of induction and trans-complementation with BALF2 (DNA replication restored) but in absence of BcRF1 (Fig 8A, I + BALF2 condition), levels of VCAp23 were not further increased. However, expressed levels of VCAp23 were increased when DNA replication was restored in presence of a functional vPIC (Fig 8A, I + BALF2 + BcRF1 condition), consistent with the hypothesis that the lytic DNA replication dependence of leaky late genes is indeed due to superimposition of canonical late transcription on a basal level of early transcription.
Fig 7. Distinct BLRF1 transcription start sites (TSS) exhibit different kinetics.
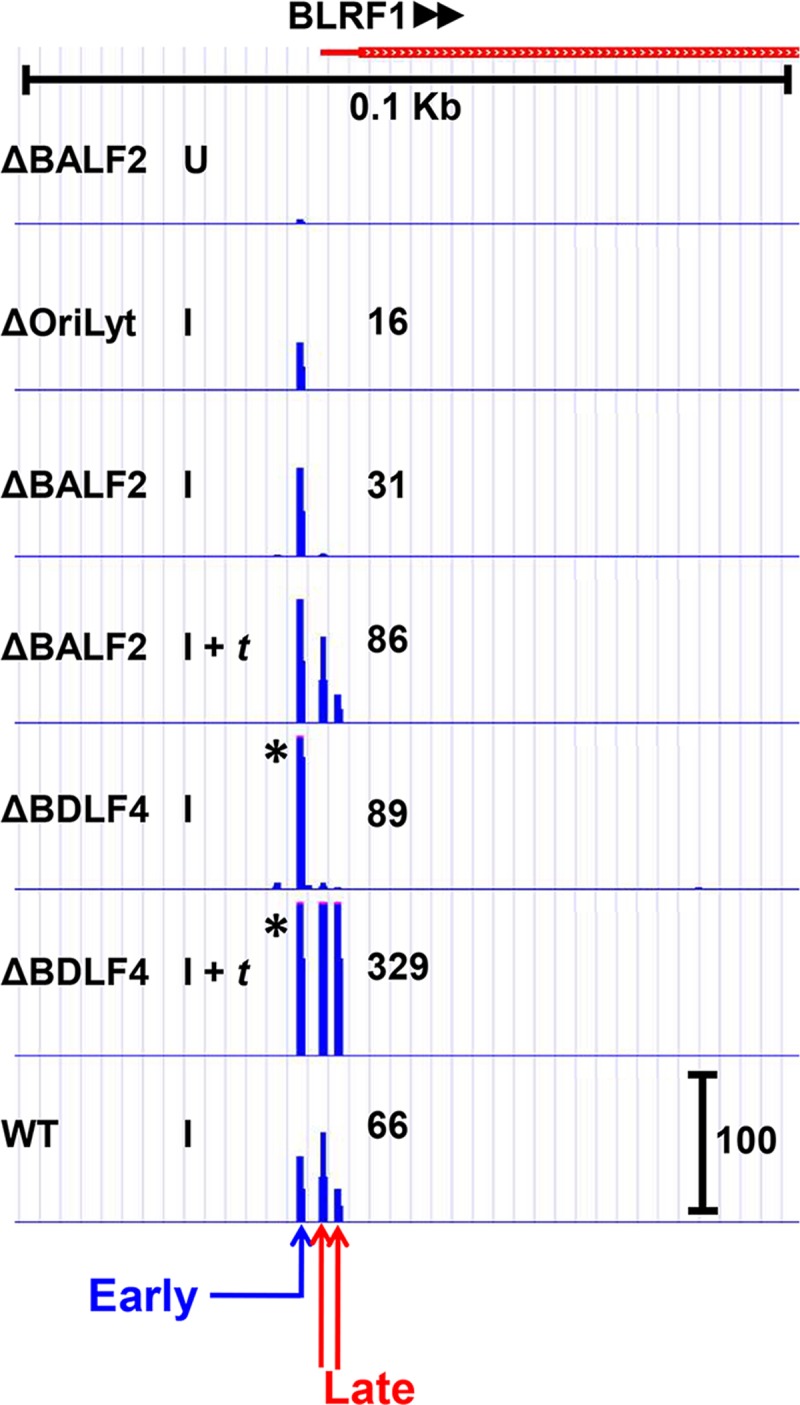
CAGE-seq tracks for the BLRF1 transcript demonstrate that transcription starts in at least 3 unique locations. One TSS is independent of DNA replication and the βγ-encoded vPIC (EARLY), whereas the other two are dependent on both DNA replication and vPIC (LATE). Treatment conditions are similar as described for Fig 2. Scale marker in the WT track indicates track heights depicted in all tracks for the same TSS cluster. * indicates TSS signals surpassing the indicated scale. Full tracks are available to view in an interactive viewer (https://go.wisc.edu/58sxkb).
Fig 8. BLRF2’s partial dependence on DNA replication requires vPIC.

A) Western blot showing levels of the BLRF2 product VCAp23 in EBV ΔBALF2/ΔBcRF1 HEK293 cells. Cells were either uninduced (U), induced by transfection of Rta and Zta expression plasmids (I), induced by transfection of Rta and Zta expression plasmids and trans-complemented by transfection of either only BALF2 (I + BALF2) or both BALF2 and the essential vPIC component BcRF1 (I + BALF2 + BcRF1). B) CAGE-seq tracks corresponding to BLRF2 TSS levels. Treatment conditions are described in Fig 2. Scale marker in the WT track indicates track heights depicted in all tracks for the same TSS cluster. * indicates TSS signal surpassing the indicated scale. Full tracks are available to view in an interactive viewer (https://go.wisc.edu/58sxkb).
Discussion
In this study, we present a comprehensive analysis of the kinetics of EBV lytic gene transcription based on dependence on lytic DNA replication and expression of BDLF4 (a βγ gene encoding an essential component of the viral late gene pre-initiation complex (vPIC) [30,31]). We chose CAGE-seq to accurately quantify transcription start sites in a strand-specific manner. By using this approach, we were able to avoid measurement of overlapping transcripts which confounds most other genome-wide attempts to analyze EBV lytic gene expression. CAGE-seq identified 32 TSS clusters that were expressed independently of lytic DNA replication (i.e. early kinetics) and 16 TSS that exhibited late kinetics defined by their strict dependence upon lytic DNA replication. In addition, 16 TSS were identified that exhibited partial dependence on lytic DNA replication (referred to here as leaky late, but also termed early-late or γ1). Although BALF2 has been suggested to play a role in recruiting Zta to the BHLF1 promoter [47], we consistently found that genes requiring BALF2 for their expression also required OriLyt (S1 Table). Thus, for each TSS, BALF2-dependence reflects lytic DNA replication dependence.
Based on this measure, we propose reclassification (see reference [33]) of 13 genes previously identified as late (BORF1, BLRF1, BLRF2, BRRF2, BKRF2, BBRF1, BBRF2, BBRF3, BdRF1, BALF4, BXLF2, BDLF3, BBLF1) to leaky late and 4 putative late genes (BMRF2, BSRF1, BKRF4, and BVRF1) as early. We also suggest reclassification of 3 early genes (BcRF1, BALF3, and BFLF1) as leaky late and BFRF1 from early to late. It bears mentioning that the calculated BALF2 ratios form a nearly continuous spectrum; our subdivision into three kinetic classes (early, leaky late, and late) is necessarily reductive and the BALF2 ratios themselves represent the most accurate description of a given gene’s kinetics. As anticipated, the majority of late genes encode structural proteins, including 3 capsid, 3 tegument, and 3 virion glycoproteins. This was also true for leaky late genes which encode 3 capsid, 3 tegument, and 6 virion glycoproteins. It is interesting to note that BcRF1, which encodes a vPIC component exhibited leaky late kinetics; however it did not depend upon vPIC for its expression based on its BDLF4 ratio.
In an effort to evaluate important TSS not captured by our bioinformatics pipeline, we manually examined the dependence of well-annotated transcripts. Including the results reported in Table 3, in total, we identified 37 early, 17 late, and 16 leaky late TSS. BILF1 was the only transcript for which we were unable to identify a corresponding TSS signal via CAGE-seq. Consistent with prior reports, we found a large number of unannotated TSS [37,38]. We chose to focus our analysis on well-annotated protein-coding transcripts and assigned TSS to specific genes based on prior annotated start sites or, when necessary, proximity to the ORF. However, it is possible that some “unknown” TSS represent additional transcripts for annotated ORFs with long 5’ UTRs. We also do not exclude the possibility that additional transcripts initiating at TSS reported in S1 Table will prove to have biologically significant roles in the EBV life cycle.
Our results revealed that the majority of late genes require BDLF4 for their expression and are thus vPIC-dependent, canonical late genes. This phenomenon extends to most leaky late genes which were found to exhibit a dependence on BDLF4 proportional to their dependence on BALF2 (Fig 4). We found that two late genes, BCRF1 (vIL10) and BDLF2, while dependent on lytic DNA replication are vPIC-independent, and thus non-canonical late genes. Furthermore, the leaky late genes BDLF3, BcRF1, and BALF3 appear to be less dependent upon vPIC, suggesting non-canonical late mechanisms may also contribute to their transcription. Previously, using a BGLF3 siRNA knockdown approach, BCRF1 (vIL10) was identified as a non-canonical late gene [36]. Other candidates identified by this approach (BPLF1, BSRF1, or BTRF1) [36] could not be confirmed in our study. Based on calculated BALF2 ratios, our data suggests that BSRF1 and BTRF1 should be classified as early genes. In contrast, because BPLF1 exhibits strict dependence on lytic DNA replication and vPIC, we classified it as a canonical late gene. It is important to note that except for BSRF1, we found that all of these genes were expressed at low levels during EBV replication which are likely to magnify the effect of any errors in measurement. Despite our attempts to minimize detection errors due to transcript overlap by using CAGE-seq as well as using highly specific mutant BACmids instead of siRNA knockdowns, independent confirmation of the kinetics of these genes by other investigators will be important given the existing apparent discrepancies. Nevertheless, we agree that the existence of non-canonical late genes represents an important exception to the existing model. Thus, deciphering the vPIC-independent mechanism linking late gene expression to lytic DNA replication and the extent to which this mechanism mirrors what is observed in α-herpesviruses will be an important step toward advancement of our understating of late gene transcription.
What additional factors are required for true late gene expression? It is now generally accepted that an OriLyt in cis and the βγ gene-encoded vPIC are central to late gene transcription. Given the divergence of late gene transcription from host transcription mechanisms, it is highly probable that additional viral factors are required. Recently, the BMRF1-encoded processivity factor has been suggested to play a role in transcription of some late (as well as some latent and early) genes separate from its role in DNA replication [48]. However, the gene array used to measure EBV transcripts in that study [48] is also susceptible to errors due to overlapping transcripts and must be interpreted with caution. Nevertheless, it is notable that BMRF1 (EA-D) is present in quantities vastly exceeding that of the BALF5 catalytic subunit [49], suggesting potential functionality beyond its well-defined role in DNA synthesis [50]. An intriguing possibility is that BMRF1 may serve as a docking site for vPIC–similar to what is seen in the bacteriophage T4 where the gp55/gp33 late gene specific sigma factor links the RNA-polymerase to DNA replication via an interaction with the gp45 processivity factor [51].
The EBV kinase BGLF4 is also implicated in late gene expression [52]. Although BGLF4 has been observed to be localized to the replication compartments [53], its precise role in late gene transcription remains to be defined. Another factor that appears to be important for late gene expression is SM. Specifically, an EBV genome deleted for SM was deficient for expression of a subset of late genes, suggesting that it may play a role in ensuring that their mRNAs are properly exported or otherwise processed as they emerge from replication compartments [54]. Interestingly, disruption of the KSHV SM homolog (ORF57) did not exhibit a late gene defect [55], suggesting that even though SM homologs are present in all herpesviruses, their role in late gene transcription may be unique to EBV.
Finally, a role for Rta in late gene expression has been postulated by many investigators. Rta binding sites are found throughout the EBV genome, potentially allowing activation of nearly any lytic gene promoter [36,44]. Reporter assays; however, offer conflicting evidence regarding their functional significance. Several studies, including our own, have documented RRE-dependent activation of late (or leaky late) promoters [44,56,57]. Aubry et al. have shown that the βγ-encoded vPIC (without presence of Rta) can activate a minimal TATT reporter in EBV-negative HEK293 cells [31]. However, in light of the mounting evidence supporting the central role of replication compartments in both DNA replication and late gene expression [50,58,59], it is unclear the extent to which reporter assays performed in cells not undergoing viral DNA replication (i.e., lacking replication compartments) can be used as models of late gene transcription. Although Rta is undoubtedly essential for late gene expression, what is less clear is whether it plays a direct role or merely supports late gene transcription by fulfilling its essential roles in activation of early gene expression and DNA replication [60]. Experiments to resolve this issue, while extremely important, are difficult to design. If, however, Rta plays only an indirect role in late gene transcription, it would provide an explanation for the role of LF2. Our laboratory has previously shown that LF2 can block EBV replication by sequestration of Rta in the cytoplasm [61,62]. In those experiments, LF2 was transfected simultaneously with Rta. During EBV replication induced by physiologic stimuli, LF2 protein levels would accumulate more slowly and could eventually shutoff Rta-dependent early gene transcription while vPIC dependent late transcription continues. We performed our CAGE-seq analysis using a B95-8 BACmid, a laboratory strain deleted for LF2. Our results suggest that, at least in the absence of LF2, early and late gene transcription compete for limited resources. It is interesting to consider whether, in clinical EBV strains, shutdown of early transcription by LF2 could lead to increased efficiency of late gene transcription once replication compartments have formed under the influence of Rta.
In conclusion, the results reported in this study add to a growing body of evidence that EBV early and late gene transcription are governed by distinct mechanisms. Early gene expression seems to closely resemble that of host gene transcription in which transcription can occur from a chromatinized template. In contrast, we and others have found that late gene transcripts localize to replication factories which are devoid of core histones [30,58,63]. We previously proposed that the major role of the βγ-gene encoded vPIC is to serve as an adaptor complex that exclusively recruits RNA polymerase II to newly replicated viral genomes and facilitates transcription from this atypical unchromatinized template [30]. If such strict requirements are in place for linking late gene transcription to newly replicated templates, how is it then possible for some promoters to exhibit “partial” dependence on DNA replication (i.e., leaky late kinetics)? The results in this study are consistent with the hypothesis that leaky late genes are not transcribed via an entirely distinct mechanism; rather, we propose that they are transcribed by both early (from chromatinized genomes) as well as late (in replication factories) mechanisms. In the case of BLRF1, it was possible to distinguish early from late TSS and observe this superimposition directly. A much greater degree of TSS separation occurs with transcription of the CMV UL44 gene [64], where a late TSS is embedded between two early TSS. If the same TSS was used by both early and late mechanisms, each site would appear to be leaky, even if there were no distinct leaky late transcription mechanism.
A corollary of the hypothesis that a leaky late mechanism does not exist per se is that leaky late genes have hybrid promoters that support both the early and late transcription mechanisms. There is precedent for this in HSV1, where the VP5 leaky late promoter reverts to early kinetics upon mutation of an initiator element (Inr) known to be important in HSV late promoters [65]. In the case of EBV, we suggest that a leaky late promoter would consist of upstream elements that bind cell transcription factors, an Rta responsive element (RRE) and/or a Zta responsive element (ZRE) with a TATA box capable of binding both hTBP and BcRF1 (vTBP) (model shown in Fig 9). Despite the preference of hTBP and vTBP for TATA boxes with A and T at the fourth position, respectively, they each can bind to the other’s optimal sequence [28,46]. The majority of leaky late gene promoters identified in this study had a T at the fourth position (TATT). It is likely that the presence of strong upstream activators can overcome the reduced affinity of hTBP for TATT, allowing early transcription to also occur at promoters with “late” TATA boxes. Our results demonstrate this directly for the BLRF2 gene which is transcribed in the absence of BcRF1 (vTBP) with early kinetics, demonstrating the ability of hTBP to bind to its TATT box. In the presence of both DNA replication and BcRF1 (vTBP) leaky late kinetics are observed due to superimposition of early and late transcription. We predict that leaky late transcripts arise from both replication compartments (late) and chromatinized DNA (early) and plan to investigate this hypothesis further using live cell imaging.
Fig 9. Model for EBV leaky late gene transcription.
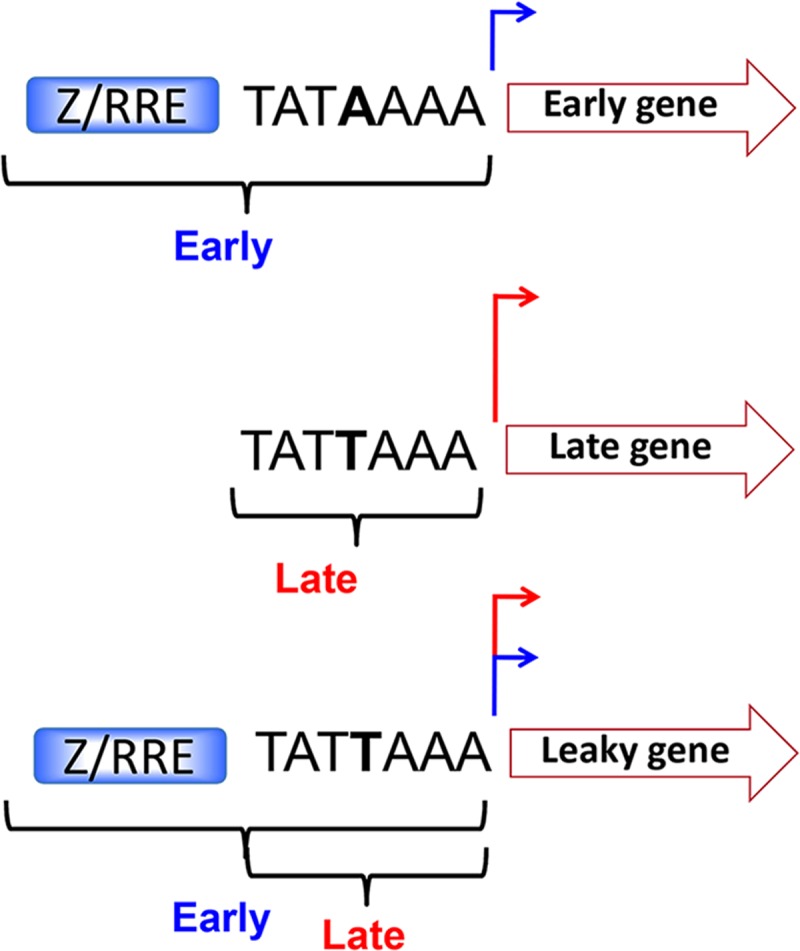
Current model illustrating early promoters consisting of canonical TATA boxes in addition to upstream Zta and/or Rta response elements (ZREs and/or RREs respectively), true late genes with non-canonical TATT boxes; and hybrid leaky late genes consisting of elements found in both early and late promoters (i.e. ZRE/RRE + TATT).
Materials and methods
Cell lines and culture
All cell lines were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. The EBV-negative HEK293 cell line, used for infection with all EBV BACmids, was obtained from Bill Sugden (University of Wisconsin-Madison).
EBV mutant genomes and derivation of EBV-positive HEK293 cell lines
The EBV p2089 BACmid contains the complete genome of the B95.8 strain of EBV in addition to a cassette containing the prokaryotic F-factor as well as the green fluorescent protein (GFP) and Hygromycin B resistance genes in the B95.8 deletion as previously described [66]. The parental EBV Wild-Type (WT) BACmid used in these studies is a modification of the p2089 BACmid lacking a functional GFP ORF. The WT, ΔBcRF1 (MI-27), and ΔBDLF4 (MI-84) BACmids were part of a comprehensive library of mutant EBV genomes [67]. ΔBALF2/HA-BcRF1 (referred to as ΔBALF2 elsewhere in this text), and ΔOriLyt BACmids have been previously described [30]. ΔBALF2/ΔBcRF1 double mutant was constructed using the GS1783 E. coli–based En Passant method [68,69] by inserting a stop codon in the BALF2 sequence similar to that present in ΔBALF2/HA-BcRF1 [30] in context of the ΔBcRF1 (MI-27) BACmid described above. All EBV-positive HEK293 cell lines were derived as described previously [30].
Plasmids
pcDNA3-Rta [70], pSG5-Zta (or pSVNaeZ) [71], pMSCV-F-HA-BDLF4 [72], pcDNA3-HA-BcRF1 [30] and pSG5-HA-BALF2 [30] have been previously described.
Immunoblotting
Total cell lysates were harvested in RIPA Buffer, separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to nitrocellulose membrane. Membranes were blocked in Tris-buffered saline (TBS) containing 5% milk and 0.1% Tween 20 and incubated with appropriate primary antibodies overnight at 4°C. The following primary antibodies were used: anti-EBV EA-D p52/50 (EMD Millipore, MAB8186; 1:3,000), anti-EBV VCAp18 (Thermo Scientific, PA1-73003; 1:1,000), anti-α-tubulin (Sigma, T6074; 1:1,000) and rabbit anti BLRF2 (SLO25-1, generous gift from Ayman El-Guindy; 1:200). Following treatment with primary antibodies, membranes were washed with TBS containing 0.1% tween and incubated with appropriate secondary antibodies for 1 hour at room temperature. The following secondary antibodies were used: goat anti-mouse poly-HRP (Fisher Scientific), goat anti-rabbit poly-HRP (Fisher Scientific), and donkey anti-goat (Fisher Scientific). Membranes were washed again and visualized using ECL chemiluminescent kit (Thermo Scientific) according to manufacturer’s protocol.
RNA isolation, reverse transcription and quantification by real-time (RT) PCR
EBV-positive HEK293 cells were induced in 12-well plates using 125 ng each of Rta and Zta expression plasmids along with 250 ng of the trans-complementing plasmid. 48 hours post lytic induction, cells were washed with phosphate-buffered saline (PBS), and RNA was extracted using GeneJET RNA purification kit (Thermo Scientific) according to manufacturer’s protocol with the following modification: after lysis and before loading on column, lysates was passed through a QIAshredder cell and tissue homogenizer (Qiagen). The eluted RNA was then treated with DNase (1 unit/µg DNA), DNase was deactivated by incubation at 65°C and the treated RNA (~ 1 µg) was reverse transcribed using the ImProm-II Reverse Transcription System (Promega). Purified cDNA was subjected to RT-qPCR with a 7900HT Fast Real-Time PCR system (Applied Biosciences) using SYBR Green Real-Time PCR Master Mix (Biorad). Primers used for detection of β-actin and EBV “BFRF3” (false positive from Fig 1B) are as follows: β-Actin-cDNA-Fwd (GCCGGGACCTGACTGACTAC), β-Actin-cDNA-Rev (TTCTCCTTAATGTCACGCACGAT); FR3-cDNA-qPCR-F (CGGGAGGCTCAAAGAAGTTA), and FR3-cDNA-qPCR-R (GCTCTCTGCCTCTTGTCTATG). All values are reported relative to β-actin mRNA using the 2-ΔΔCT method described previously [73].
non-Amplified non-Tagging Illumina Cap Analysis of Gene Expression (nAnT-iCAGE, referred to as CAGE-seq in text) library preparation and sequencing
Library preparation and sequencing were performed by DNAFORM (Yokohama, Janagawa, Japan) as previously described [34]. Briefly, after RNA assessment by Bioanalyzer (Agilent), first strand cDNAs were transcribed to the 5' end of capped RNAs and attached to CAGE "barcode" tags. Libraries were then sequenced on a NextSeq (Illumina). The raw CAGE-seq data from this study has been deposited in the NCBI SRA under bioproject PRJNA471349.
Alignment to EBV genome
Reads from CAGE-seq were aligned to the GDC GRCh38 based genome index using STAR [74] version 2.5.1b. Reads were sorted and filtered for primary alignments and mapping quality greater than 30 using samtools version 1.5. Coordinates were converted to Genbank accession V01555.2 coordinates using CrossMap [75].
Identification of transcription start sites (TSS) clusters and their assignment to annotated ORFs
To identify transcription start sites from CAGE-seq data, reads from bam files containing V01555 coordinates were converted to wigs using a custom python script. A modified version of Paraclu [35] allowing singletons was used to call peaks on the three (pseudo-)wildtypes (WT, BALF2 I + t, and BDFL4 I + t) with the following parameters: reads per cluster ≥15 and cluster length ≤20. Consensus clusters were identified using a custom python script using Python3, Jupyter Notebook, and Pandas. Briefly, clusters meeting cutoff criteria in at least 2 of the 3 (pseudo-)wildtypes were added to a list of consensus clusters. Partially overlapping clusters were merged. Coordinates of consensus clusters were then applied to calculate the number of tags mapping to each consensus cluster in each sample. Cluster values were normalized to Tags Per Million (TPM) by dividing the number of reads assigned to each cluster by the total number of mapped reads in each sample (S1 Table). Subsequently, clusters were annotated to known EBV gene products. In cases where more than one cluster was annotated to the same gene, we reported (Table 2) the major cluster (provided all minor clusters were <10% of the major TSS signal) or merged the clusters into a single larger cluster (in cases with multiple TSS lacked a single dominant TSS). Full tracks are available to view in an interactive viewer (https://go.wisc.edu/58sxkb).
MEME (Multiple EM for Motif Elicitation) analysis of late and leaky late promoters
The MEME software (meme-suite.org; [45]) was used to search for enriched motifs up to 500 bp upstream of late and leaky late TSS (as determined by BALF2 ratios in this study, see Table 2).
Supporting information
MEME analysis showing positions of TATTWAA motif identified in A) late and B) leaky late promoters for 16 late and 15 out 16 leaky late TSS identified via CAGE-seq. BcRF1 is systematically excluded by MEME-suite due to significant deviation from consensus.
(TIF)
Shown are all EBV lytic transcripts identified by CAGE-seq, including strong TSS signals not corresponding to previously annotated ORFs. For each transcription start site (TSS) cluster the table specifies: the location (B95-8, V01555), strand (+ or -), kinetic class based on BALF2 ratio, the gene or promoter corresponding to the observed CAGE-seq TSS cluster, the corresponding annotation for each ORF/promoter, transcript abundance in tags per million (TPM) for each condition, BALF2 ratio (ratio of signal in I / I + t for ΔBALF2), and BDLF4 ratio (ratio of signal in I / I + t for ΔBDLF4. Clusters that did not correspond to well annotated ORF/promoters are labeled as unknown. Transfection of Rta and Zta resulted in a large number of clusters in this region and, accordingly, have been labeled as artifacts.
(XLS)
Acknowledgments
We would like to thank Drs. Bill Sugden, Shannon Kenney, Janet Mertz, Erik Flemington and members of their laboratories for helpful suggestions and discussions. We are grateful to Dr. Ayman El-Guindy for sharing unpublished reagents and the generous gift of the BLRF2 antibody.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by National Institute of Dental and Craniofacial Research (http://www.nih.gov/) grant R01-DE023939 to EJ, National Cancer Institute (http://www.nih.gov/) grants P01-CA022443 and P30CA014520-43 to EJ, and the National Institute of Allergy and Infectious Diseases (http://www.nih.gov/) grant T32-AI-078985 grant to RD. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Rickinson AB, Kieff E. Epstein-Barr virus Fields Virology. 5th ed Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2007. pp. 2655–2700. [Google Scholar]
- 2.Henle W. Role of Epstein-Barr virus in infectious mononucleosis and malignant lymphomas in man. Fed Proc. 1972;31: 1674 [PubMed] [Google Scholar]
- 3.Thorley-Lawson DA, Allday MJ. The curious case of the tumour virus: 50 years of Burkitt’s lymphoma. Nat Rev Microbiol. 2008;6: 913–924. 10.1038/nrmicro2015 [DOI] [PubMed] [Google Scholar]
- 4.zur Hausen H, Schulte-Holthausen H, Klein G, Henle W, Henle G, Clifford P, et al. EBV DNA in biopsies of Burkitt tumours and anaplastic carcinomas of the nasopharynx. Nature. 1970;228: 1056–1058. [DOI] [PubMed] [Google Scholar]
- 5.Kutok JL, Wang F. Spectrum of Epstein-Barr virus-associated diseases. Annu Rev Pathol. 2006;1: 375–404. 10.1146/annurev.pathol.1.110304.100209 [DOI] [PubMed] [Google Scholar]
- 6.Thompson MP, Kurzrock R. Epstein-Barr virus and cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2004;10: 803–821. [DOI] [PubMed] [Google Scholar]
- 7.Hong GK, Gulley ML, Feng W-H, Delecluse H-J, Holley-Guthrie E, Kenney SC. Epstein-Barr virus lytic infection contributes to lymphoproliferative disease in a SCID mouse model. J Virol. 2005;79: 13993–14003. 10.1128/JVI.79.22.13993-14003.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jochum S, Moosmann A, Lang S, Hammerschmidt W, Zeidler R. The EBV immunoevasins vIL-10 and BNLF2a protect newly infected B cells from immune recognition and elimination. PLoS Pathog. 2012;8: e1002704 10.1371/journal.ppat.1002704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Altmann M, Hammerschmidt W. Epstein-Barr virus provides a new paradigm: a requirement for the immediate inhibition of apoptosis. PLoS Biol. 2005;3: e404 10.1371/journal.pbio.0030404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thorley-Lawson DA, Hawkins JB, Tracy SI, Shapiro M. The pathogenesis of Epstein-Barr virus persistent infection. Curr Opin Virol. 2013;3: 227–232. 10.1016/j.coviro.2013.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Israel B, Kenney S. EBV lytic infection In: Robertson E, editor. Epstein-Barr virus. Philadelphia: Caister Academic Press; 2005. pp. 571–611. [Google Scholar]
- 12.Chevallier-Greco A, Manet E, Chavrier P, Mosnier C, Daillie J, Sergeant A. Both Epstein-Barr virus (EBV)-encoded trans-acting factors, EB1 and EB2, are required to activate transcription from an EBV early promoter. EMBO J. 1986;5: 3243–3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Countryman J, Miller G. Activation of expression of latent Epstein-Barr herpesvirus after gene transfer with a small cloned subfragment of heterogeneous viral DNA. Proc Natl Acad Sci U S A. 1985;82: 4085–4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takada K, Shimizu N, Sakuma S, Ono Y. trans activation of the latent Epstein-Barr virus (EBV) genome after transfection of the EBV DNA fragment. J Virol. 1986;57: 1016–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zalani S, Holley-Guthrie E, Kenney S. Epstein-Barr viral latency is disrupted by the immediate-early BRLF1 protein through a cell-specific mechanism. Proc Natl Acad Sci U S A. 1996;93: 9194–9199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seibl R, Motz M, Wolf H. Strain-specific transcription and translation of the BamHI Z area of Epstein-Barr Virus. J Virol. 1986;60: 902–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hardwick JM, Lieberman PM, Hayward SD. A new Epstein-Barr virus transactivator, R, induces expression of a cytoplasmic early antigen. J Virol. 1988;62: 2274–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rooney CM, Rowe DT, Ragot T, Farrell PJ. The spliced BZLF1 gene of Epstein-Barr virus (EBV) transactivates an early EBV promoter and induces the virus productive cycle. J Virol. 1989;63: 3109–3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kenney S, Kamine J, Holley-Guthrie E, Lin JC, Mar EC, Pagano J. The Epstein-Barr virus (EBV) BZLF1 immediate-early gene product differentially affects latent versus productive EBV promoters. J Virol. 1989;63: 1729–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arumugaswami V, Wu T-T, Martinez-Guzman D, Jia Q, Deng H, Reyes N, et al. ORF18 is a transfactor that is essential for late gene transcription of a gammaherpesvirus. J Virol. 2006;80: 9730–9740. 10.1128/JVI.00246-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong E, Wu T-T, Reyes N, Deng H, Sun R. Murine gammaherpesvirus 68 open reading frame 24 is required for late gene expression after DNA replication. J Virol. 2007;81: 6761–6764. 10.1128/JVI.02726-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu T-T, Park T, Kim H, Tran T, Tong L, Martinez-Guzman D, et al. ORF30 and ORF34 are essential for expression of late genes in murine gammaherpesvirus 68. J Virol. 2009;83: 2265–2273. 10.1128/JVI.01785-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kayhan B, Yager EJ, Lanzer K, Cookenham T, Jia Q, Wu T-T, et al. A replication-deficient murine gamma-herpesvirus blocked in late viral gene expression can establish latency and elicit protective cellular immunity. J Immunol Baltim Md 1950. 2007;179: 8392–8402. [DOI] [PubMed] [Google Scholar]
- 24.Isomura H, Stinski MF, Murata T, Yamashita Y, Kanda T, Toyokuni S, et al. The human cytomegalovirus gene products essential for late viral gene expression assemble into prereplication complexes before viral DNA replication. J Virol. 2011;85: 6629–6644. 10.1128/JVI.00384-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perng Y-C, Qian Z, Fehr AR, Xuan B, Yu D. The human cytomegalovirus gene UL79 is required for the accumulation of late viral transcripts. J Virol. 2011;85: 4841–4852. 10.1128/JVI.02344-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Omoto S, Mocarski ES. Transcription of true late (γ2) cytomegalovirus genes requires UL92 function that is conserved among beta- and gammaherpesviruses. J Virol. 2014;88: 120–130. 10.1128/JVI.02983-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Omoto S, Mocarski ES. Cytomegalovirus UL91 is essential for transcription of viral true late (γ2) genes. J Virol. 2013;87: 8651–8664. 10.1128/JVI.01052-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gruffat H, Kadjouf F, Mariamé B, Manet E. The Epstein-Barr virus BcRF1 gene product is a TBP-like protein with an essential role in late gene expression. J Virol. 2012;86: 6023–6032. 10.1128/JVI.00159-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wyrwicz LS, Rychlewski L. Identification of Herpes TATT-binding protein. Antiviral Res. 2007;75: 167–172. 10.1016/j.antiviral.2007.03.002 [DOI] [PubMed] [Google Scholar]
- 30.Djavadian R, Chiu Y-F, Johannsen E. An Epstein-Barr Virus-Encoded Protein Complex Requires an Origin of Lytic Replication In Cis to Mediate Late Gene Transcription. PLoS Pathog. 2016;12: e1005718 10.1371/journal.ppat.1005718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aubry V, Mure F, Mariamé B, Deschamps T, Wyrwicz LS, Manet E, et al. Epstein-Barr virus late gene transcription depends on the assembly of a virus-specific preinitiation complex. J Virol. 2014;88: 12825–12838. 10.1128/JVI.02139-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watanabe T, Narita Y, Yoshida M, Sato Y, Goshima F, Kimura H, et al. Epstein-Barr virus BDLF4 Gene Is Required for Efficient Expression of Viral Late Lytic Genes. J Virol. 2015; 10.1128/JVI.01604-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuan J, Cahir-McFarland E, Zhao B, Kieff E. Virus and cell RNAs expressed during Epstein-Barr virus replication. J Virol. 2006;80: 2548–2565. 10.1128/JVI.80.5.2548-2565.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murata M, Nishiyori-Sueki H, Kojima-Ishiyama M, Carninci P, Hayashizaki Y, Itoh M. Detecting expressed genes using CAGE. Methods Mol Biol Clifton NJ. 2014;1164: 67–85. 10.1007/978-1-4939-0805-9_7 [DOI] [PubMed] [Google Scholar]
- 35.Frith MC, Valen E, Krogh A, Hayashizaki Y, Carninci P, Sandelin A. A code for transcription initiation in mammalian genomes. Genome Res. 2008;18: 1–12. 10.1101/gr.6831208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McKenzie J, Lopez-Giraldez F, Delecluse H-J, Walsh A, El-Guindy A. The Epstein-Barr Virus Immunoevasins BCRF1 and BPLF1 Are Expressed by a Mechanism Independent of the Canonical Late Pre-initiation Complex. PLOS Pathog. 2016;12: e1006008 10.1371/journal.ppat.1006008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Grady T, Wang X, Höner Zu Bentrup K, Baddoo M, Concha M, Flemington EK. Global transcript structure resolution of high gene density genomes through multi-platform data integration. Nucleic Acids Res. 2016;44: e145 10.1093/nar/gkw629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Grady T, Cao S, Strong MJ, Concha M, Wang X, Splinter Bondurant S, et al. Global bidirectional transcription of the Epstein-Barr virus genome during reactivation. J Virol. 2014;88: 1604–1616. 10.1128/JVI.02989-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bencun M, Klinke O, Hotz-Wagenblatt A, Klaus S, Tsai M-H, Poirey R, et al. Translational profiling of B cells infected with the Epstein-Barr virus reveals 5’ leader ribosome recruitment through upstream open reading frames. Nucleic Acids Res. 2018;46: 2802–2819. 10.1093/nar/gky129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Farrell PJ. Epstein-Barr Virus Epstein-Barr Virus Protocols Methods in Molecular Biology. Humana Press; 2001. Available: http://www.springer.com/us/book/9780896036901 [DOI] [PubMed] [Google Scholar]
- 41.Johannsen E, Luftig M, Chase MR, Weicksel S, Cahir-McFarland E, Illanes D, et al. Proteins of purified Epstein-Barr virus. Proc Natl Acad Sci U S A. 2004;101: 16286–16291. 10.1073/pnas.0407320101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Färber I, Hinderer W, Rothe M, Lang D, Sonneborn HH, Wutzler P. Serological diagnosis of Epstein-Barr virus infection by novel ELISAs based on recombinant capsid antigens p23 and p18. J Med Virol. 2001;63: 271–276. [DOI] [PubMed] [Google Scholar]
- 43.Ragoczy T, Miller G. Role of the epstein-barr virus RTA protein in activation of distinct classes of viral lytic cycle genes. J Virol. 1999;73: 9858–9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heilmann AMF, Calderwood MA, Portal D, Lu Y, Johannsen E. Genome-wide analysis of Epstein-Barr virus Rta DNA binding. J Virol. 2012;86: 5151–5164. 10.1128/JVI.06760-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994;2: 28–36. [PubMed] [Google Scholar]
- 46.Wobbe CR, Struhl K. Yeast and human TATA-binding proteins have nearly identical DNA sequence requirements for transcription in vitro. Mol Cell Biol. 1990;10: 3859–3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.El-Guindy A, Heston L, Miller G. A subset of replication proteins enhances origin recognition and lytic replication by the Epstein-Barr virus ZEBRA protein. PLoS Pathog. 2010;6: e1001054 10.1371/journal.ppat.1001054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Su M-T, Wang Y-T, Chen Y-J, Lin S-F, Tsai C-H, Chen M-R. The SWI/SNF Chromatin Regulator BRG1 Modulates the Transcriptional Regulatory Activity of the Epstein-Barr Virus DNA Polymerase Processivity Factor BMRF1. J Virol. 2017;91 10.1128/JVI.02114-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ersing I, Nobre L, Wang LW, Soday L, Ma Y, Paulo JA, et al. A Temporal Proteomic Map of Epstein-Barr Virus Lytic Replication in B Cells. Cell Rep. 2017;19: 1479–1493. 10.1016/j.celrep.2017.04.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sugimoto A, Kanda T, Yamashita Y, Murata T, Saito S, Kawashima D, et al. Spatiotemporally Different DNA Repair Systems Participate in Epstein-Barr Virus Genome Maturation▿. J Virol. 2011;85: 6127–6135. 10.1128/JVI.00258-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Geiduschek EP, Kassavetis GA. Transcription of the T4 late genes. Virol J. 2010;7: 288 10.1186/1743-422X-7-288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.El-Guindy A, Lopez-Giraldez F, Delecluse H-J, McKenzie J, Miller G. A locus encompassing the Epstein-Barr virus bglf4 kinase regulates expression of genes encoding viral structural proteins. PLoS Pathog. 2014;10: e1004307 10.1371/journal.ppat.1004307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang J-T, Yang P-W, Lee C-P, Han C-H, Tsai C-H, Chen M-R. Detection of Epstein-Barr virus BGLF4 protein kinase in virus replication compartments and virus particles. J Gen Virol. 2005;86: 3215–3225. 10.1099/vir.0.81313-0 [DOI] [PubMed] [Google Scholar]
- 54.Thompson J, Verma D, Li D, Mosbruger T, Swaminathan S. Identification and Characterization of the Physiological Gene Targets of the Essential Lytic Replicative Epstein-Barr Virus SM Protein. J Virol. 2015;90: 1206–1221. 10.1128/JVI.02393-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Verma D, Li D-J, Krueger B, Renne R, Swaminathan S. Identification of the physiological gene targets of the essential lytic replicative Kaposi’s sarcoma-associated herpesvirus ORF57 protein. J Virol. 2015;89: 1688–1702. 10.1128/JVI.02663-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen L-W, Chang P-J, Delecluse H-J, Miller G. Marked variation in response of consensus binding elements for the Rta protein of Epstein-Barr virus. J Virol. 2005;79: 9635–9650. 10.1128/JVI.79.15.9635-9650.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chua H-H, Lee H-H, Chang S-S, Lu C-C, Yeh T-H, Hsu T-Y, et al. Role of the TSG101 gene in Epstein-Barr virus late gene transcription. J Virol. 2007;81: 2459–2471. 10.1128/JVI.02289-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chiu Y-F, Sugden AU, Sugden B. Epstein-Barr viral productive amplification reprograms nuclear architecture, DNA replication, and histone deposition. Cell Host Microbe. 2013;14: 607–618. 10.1016/j.chom.2013.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Daikoku T, Kudoh A, Fujita M, Sugaya Y, Isomura H, Shirata N, et al. Architecture of replication compartments formed during Epstein-Barr virus lytic replication. J Virol. 2005;79: 3409–3418. 10.1128/JVI.79.6.3409-3418.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Su M-T, Liu I-H, Wu C-W, Chang S-M, Tsai C-H, Yang P-W, et al. Uracil DNA glycosylase BKRF3 contributes to Epstein-Barr virus DNA replication through physical interactions with proteins in viral DNA replication complex. J Virol. 2014;88: 8883–8899. 10.1128/JVI.00950-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heilmann AMF, Calderwood MA, Johannsen E. Epstein-Barr virus LF2 protein regulates viral replication by altering Rta subcellular localization. J Virol. 2010;84: 9920–9931. 10.1128/JVI.00573-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Calderwood MA, Holthaus AM, Johannsen E. The Epstein-Barr virus LF2 protein inhibits viral replication. J Virol. 2008;82: 8509–8519. 10.1128/JVI.00315-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sugimoto A, Sato Y, Kanda T, Murata T, Narita Y, Kawashima D, et al. Different Distributions of Epstein-Barr Virus Early and Late Gene Transcripts within Viral Replication Compartments. J Virol. 2013;87: 6693–6699. 10.1128/JVI.00219-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Leach FS, Mocarski ES. Regulation of cytomegalovirus late-gene expression: differential use of three start sites in the transcriptional activation of ICP36 gene expression. J Virol. 1989;63: 1783–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lieu PT, Wagner EK. Two leaky-late HSV-1 promoters differ significantly in structural architecture. Virology. 2000;272: 191–203. 10.1006/viro.2000.0365 [DOI] [PubMed] [Google Scholar]
- 66.Delecluse HJ, Hilsendegen T, Pich D, Zeidler R, Hammerschmidt W. Propagation and recovery of intact, infectious Epstein-Barr virus from prokaryotic to human cells. Proc Natl Acad Sci U S A. 1998;95: 8245–8250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chiu Y-F, Tung C-P, Lee Y-H, Wang W-H, Li C, Hung J-Y, et al. A comprehensive library of mutations of Epstein Barr virus. J Gen Virol. 2007;88: 2463–2472. 10.1099/vir.0.82881-0 [DOI] [PubMed] [Google Scholar]
- 68.Braman J, editor. In Vitro Mutagenesis Protocols [Internet]. Totowa, NJ: Humana Press; 2010. Available: http://link.springer.com/10.1007/978-1-60761-652-8 10.1038/nprot.2009.245 [DOI] [Google Scholar]
- 69.Tischer BK, Smith GA, Osterrieder N. En passant mutagenesis: a two step markerless red recombination system. Methods Mol Biol Clifton NJ. 2010;634: 421–430. 10.1007/978-1-60761-652-8_30 [DOI] [PubMed] [Google Scholar]
- 70.Calderwood MA, Venkatesan K, Xing L, Chase MR, Vazquez A, Holthaus AM, et al. Epstein-Barr virus and virus human protein interaction maps. Proc Natl Acad Sci U S A. 2007;104: 7606–7611. 10.1073/pnas.0702332104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Swaminathan S, Tomkinson B, Kieff E. Recombinant Epstein-Barr virus with small RNA (EBER) genes deleted transforms lymphocytes and replicates in vitro. Proc Natl Acad Sci U S A. 1991;88: 1546–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rozenblatt-Rosen O, Deo RC, Padi M, Adelmant G, Calderwood MA, Rolland T, et al. Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature. 2012;487: 491–495. 10.1038/nature11288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Livak KJ, Schmittgen TD. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods. 2001;25: 402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 74.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinforma Oxf Engl. 2013;29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhao H, Sun Z, Wang J, Huang H, Kocher J-P, Wang L. CrossMap: a versatile tool for coordinate conversion between genome assemblies. Bioinforma Oxf Engl. 2014;30: 1006–1007. 10.1093/bioinformatics/btt730 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MEME analysis showing positions of TATTWAA motif identified in A) late and B) leaky late promoters for 16 late and 15 out 16 leaky late TSS identified via CAGE-seq. BcRF1 is systematically excluded by MEME-suite due to significant deviation from consensus.
(TIF)
Shown are all EBV lytic transcripts identified by CAGE-seq, including strong TSS signals not corresponding to previously annotated ORFs. For each transcription start site (TSS) cluster the table specifies: the location (B95-8, V01555), strand (+ or -), kinetic class based on BALF2 ratio, the gene or promoter corresponding to the observed CAGE-seq TSS cluster, the corresponding annotation for each ORF/promoter, transcript abundance in tags per million (TPM) for each condition, BALF2 ratio (ratio of signal in I / I + t for ΔBALF2), and BDLF4 ratio (ratio of signal in I / I + t for ΔBDLF4. Clusters that did not correspond to well annotated ORF/promoters are labeled as unknown. Transfection of Rta and Zta resulted in a large number of clusters in this region and, accordingly, have been labeled as artifacts.
(XLS)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.