

Abstract
Background
Movement disorders are a significant clinical problem in lysosomal storage diseases (LSD) and account for substantial morbidity. The spectrum of movement disorders in childhood‐onset LSD, however, remains poorly defined.
Objectives
To define the spectrum of movement disorders in a well‐characterized cohort of children with LSD.
Methods
A retrospective chart review at a single tertiary care center (Boston Children's Hospital). Patients up to the age of 18 years with a clinical, genetic, and/or biochemical diagnosis of an LSD and at least one predefined movement disorder (parkinsonism, dystonia, ataxia, tremor, chorea, myoclonus, ballism, restless leg syndrome) were included.
Results
Ninety‐six patients were identified and 76 patients had a sufficiently document biochemical and/or genetic diagnosis. Of these, 18 patients met inclusion criteria (mean age: 10.3 ± 5.8 [SD] years, range: 3–18 years; 72% male). The most common LSD associated with a movement disorder was Niemann‐Pick disease type C (NPC), followed by several types of neuronal ceroid lipofuscinosis (NCL), and different mucopolysaccharidoses. The most common movement disorder was ataxia followed by rest tremor, dystonia, and myoclonus. The other predefined movement disorders were rare. The majority of patients presented with more than one movement disorder. The movement disorder was slowly progressive in all patients. Brain MRI changes included diffuse cerebral volume loss, white matter abnormalities with thinning of the corpus callosum, and cerebellar atrophy.
Conclusions
Movement disorders develop in a significant number of LSD patients. Ataxia, often in patients with NPC and NCL, is the most common phenotype but significant heterogeneity exists within and between different LSD.
Keywords: ataxia, lysosomal storage diseases, neurogenetics, neuronal ceroid lipofuscinosis, Niemann‐Pick disease type C
Lysosomal storage diseases (LSD) are a heterogeneous group of inborn errors of metabolism with a combined prevalence of about 1 in 5000 births.1, 2 Around 60 different LSD have been described, each sharing a genetic defect that leads to progressive substrate accumulation in many tissues, including the central nervous system. Most LSD affecting the brain are characterized by progressive neurologic dysfunction, often with onset in childhood. Movement disorders are a significant clinical problem in LSD, accounting for a substantial part of the morbidity,3 yet they remain poorly characterized. For example, generalized dystonia in certain LSD can be extremely difficult to treat and may lead to significant disability. Several types of movement disorders have been described in single case reports and smaller case series of individual disorders, mostly in adult patients.4, 5, 6, 7 However, systematic approaches are lacking. Clinical, genetic, and radiological features that associate with movement disorders in LSD remain poorly understood, although many LSD are recognized as genetic causes of movement disorders8 and some are treatable.9 Here, we systematically investigate a large and well‐characterized cohort of patients with childhood‐onset LSD and provide a description of associated movement disorders.
Methods
This study was approved by the Institutional Review Board at Boston Children's Hospital (#IRB‐P00023935). Patients were identified through a retrospective chart review at a single tertiary care center (Boston Children's Hospital) covering the time period from 2010 to 2017. Patients with (1) a clinical, genetic, and/or biochemical diagnosis of an LSD and (2) at least one predefined movement disorder (parkinsonism, dystonia, ataxia, tremor, chorea, myoclonus, ballism, restless leg syndrome) were included. Ninety‐six patients were initially identified and 76/96 patients were found to have a sufficiently documented genetic or biochemical diagnosis (Fig. 1). In total, 18/76 patients presented with at least one of the predefined movement disorders and thus met both inclusion criteria (Fig. 1).
Figure 1.
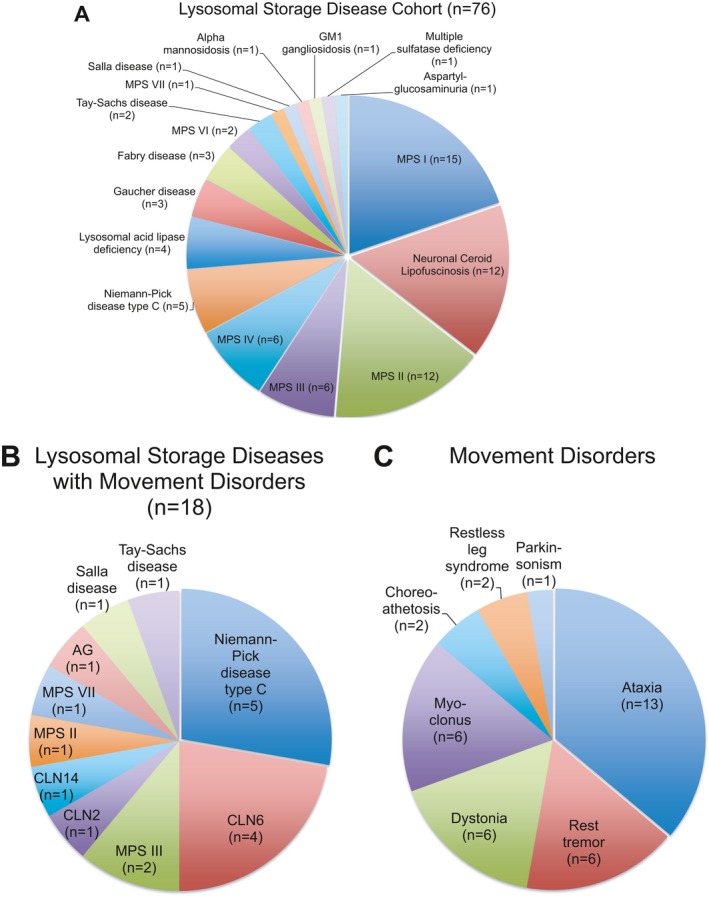
A: Distribution of different lysosomal storage diseases (LSD) in this cohort of 76 patients with a clinical, genetic and/or biochemical diagnosis of a LSD. B: Summary of 18 LSD patients who presented with at least one of the predefined movement disorders (parkinsonism, dystonia, ataxia, tremor, chorea, myoclonus, ballism, restless leg syndrome). C: Distribution of 36 movement disorders in these 18 patients. Abbreviations: AG, aspartylglycosaminuria; CLN, ceroid lipofuscinosis; MPS, mucopolysaccharidosis.
Results
A detailed review of our cohort of 76 children with a sufficiently documented clinical and biochemical or genetically‐confirmed diagnosis of a LSD (Fig. 1A) revealed a majority of patients with a mucopolysaccharidosis (n = 43) followed by the neuronal ceroid lipofuscinoses (NCL, n = 12), various sphingolipidoses (n = 9), Niemann‐Pick disease type C (NPC, n = 5), lysosomal acid lipase deficiency (n = 4), and various forms of oligosaccharidoses and mucolipidoses (n = 3). This likely reflects the distribution of LSD in our pediatric population but also the referral pattern to our tertiary care center.
In this cohort, we identified a total of 18 patients from 16 pedigrees with at least one movement disorder, and thus met both inclusion criteria. Demographic and clinical data are summarized in Table 1 and Table S1. Mean age at last follow up was 10.3 ± 5.8 (SD) years (range: 3–18 years; 72% male) with a mean age at diagnosis of a LSD of 5.4 ± 5.4 (SD) years and a mean age at onset for the predominant movement disorder of 6.9 ± 4.2 (SD) years. Two patients were deceased at the time of study completion. Most patients were of North American/European descent (50%), followed by Hispanics (22%), Arabs (17%), and other ethnic backgrounds (Table S1). No patient reported Ashkenazi Jewish ancestry. Consanguinity was reported in 6/18 patients (Table S1) and a positive family history was documented in three related cases from a single pedigree (P6, P7, P8).
Table 1.
Demographic, Clinical, and Genetic Characteristics
| Pt./Sex | LSD | Genetic/biochemical Dx | Annotation | F/U, AoO, ADx (yr) | Treatment LSD | Main MDx (AoO) | MDx description | Brain MRI findings |
|---|---|---|---|---|---|---|---|---|
| P1/M | NPC | NPC1 [c.743G>T/c.3410_3411 insA]/filipin staining + | Chr18:21152107; Chr18:21115480‐21115481 | 18/14/15 | Cyclodextrin, miglustat | Ataxia (14 yr) | Dystonia (mainly b/l arms and legs), truncal and limb ataxia | Normal |
| P2/Ma | NPC | NPC1 [c.2008_2011delTGCT/c.3565_3566insG]/filipin staining + |
Patho/patho Chr18:21124246‐21224249; Chr18:21114434‐21114435 |
7/1/1 | Cyclodextrin, miglustat | Ataxia (n.a.) | Dystonia (mainly b/l arms and legs), truncal and limb ataxia, gelastic cataplexy | Age 3 yr: delayed myelination, frontal lobe atrophy, thin CC, small optic nerves |
| P3/F | NPC | NPC1 [c.743G>T/c.3182T>C]/filipin staining +/− | Patho/patho Chr18:21140377; Chr18:21116700 | 18/8/16 | Cyclodextrin, vorinostat | Dystonia (11 yr) | Facial/orolingual and b/l foot dystonia, akanthisia, possible RLS | Age 18 yr: normal |
| P4/M | NPC |
NPC1 [c.3182T>C/c.1319T>C] Filipin staining + |
Patho/patho Chr18:21116700; Chr18:21136250 |
18/10/10 | Symptomatic | Ataxia (10 yr) | Dystonia (mainly b/l arms and legs), truncal and limb ataxia | n.a. |
| P5/M | NPC | Filipin staining + | n.a. | 6/1/1 | Miglustat | Ataxia (n.a) | Generalized ataxia, gelastic cataplexy | Age 6 yr: diffuse WM abnormalities |
| P6/Mb | CLN6 | CLN6 [c.793‐795delTCC/c.793‐795delTCC] |
Likely patho Chr15: 68500617‐68500619 |
7/4/4.5 | Symptomatic | Ataxia (4.5 yr) | Generalized ataxia, generalized dystonia (b/l arms and legs, trunk, neck), myoclonus | Age 5 yr: diffuse cerebral, brainstem and cerebellar atrophy, WM abnormalities |
| P7/Fb | CLN6 | CLN6 [c.793‐795delTCC/c.793‐795delTCC] |
Likely patho Chr15: 68500617‐68500619 |
6/4/5 | Symptomatic | Ataxia (5 yr) | Generalized ataxia, rest tremor, myoclonus | Age 6 yr: diffuse cerebral and cerebellar atrophy, WM abnormalities |
| P8/Fb | CLN6 | CLN6 [cc.793‐795delTCC/c.793‐795delTCC] |
Likely patho Chr15: 68500617‐68500619 |
6/4.5/4.5 | Symptomatic | Ataxia (4.5 yr) | Generalized ataxia, rest tremor, myoclonus | Age 6 yr: diffuse cerebral and cerebellar atrophy, WM abnormalities |
| P9/M | CLN6 | CLN6 [c.794_796delCCT/c.794_796delCCT] |
Patho Chr15: 68500618‐68500620 |
7/2/5 | Symptomatic | Ataxia (5 yr) | Generalized ataxia | Age 6 yr: diffuse cerebral and cerebellar atrophy, WM abnormalities |
| P10/M | CLN2 | TPP1 [c.1093C>T/c.1600C>T] |
Patho/patho Chr11:6637288; Chr11:6635849 |
6/3/3 | ERT (clinical trial) | Tremor (3 yr) | B/l rest and intention tremor | Age 3 yr: normal |
| P11/F | MPS IIIb |
NAGLU [c.192delC/c.192delC] alpha‐N‐acetylglucosaminidase activity not detectable |
Chr17: 40688482 | 18/1/1 | Symptomatic | Myoclonus (n.a.) | Generalized myoclonus, dystonia (b/l arms and legs), choreoathetosis | n.a. |
| P12/F | MPS IIIa |
SGSH [c.877C>T/c.949G>C] skin biopsy + |
Patho/likely patho Chr17:78185942; Chr17: 78185999 |
13/1.5/1.5 | Allogenic SCT | Tremor (n.a.) | Rest and intention tremor, choreoathetosis | Age 13 yr: diffuse WM loss, thin CC |
| P13/M | AG |
AGA [c.677G>A/c.677G>A] Low aspartylglucosaminidase activity |
Patho Chr4:178357451 |
18/0.75/17 | Symptomatic | Ataxia (n.a.) | Gait ataxia, bilateral extension tremors | n.a. |
| P14/M | SD | SLC17A5 [c.406>G/c.406>G] |
Patho/likely patho Chr6:74351533 |
6/0.25/5 | Symptomatic | Ataxia (n.a.) | Mild ataxia, b/l intention tremor | Age 3 yr: hypomyelination, thin CC |
| P15/M | MPS II |
IDS [c.410_411delTT/c.410_411delTT] Elevated GAG in urine |
Likely patho ChrX: 14854848‐14854849 |
8/1/1 | Symptomatic | Ataxia (5.5 yr) | Progressive generalized ataxia | Age 8 yr: VM, vermis hypoplasia, basal encephaloceles |
| P16/M | MPS VII | GUSB [c.526C>T/c.1169A>G] |
Patho/VUS Chr7:65444769; Chr7:65439588 |
17/0.3/3 | Symptomatic | Tremor (14 yr) | Rest tremor in all extremities, restless leg syndrome | Age 12 yr: diffuse WM abnormalities, thin CC |
| P17/M | CLN14 | KCTD7 [c.334C>T/c.334C>T] | Chr7:66103259 | 3/0.8/2 | Symptomatic | Ataxia (2 yr) | Gait ataxia, myoclonus | Normal |
| P18/M | TSD | HEXA [c.533G>A/c.1073+1G>A] |
Patho/patho Chr15:72645446; Chr15:72640388 |
4/1.5/3 | Miglustat | Parkinsonism (2.5 yr) | Parkinsonism with bradykinesia, hypomimia, hypophonia, tremor, postural instability, retropulsion. Gait ataxia, stimulus‐induced myoclonus | Age 2.5 yr: mild diffuse WM loss, thin CC, PVL, diffusely abnormal cerebellar cortex |
Variant classification according to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar; Single nucleotide variants of P1, P11, and P17 are not classified) or reporting laboratory (P12). Genomic location is shown according to on Assembly GRCh37. The movement disorder in all patients was described as slowly progressive with the exception of P18 who showed non‐progressive parkinsonism but slowly progressive ataxia and myoclonus.
Deceased.
First‐degree cousins.
Abbreviations: ADx, age at diagnosis; AoO, age at onset; AG, aspartylglucosaminuria; CC, corpus callosum; Dx, diagnosis; ERT, enzyme replacement therapy; F, female; F/U, follow up; GAG, glycosaminoglycans; LSD, lysosomal storage disease; M, male; MDx, movement disorder; MPS, mucopolysaccharidosis; NCL, neuronal ceroid lipofuscinosis; NPC, Niemann‐Pick Type C disease; patho, pathogenic; PVL, periventricular leukomalacia; SCT, stem cell transplantation; SD, Salla disease; TSD, Tay‐Sachs disease; VM, ventriculomegaly; VUS, variant of unknown significance; WM, white matter; yr, years.
The most common LSD associated with a movement disorder was NPC, followed by several types of NCL and mucopolysaccharidoses (Fig. 1B). The most common predominant movement disorder was ataxia, which was found in 12/18 patients, followed by rest tremor (6/18), dystonia (6/18), and myoclonus (6/18; Fig. 1C). The majority of patients (15/18) presented with more than one movement disorder, commonly a combination of ataxia and either dystonia, myclonus, or tremor. The movement disorder phenotype was slowly progressive in all patients. Brain MRI data were available for 15/18 of patients. Magnetic resonance imaging most commonly revealed diffuse cerebral volume loss (8/15) and white matter abnormalities (9/15) with thinning of the corpus callosum (5/15) as well as cerebellar atrophy (6/15). A correlation between imaging abnormalities and movement disorder severity was not readily apparent. Treatment of movement disorders was symptomatic, but 11/18 patients received treatment targeted at their LSD, including substrate‐reduction therapy, enzyme replacement, allogenic stem cell transplantation, and novel treatments as part of clinical trials (Table 1). Levodopa was not systematically trialed in patients with generalized dystonia, mainly due to the predominance of ataxia (P6) and rapid disease progression in individuals with multiple neurological and medical comorbidities (P2, P6, P18).
Niemann‐Pick Disease Type C
All five patients with NPC in our cohort presented with a prominent movement disorder. The predominant movement disorder was ataxia in four patients and dystonia in one (P3). Onset of the movement disorder was between 10 and 14 years of age and on a slowly progressive course. The ataxia involved the trunk and limbs, while dystonic movements were generalized and often severe. Facial and orolingual dystonia was described in one individual (P3). Gelastic cataplexy was present in two individuals (P2 and P5).
Neuronal Ceroid Lipofuscinoses (CLN6, CLN2, CLN14)
Four patients with a molecularly confirmed diagnosis of CLN6, and a single patient with CLN2 and CLN14, with a prominent movement disorder, were identified. This included three CLN6 patients from a single consanguineous Arab pedigree (P6, P7, P8). All individuals with CLN6 presented with generalized ataxia. Rest tremor and myoclonus were found in two patients (P7 and P8). The CLN2 patient had a milder movement disorder with a prominent bilateral rest and intention tremor, while the patient with CLN14 presented with early‐onset gait ataxia and myoclonus around the age of 2 years.
Mucopolysaccharidoses (MPS‐III, MPS‐II, MPS‐VII)
The spectrum of mucopolysaccharidoses (MPS) manifesting with movement disorders in our cohort included two individuals with MPS‐III (Sanfilippo syndrome), one individual with MPS‐II (Hunter syndrome), and one individual with MPS‐VII (Sly syndrome). The spectrum of movement disorders was heterogeneous. Interestingly, the two individuals with MPS‐III (P11 and P12) both displayed prominent choreoathethoid movements, which were otherwise rare in our study population.
Other Lysosomal Storage Diseases
Other LSD that presented with movement disorders included an 18‐year‐old boy with aspartylglucosaminuria who presented with ataxia and intention tremors around 17 years of age (P13), a 6‐year‐old boy with Salla disease who developed mild ataxia with bilateral intention tremors (P14), and a 4‐year‐old boy with Tay‐Sachs disease (P18) who presented with gait ataxia, myoclonus, and parkinsonism manifesting with bradykinesia, hypomimia, hypophonia, rest tremors, and postural instability with prominent retropulsion (Video S1).
Discussion
With exciting new genetic and clinical links between LSD and adult‐onset neurodegenerative diseases such as sporadic Parkinson's disease,10, 11 it is imperative to systematically define the spectrum of movement disorders in childhood‐onset LSD. Here we systematically investigated the presence of movement disorders in a large cohort of children with genetically or biochemically confirmed LSD. Limitations of our study include its retrospective nature and the design as a single center study with a potential referral bias. We identified a total of 18 patients covering a wide spectrum of LSD, including NPC, several forms of NCL, and mucopolysaccharidoses as well as individual cases of aspartylglucosaminuria, Salla disease, and Tay‐Sachs disease. Although the phenotypic spectrum for these disorders has been characterized, the spectrum of movement disorders has not been clearly defined in most. NPC is a very heterogeneous disorder both with regards to age of onset and clinical manifestations. Ataxia is a recognized manifestation, particularly in adult cases,6, 12, 13 and was also common in our cohort. Dystonia was found in 4/5 of our pediatric and adolescent patients and was thus more frequent compared to previously published cohorts of older adolescent and adult patients.6, 13 Myoclonus has been reported as a variable manifestation in juvenile and adult cases, but was not prevalent in our pediatric cases. It is important to recognize that movement disorders, although not a presenting symptom in our patients, are among the most frequent initial symptoms, particularly in adolescents and adults, where a combination of two or more movement disorders, or a combination of psychiatric symptoms and movement disorders, should raise suspicion for NPC or other less common LSD, such as Tay‐Sachs disease.14 CLN6, a “variant” late infantile‐onset from of NCL, is characterized by developmental delay and regression, vision loss, seizures, myoclonus, as well as cerebellar dysfunction with ataxia and dysarthria.15 This was replicated in our four patients who developed a generalized ataxia around the age of 3 to 4 years. Myoclonus and tremor were also present in the majority of patients, consistent with published case reports.15 CLN2, the classic late‐infantile onset form, is characterized by rapid neurological decline with seizures, vision loss, and motor decline.16 Prominent truncal and peripheral ataxia can be initial symptoms. In our patient, movement disorders were more subtle and consisted of rest and cerebellar tremors. The mucopolysaccharidoses cause a wide spectrum of neurological dysfunction, but movement disorders have not been clearly defined. Our cohort revealed two patients with MPS‐III, including an 18‐year‐old young woman with MPS‐IIIb, who manifested with generalized myoclonus, dystonia, and choreoathetosis and a 13‐year‐old girl with rest and intention tremors and choreoathetoid movements. To our knowledge, our accounts of movement disorders in patients with MPS‐II, MPS‐VII, aspartylglucosaminuria, and Salla disease are the first in literature. Parkinsonism has been described in adult‐onset cases of GM2 gangliosidosis17, 18 and dystonia has been documented in juvenile‐onset cases.7, 19 However, in our cohort, we identified a 4‐year‐old boy with genetically confirmed Tay‐Sachs disease who presented with a peculiar picture of parkinsonism with prominent postural instability and retropulsion (Video S1). This is, to our knowledge, the first description of parkinsonism in a child with Tay‐Sachs disease.
In summary, our study reveals the presence of movement disorders in a significant portion of LSD patients. Ataxia is the most common phenotype, signifying a common cerebellar involvement in LSD, followed by tremors, dystonia, and myoclonus. Significant heterogeneity exists within and between different LSD. Additional longitudinal studies with larger sample sizes are needed to further delineate the genetic and imaging characteristics associated with movement disorders in LSD. With emerging treatments for LSD and an expanding array of therapies available to treat movement disorders, understanding the spectrum and clinical challenges of movement disorders in LSD will help guide clinical management and benefit patients.
Author Roles
1. Research Project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript Preparation: A. Writing the First Draft, B. Review and Critique.
D.E.‐F.: 1A, 1B, 1C, 2A, 2B, 2C, 3A, 3B
C.H.: 1B, 1C, 2B, 2C, 3B
P.E.D.: 1C, 2C, 3B
L.H.R.: 1C, 2C, 3B
I.A.: 1C, 2C, 3B
O.B.: 1A, 1B, 1C, 2A, 2B, 2C, 3B
Disclosures
Ethical Compliance Statement: We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest: This work was supported by the Fred Lovejoy Research and Education Fund (to D.E.‐F.).
Financial Disclosure for previous 12 months: D.E.F. is employed by Boston Children's Hospital and received support from the Fred‐Lovejoy Award of the Fred Lovejoy Research and Education Fund and the DESITIN Investigator Award of the German Society of Pediatric Neurology, and CureSPG47, Inc. C.H. is employed by Boston Children's Hospital and reports no conflict of interest. P.E.D. is employed by Boston Children's Hospital and received support from the Neurology Resident Research Education Program NIH NINDS (5R25NS070682‐07), Boston Children's Hospital Office of Faculty Development, and the Tuberous Sclerosis Alliance. L.H.R. is employed by Boston Children's Hospital and received support from the NIH Undiagnosed Diseases Network (UDN). I.A. is employed by Boston Children's Hospital and received support from Lumos Pharma and the North American Mitochondrial Disease Consortium. O.B. is employed by Boston Children's Hospital and is the site PI for VTS301, a phase 2/3 trial for the evaluation of intrathecal cyclodextrin in Niemann‐Pick disease type C; has stock ownership from Aptevo Therapeutics, Emergent Biosolutions; is a consult for Genzyme‐Sanofi and Shire, ELOXX, NPKUA, Sucampo, FDNA, and Perkin Elmer; received a grant from Moderna; holds a patent (PCT/US2001/030965); and received royalties from UptoDate.
Supporting information
A video accompanying this article is available in the supporting information here.
Table S1. Detailed demographic, clinical and genetic characteristics of 18 patients with a lysosomal storage disease and movement disorder
Video S1. Patient #18 with Tay‐Sachs disease. Examined at age 31 months. The patient is showing features of parkinsonism with bradykinesia, hypomimia, hypophonia, tremor, postural instability, and prominent retropulsion. Gait ataxia and stimulus‐induced myoclonus developed later and are not shown here.
Acknowledgments
The authors thank the patients and their families for supporting this study. The authors are also grateful to B. Aravamuthan, G. Berry, D. Coulter, K. Hawash Kuemmerle, A. Kritzer, L. Lehman, C. Moufawad El Achkar, E. Neilan, J. Picker, A. Rotenberg, M. Takeoka, W.‐H. Tan, A. Torres, and J. L. Waugh (Boston Children's Hospital) who participated in the care of the patients included in this study.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Kingma SD, Bodamer OA, Wijburg FA. Epidemiology and diagnosis of lysosomal storage disorders; challenges of screening. Best Pract Res Clin Endocrinol Metab 2015;29:145–157. [DOI] [PubMed] [Google Scholar]
- 2. Boustany RM. Lysosomal storage diseases–the horizon expands. Nat Rev Neurol 2013;9:583–598. [DOI] [PubMed] [Google Scholar]
- 3. Stampfer M, Theiss S, Amraoui Y, et al. Niemann‐Pick disease type C clinical database: cognitive and coordination deficits are early disease indicators. Orphanet J Rare Dis 2013;8:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Muthane U, Chickabasaviah Y, Kaneski C, et al. Clinical features of adult GM1 gangliosidosis: report of three Indian patients and review of 40 cases. Mov Disord 2004;19:1334–1341. [DOI] [PubMed] [Google Scholar]
- 5. Roze E, Paschke E, Lopez N, et al. Dystonia and parkinsonism in GM1 type 3 gangliosidosis. Mov Disord 2005;20:1366–1369. [DOI] [PubMed] [Google Scholar]
- 6. Anheim M, Lagha‐Boukbiza O, Fleury‐Lesaunier MC, et al. Heterogeneity and frequency of movement disorders in juvenile and adult‐onset Niemann‐Pick C disease. J Neurol 2014;261:174–179. [DOI] [PubMed] [Google Scholar]
- 7. Nardocci N, Bertagnolio B, Rumi V, Angelini L. Progressive dystonia symptomatic of juvenile GM2 gangliosidosis. Mov Disord 1992;7:64–67. [DOI] [PubMed] [Google Scholar]
- 8. Marras C, Lang A, van de Warrenburg BP, et al. Nomenclature of genetic movement disorders: recommendations of the International Parkinson and Movement Disorder Society Task Force. Mov Disord 2016;31:436–457. [DOI] [PubMed] [Google Scholar]
- 9. Jinnah HA, Albanese A, Bhatia KP, et al. Treatable inherited rare movement disorders. Mov Disord 2017. 10.1002/mds.27140. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aflaki E, Westbroek W, Sidransky E. The complicated relationship between Gaucher disease and parkinsonism: insights from a rare disease. Neuron 2017;93:737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lloyd‐Evans E, Haslett LJ. The lysosomal storage disease continuum with ageing‐related neurodegenerative disease. Ageing Res Rev 2016;32:104–121. [DOI] [PubMed] [Google Scholar]
- 12. Koens LH, Kuiper A, Coenen MA, et al. Ataxia, dystonia and myoclonus in adult patients with Niemann‐Pick type C. Orphanet J Rare Dis 2016;11:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sevin M, Lesca G, Baumann N, et al. The adult form of Niemann‐Pick disease type C. Brain 2007;130(Pt 1):120–133. [DOI] [PubMed] [Google Scholar]
- 14. Liguori M, Tagarelli G, Romeo N, Bagala A, Spadafora P. Identification of a patient affected by “Juvenile‐chronic” Tay Sachs disease in South Italy. Neurol Sci 2016;37:1883–1885. [DOI] [PubMed] [Google Scholar]
- 15. Canafoglia L, Gilioli I, Invernizzi F, et al. Electroclinical spectrum of the neuronal ceroid lipofuscinoses associated with CLN6 mutations. Neurology 2015;85:316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Williams RE, Adams HR, Blohm M, et al. Management strategies for CLN2 disease. Pediatr Neurol 2017;69:102–112. [DOI] [PubMed] [Google Scholar]
- 17. Oates CE, Bosch EP, Hart MN. Movement disorders associated with chronic GM2 gangliosidosis. Case report and review of the literature. Eur Neurol 1986;25:154–159. [DOI] [PubMed] [Google Scholar]
- 18. Inzelberg R, Korczyn AD. Parkinsonism in adult‐onset GM2 gangliosidosis. Mov Disord 1994;9:375–377. [DOI] [PubMed] [Google Scholar]
- 19. Meek D, Wolfe LS, Andermann E, Andermann F. Juvenile progressive dystonia: a new phenotype of GM2 gangliosidosis. Ann Neurol 1984;15:348–352. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A video accompanying this article is available in the supporting information here.
Table S1. Detailed demographic, clinical and genetic characteristics of 18 patients with a lysosomal storage disease and movement disorder
Video S1. Patient #18 with Tay‐Sachs disease. Examined at age 31 months. The patient is showing features of parkinsonism with bradykinesia, hypomimia, hypophonia, tremor, postural instability, and prominent retropulsion. Gait ataxia and stimulus‐induced myoclonus developed later and are not shown here.