

Abstract
Several mechanisms of action have been proposed for DNA methyltransferase and histone deacetylase inhibitors (DNMTi and HDACi); mainly based on candidate gene approaches. However, less is known about their genome-wide transcriptional and epigenomic consequences. By mapping global transcription start site (TSS) and chromatin dynamics, we observed the cryptic transcription of thousands of treatment-induced non-annotated TSSs (TINATs) following DNMTi and/or HDACi treatment. The resulting transcripts frequently splice into protein-coding exons and encode truncated or chimeric open reading frames translated into products with predicted abnormal or immunogenic functions. TINAT transcription after DNMTi coincided with DNA hypomethylation and gain in classical promoter histone marks, while HDACi specifically induced a subset of TINATs in association with H2AK9ac, H3K14ac, and H3K23ac. Despite this mechanistic difference, both inhibitors convergently induced transcription from identical sites since we found TINATs to be encoded in solitary long-terminal repeats of the LTR12 family, epigenetically repressed in virtually all normal cells.
In contrast to genetic mutations, epigenetic changes are potentially reversible, which is deeming them an attractive target for cancer treatment. Inhibitors directed against DNA methyltransferases (DNMTi) and histone deacetylases (HDACi) are used for the treatment of several haematopoietic malignancies1,2. However, despite their clinical use for several years, there is still a lack of knowledge regarding the mode of action3. Two previous studies on DNMTi in cancer cell lines reported the up-regulation of double stranded RNA (dsRNA) molecules originating from codogenic endogenous retroviruses (ERV) followed by an interferon response and the induction of viral defense genes4,5. However, it remains unclear how other classes of epigenetic drugs integrate into these findings and whether there are additional effects, potentially missed by candidate gene approaches. Here, we globally mapped DNMTi and HDACi-induced transcriptomic and epigenomic changes by using whole-genome profiling technologies (Supplementary Fig. 1 and Supplementary Table 1) and show that the vast majority of TSSs that transcriptionally responded towards epigenetic modulation were cryptic, currently non-annotated TSSs encoded in solitary long-terminal repeats (LTRs).
Results
Epigenetic drugs activate cryptic TSSs in the DAPK1 gene
To efficiently measure the effects of epigenetic drugs on endogenous gene expression, we engineered the lung cancer cell line NCI-H1299 by introducing a dual fluorescence/resistance (EGFP-NEO) reporter into intron 3 of DAPK1 which is epigenetically silenced in association with CpG island hypermethylation (Fig. 1a and Supplementary Fig. 2a,b). Upon treatment with the DNMTi, 5-aza-2′deoxycytidine (DAC) or with siRNAs/shRNAs targeting DNMT1 mRNA, the DAPK1 promoter loses methylation and a fusion transcript consisting of exons 1-3 and the EGFP-NEO reporter is expressed (Supplementary Fig. 2c-f). Consequently, DAPK1 reactivated cells can be further enriched and quantified by G418 selection or FACS-sorting (Fig. 1b). To determine the suitability of this cell line to screen for epigenetically active substances, we tested several compounds that are known to affect various epigenetic enzyme classes. Epigenetic reactivation was read out in a G418 resistance screen, where cell viability increased mainly following the treatments with DNMTi and HDACi (Fig. 1c and Supplementary Fig. 2g). We confirmed reporter gene expression after DNMTi or HDACi by qRT-PCR (Fig. 1d, left). To our surprise, however, the canonical DAPK1 mRNA was induced only upon DAC treatment but not after HDACi (Fig. 1d, right). We hypothesized that HDACi activates alternative TSSs located upstream of the EGFP-NEO sequence, thus giving rise to a truncated transcript lacking the 5′ region of the DAPK1 mRNA. By performing 5′ rapid amplification of cDNA ends (5′-RACE) on RNA extracted from treated cells, we identified three distinct transcript isoforms originating from cryptic (currently non-annotated) TSSs located within DAPK1 intron 2 (termed: TSSs α, β, and γ), all of which were spliced into DAPK1 exon 3 (Fig. 1e and Supplementary Fig. 2h). These transcripts contain novel sequences towards their 5′ end (α, β, or γ) in place of the canonical first two exons which harbor the regular DAPK1 start codon, and thus comprise an alternative open reading frame (ORF). We confirmed the existence of these transcripts by qRT-PCR (Fig. 1f). In response to DNMTi and HDACi, the γ-transcript was also found in wild-type NCI-H1299 cells as well as in various other cancer cell lines (Fig. 1g), indicating that its activation is neither cell-line specific nor a consequence of genomic editing.
Figure 1. Novel DAPK1 intronic TSSs arise upon epigenetic drug treatment.
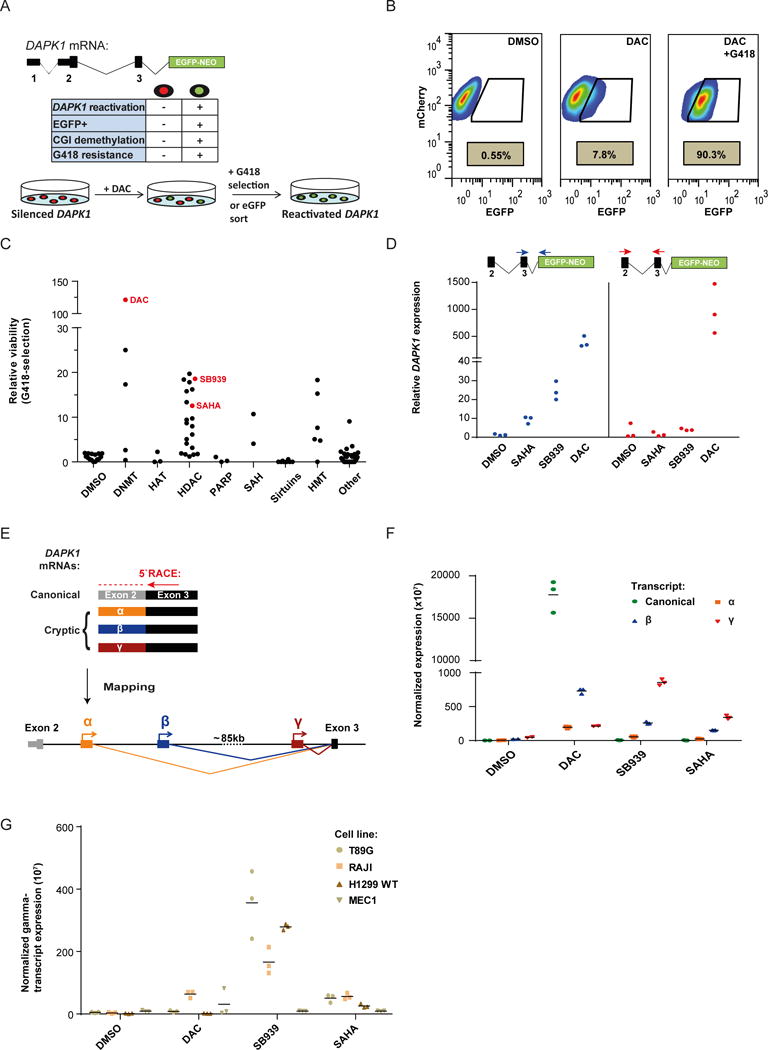
a) A fluorescence/resistance marker was introduced into one allele of the DAPK1 locus epigenetically silenced in NCI-H1299 cells. Administration of the DNA demethylating agent DAC reactivates a subpopulation of cells (green coloring).The key characteristics of DAPK1 silenced (red) and reactivated (green) cells are shown in the central table. CGI = CpG island.
b) FACS analysis showing the percentage of EGFP positive reporter cells before (left) and after DAC treatment with (right) or without (middle) additional G418 selection.
c) NCI-H1299 reporter cell viability after epigenetic compound treatment and G418 selection relative to DMSO controls. Data is sorted by inhibitor class: DNMT=DNA methyltransferase; HAT=Histone acetyltransferase; HDAC=Histone deacetylase; PARP=Poly(ADP-ribose)-Polymerase; SAH=S-Adenosyl-L-homocysteine; SIRT=Sirtuins; HMT=Histone methyltransferase.
d) DAPK1 expression after DNMTi and HDACi treatment of NCI-H1299 reporter cells relative to DMSO. qRT-PCR analysis was performed using primers located either in DAPK1 exon 2 and 3 (blue) or in exon 3 and the fluorescence/resistance marker (red).
e) Three cryptic 5′ exons (α, β and γ) were identified by 5′RACE performed on RNA from HDACi treated cells. All cryptic transcripts spliced to the canonical DAPK1 exon 3. γ: chr9 90219272 -90219341; β: chr9 90134907 - 90135007; α: chr9 90125477 - 90125599
f) qRT-PCR expression analysis of canonical DAPK1 or cryptic transcripts(α, β, and γ) across treatments relative to housekeeping genes.Vertical line represents the mean from three independent experiments.
g) Expression of the DAPK1 γ-transcript relative to housekeeping genes in untreated and treated cell lines. Vertical line represents the mean from three independent experiments.
Global transcription from cryptic TSSs after treatment
We hypothesized that the aberrant activation of cryptic TSSs is not restricted to the DAPK1 locus but a global phenomenon following treatment. By using cap analysis of gene expression (CAGE), we mapped the genome-wide TSS usage of NCI-H1299 reporter cells treated with DNMTi (DAC), HDACi (SAHA or SB939), or both DAC+SB939 (DAC+SB) (Supplementary Data 1). CAGE overcomes technical bottlenecks associated with standard RNA-seq, such as low coverage of transcript 5′ ends and difficulties in distinguishing multiple isoforms and splice variants that often overlap with reference transcripts. As a proof of concept, the CAGE data recapitulated our previous observations, in that only DAC treatment and not HDACi alone reactivated the canonical DAPK1 TSSs (Fig. 2a, left). Moreover, we found several CAGE-tags that supported the induction of the DAPK1 γ-transcript and its respective splicing into exon 3 after treatment (Fig. 2a, right).
Figure 2. CAGE-sequencing identifies genome-wide activation of non-annotated TSSs upon treatment.
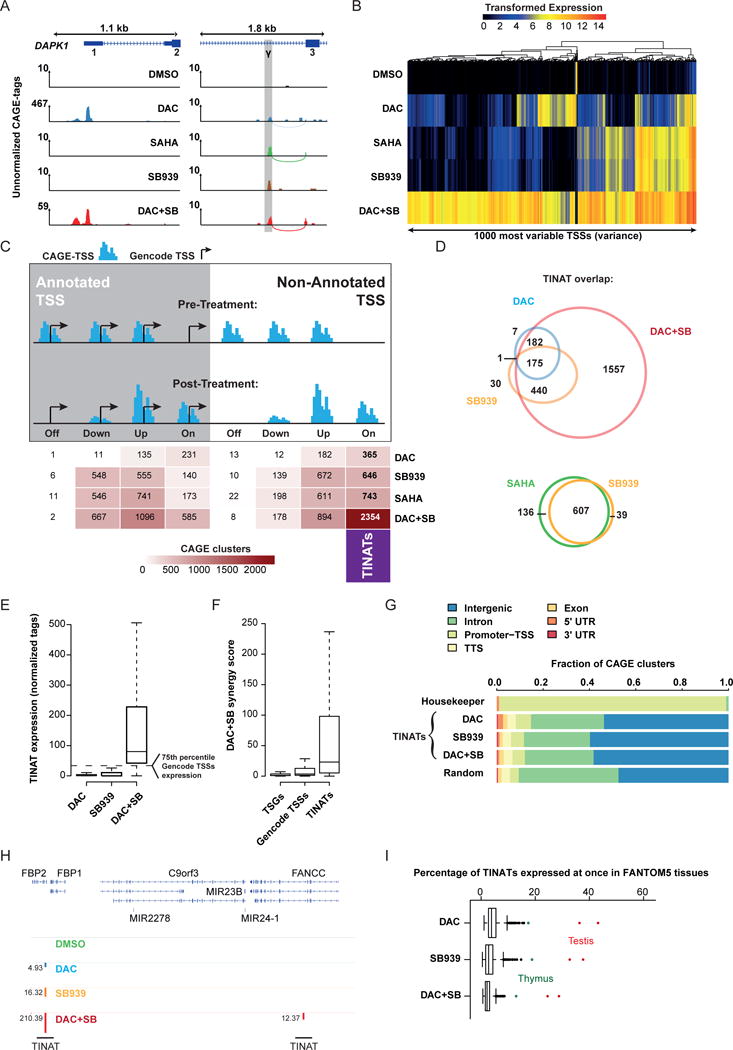
a) CAGE coverage at the canonical DAPK1 TSS (left panel) and the intronic γ-TSS (right panel, grey coloring). Curved lines indicate split-CAGE-tags. Numbers above vertical axis line denote the scale.
b) Variance stabilized expression values48 of the 1000 most variable TSSs across treatment conditions.
c) CAGE-clusters categorized into annotated (left panel) or non-annotated (right panel) peaks. CAGE-TSSs < 150 bp away from the nearest Gencode TSS were considered annotated. Silenced (Off), repressed (Down), induced (Up), and de novo transcribed (On) CAGE peaks were quantified and are shown in the table. TINATs are highlighted (right column). Arrow=Gencode TSS; blue bars=CAGE peak.
d) TINAT overlap between DAC, SB939, and DAC+SB treatment (top) as well as SB939 and SAHA (bottom). TINATs were considered overlapping if expression was significantly different from DMSO control in more than one condition.
e) Normalized DAC (blue), SB939 (orange), and DAC+SB (red) TINAT expression.
f) Synergy score for TSSs associated with lung adenocarcinoma TSGs49, de novo induced genes, and the union of TINATs. Synergy score was calculated as follows: expression DAC+SB/(expression DAC + expressionSB939). Data points beyond the extremes of the whiskers are not shown.
g) Genomic distribution of CAGE-TSS. HOMER50 was used to annotate TINATs and housekeepers (100 least variable TSSs) to genomic features. TTS = transcription termination site; UTR = untranslated region.
h) TINAT expression in the introns of the FBP2 and FANCC across treatments. Numbers next to the bar indicate normalized CAGE-tag counts.
i) Percentage of simultaneously expressed TINATs in FANTOM5 samples.
Globally, epigenetic treatment substantially changed the TSS usage of cells, with the combinatorial treatment (DAC+SB) showing the strongest effects (Fig. 2b and Supplementary Fig. 3a). We performed differential TSS expression analysis using a four-fold expression change and a false discovery rate (FDR) < 0.05 as minimal thresholds for differential expression. Epigenetic treatment caused both quantitative and qualitative expression changes at annotated TSSs (Fig. 2c, left). In line with previous reports, DAC or DAC+SB treatment significantly up-regulated cancer testis antigens (CTAs) as well as Aza-induced immune and viral defense genes (AIMs)6, which was accompanied with the transcription from codogenic ERVs that have the capability to form dsRNAs and trigger an interferon response (Supplementary Fig. 3b-d). It is important to note that neither SB939 nor SAHA significantly induced AIM expression, suggesting that HDACi exert their function independently.
Remarkably, however, we observed that all investigated drug regimens predominantly induced de novo transcription from currently non-annotated TSSs, termed here as treatment-induced non-annotated TSSs (TINATs); (Fig. 2c, right). Although DNMTi and HDACi target distinct epigenetic pathways, inhibitor treatment mostly converged on the activation of identical TINATs (Fisher′s exact test, P < 2.2 × 10−16) (Fig. 2d, top). Moreover, TSS activity after SAHA and SB939 was highly similar (r = 0.99) (Supplementary Fig. 3e and Fig. 2d, bottom). Thus, we focused on SB939 as a representative of HDACi for further analyses. In line with previous findings of the synergistic effect on gene expression by combined demethylation and HDACi7, we found multiple TINATs exclusively expressed after DAC+SB treatment. Moreover, the level of expression after combinatorial treatment was stronger than expected by the additive effect of DNMTi and HDACi alone (Fig. 2e). This synergistic effect was significantly stronger at TINATs (median synergy score = 23.2) than at the TSSs of de novo induced annotated genes (median = 3.5) or TSGs (median = 1.1) (Wilcoxon and Mann-Whitney two-sided test, P < 2.2 × 10−16; Fig. 2f). The majority of TINATs were located in either intergenic (~60%) or intronic (~20%) regions (Fig. 2g and supplementary Fig. 4) with a median distance to the nearest annotated TSS of 9.3, 9.0, and 11.6 kb for DAC, SB939, and DAC+SB, respectively. Genes in the vicinity of DAC induced TINATs were neither enriched for any biological process nor were they influenced by TINAT expression (correlation between TINAT expression and expression of nearby genes; r = 0). In contrast, genes most proximal to SB939 or DAC+SB induced TINATs were enriched for neuronal and developmental processes (Supplementary Fig. 3f) and TINAT expression was positively correlated with the expression of nearby genes [SB939, r = 0.4 (P = 2.2 × 10−9); DAC+SB, r = 0.21 (P = 3.9 × 10−29)] (Supplementary Fig. 3g). However, unlike active enhancer sites that are transcribed bidirectionally8, TINATs displayed unidirectional transcription (Supplementary Fig. 3h).
Exemplarily, Fig. 2h depicts expression of two TINATs located in the introns of FBP2 and FANCC. We further confirmed the treatment-specificity of TINATs by analyzing their expression across the FANTOM5 expression atlas8. The transcripts were generally not expressed under physiologic conditions, with the notable exception of testicular and fetal thymic tissues which concurrently expressed up to ~40% and ~20% of all TINATs, respectively (Fig. 2i and Supplementary Fig. 3i).
TINAT-exon fusion transcripts encode aberrant proteins
Based on our initial observations at the DAPK1 locus, we analyzed whether TINATs generally spliced into genic exons. Approximately 50-60% of all TINATs generated spliced transcripts of which another ~30% were spliced into protein-coding exons (Fig. 3a). These observations are exemplified at the FBP2 locus, where TINAT proximal splice sites join the cryptic TSS with exon 2 located downstream of the canonical FBP2 translation start site (Fig. 3b). We confirmed the existence of 15 TINAT-exon fusion transcript candidates in different cell lines by qPCR (Fig. 3c). Fusion candidates were manually selected based on their expression level and the number of CAGE-tags supporting the splicing event. Using StringTie9, we reconstructed 453, 744, and 3627 TINAT-exon transcript isoforms for DAC, SB939, and DAC+SB treated cells, respectively (Supplementary Table 2 and Supplementary Data 2). The exon-intron structure of the reconstructed transcripts closely matched the annotation of reference genes, as illustrated for FBP2. However, they often lacked the 3′ end of the canonical mRNA since CAGE-tag density is strongly skewed towards the 5′ end/TSS of a transcript. Around 14-21% of TINAT transcripts overlapped protein-coding genes and around 33-40% with long non-coding RNAs (lncRNAs)10. Although lncRNAs initiating from TINATs included transcripts with known function in disease, such as SChLAP111, we focused our further analyses on the protein-coding potential of TINATs. Most of the assembled fusion transcripts that contain cryptic sequence at their 5′ and native protein-coding exon sequence downstream were predicted to be coding (Fig. 3d). In silico translation showed that about one-half of the candidates encode in-frame isoforms relative to the coding DNA sequence (CDS) of the canonical mRNA, while the other half generates out-of-frame and thereby completely novel peptide sequences (Fig. 3e). Fusion transcripts that are translated in-frame with the native CDSs either give rise to the original, truncated, or chimeric isoforms, depending on whether the canonical or variant in-frame start codons are used. The truncated isoforms often lacked domains or peptide sequences important for proper protein function, localization, or binding while other functional regions remained unaffected (Fig. 3f). TINAT fusion transcripts encoding the canonical full-length sequence comprised genes with diverse biological functions, including transcription factors (TFEC, TBX4, GTF2H5), DNA damage repair and apoptosis (RAD50, SESN1, TNFRSF10B), epigenetic modifiers (HDAC4), and CTAs (MAGEB10, BRDT). Noteworthy, several other CTAs were expressed from TINATs (PRSS55, MAGEB2, and XAGE5) but the resulting fusion transcripts were predicted to encode out-of-frame peptides. While these out-of-frame transcripts are likely subjected to nonsense-mediated decay (NMD), chimeric peptide sequences encoded in TINAT fusion transcripts are potentially immunogenic based on their foreign sequence and their capability of being presented on MHC-class I molecules (Fig. 3g and Supplementary Fig. 5a). Furthermore, most of these transcripts were not expressed in the adult thymus and hence would not be expected to contribute to central tolerance. Of note, none of the potentially immunogenic peptides corresponded to known CTAs12.
Figure 3. TINAT-exon fusion transcripts encode novel protein isoforms with abnormal functions.
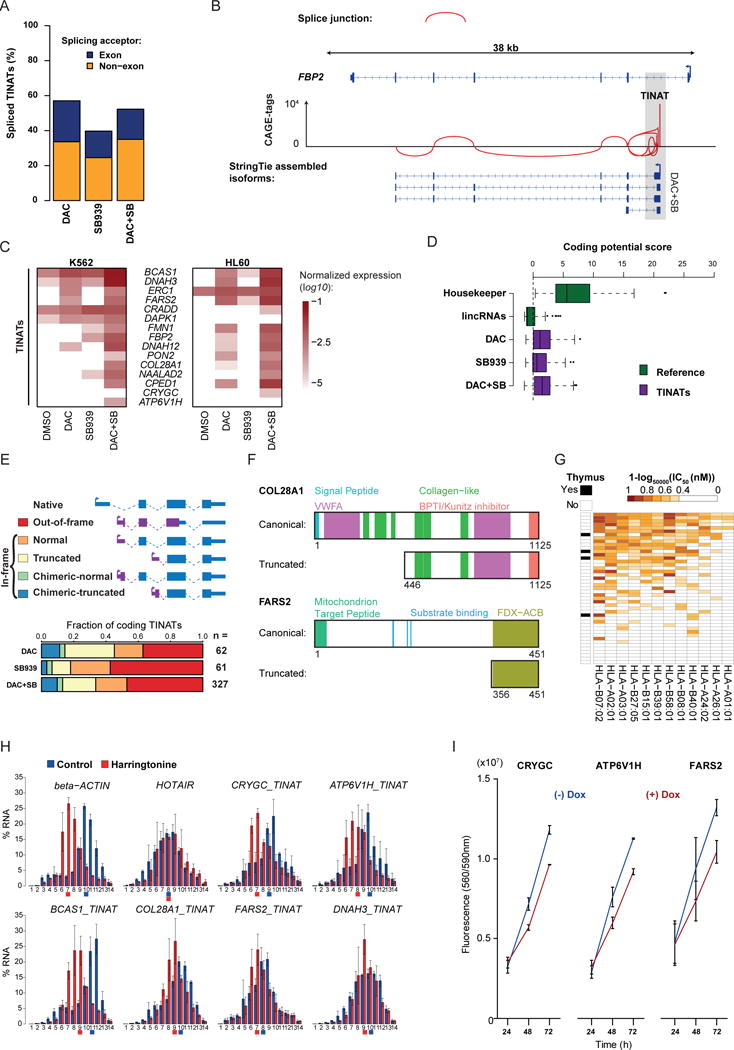
a) Fraction of TINATs having > 1% split CAGE-seq reads
b) Splice junctions at the FBP2 locus based on TINAT-derived CAGE-tags of DAC+SB treated NCI-H1299 cells.
c) TINAT-exon fusion transcript expression in K562 (left) and HL60 (right) cells. The log10 of the mean expression from three independent experiments relative to housekeepers is shown.
d) The coding potential of 100 housekeeping genes, 100 randomly selected ncRNAs, and TINATs was assessed using the coding potential calculator51. Dashed line denotes the threshold for protein-coding transcripts.
e) Schematic representation of the different scenarios for the translation of TINAT-exon fusion transcripts (upper panel). ORFs were categorized based on the criteria described in the online methods. The canonical (blue) and the novel, TINAT-derived sequence (purple) are schematically shown. Bottom panel depicts fraction of TINATs in each category.
f) COL28A1 and FARS2 protein domains for the canonical and truncated isoform are illustrated. Numbers below proteins indicate amino acid positions.
g) NetMHCpan52 was used to predict the binding affinity of 12 major HLA alleles (columns) for 45 DAC+SB chimeric peptide sequences (rows). The presence of a TINAT within the adult thymus is displayed.
h) Distribution of beta-actin, HOTAIR, and five TINAT-exon fusion transcripts along polysome fractions. Colored squares below horizontal axis line indicate the fraction where half of the mRNAs have accumulated.
i) Cell viability of NCI-H1299 reporter cells transduced with DOX-inducible TINAT-derived ORFs with or without DOX. Data from two independent experiments are shown.
To confirm the translational capacity of selected fusion transcripts, we translated the canonical CRYGC mRNA and three TINAT-exon transcripts [CRYGC (chimeric-truncated), BCAS1 (normal), and FBP2 (truncated)] in vitro. For all RNA templates, we observed translation products with the predicted sizes (Supplementary Fig. 5b). We further compared polysomal association of 15 TINAT-exon fusion candidates in DAC+SB treated cells incubated in the absence or presence of the translation inhibitor harringtonine to deplete elongating ribosomes from mRNAs13 (Supplementary Fig. 5c). As a positive control, β-actin mRNA showed highest abundance in heavy polysome fractions and the strongest release upon harringtonine treatment (Fig. 3h; others are shown in Supplementary Fig. 5d). In contrast, the lncRNA HOTAIR was barely associated with polysomes and did not respond to harringtonine. All candidates were more strongly associated with heavy polysomes than HOTAIR and we observed a harringtonine-shift for CRYGC (chimeric-truncated), ATP6V1H (out-of-frame), BCAS1 (normal), and COL28A1 (truncated) indicating their active translation. FARS2 (chimeric-truncated) and DNAH3 (truncated) displayed a weak shift and not across all replicates, thus precluding confident confirmation of their translational capacity. Additional testing of 28 TINAT-exon fusion candidates along fewer polysomal fractions identified nine additional candidates that reacted to harringtonine treatment (Supplementary Fig. 5e). Concurrently, polysome fractionation of untreated colorectal cancer cells14 showed that sporadically expressed transcripts overlapping with TINAT coordinates are preferably associated with heavy polyribosomes (Supplementary Fig. 5f). To test the impact of translated fusion-transcripts on cellular fitness, we overexpressed 11 candidate ORFs (Supplementary Table 3) in NCI-H1299 reporter cells and measured cellular proliferation. Overexpression of CRYGC, ATP6V1H, and FARS2 resulted in decreased cell growth (Fig. 3i) while the others had no effect (data not shown). Together, these observations suggest that TINATs frequently splice into protein-coding exons to create fusion transcripts that become translated into aberration protein isoforms.
DNMTi and HDACi activate TINATs via distinct mechanisms
To investigate the epigenetic reprogramming accompanying TINAT activation, we generated genome-wide maps of DNA methylation and 15 histone modifications before and after treatment (Supplementary Table 1). As expected, DAC treatment reduced global DNA methylation levels (Fig. 4a) while HDACi rapidly increased the acetylation of histone tails at various positions (Fig. 4b and Supplemenetary Fig 6).
Figure 4. DNMTi and HDACi use distinct mechanisms to activate TINATs.
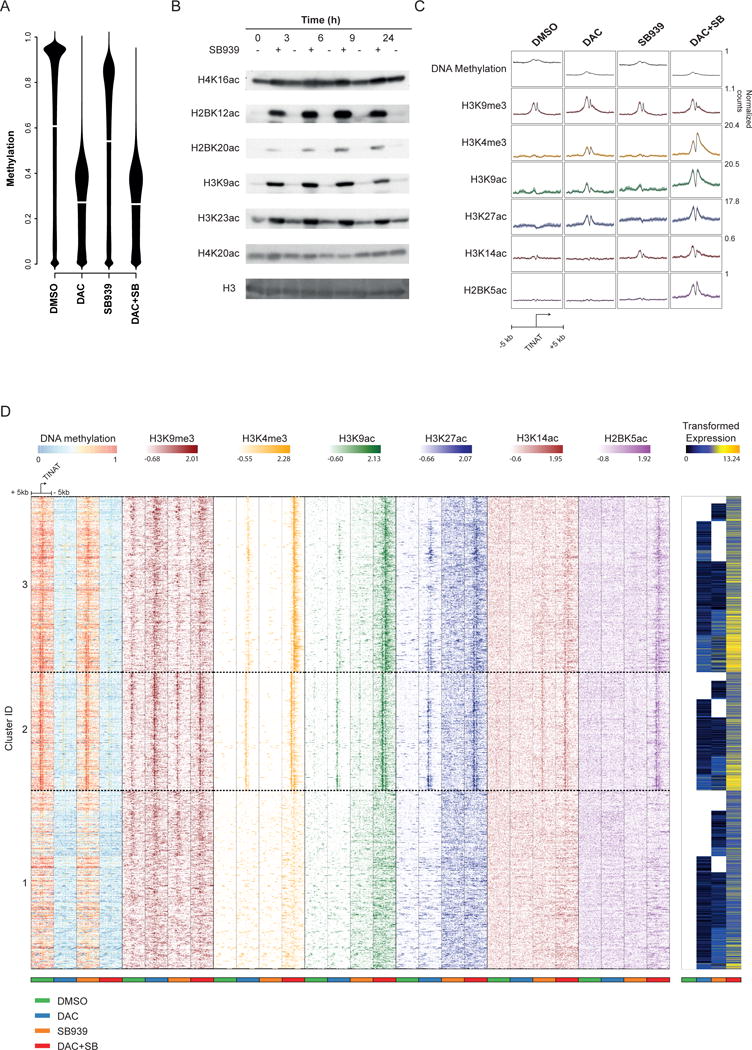
a) Beanplots showing the distribution of DNA methylation in untreated and treated NCI-H1299 cells based on whole-genome bisulfite-sequencing.
b) Western blot analysis of post-translational modifications of histones extracted from NCI-H1299 cells at different time points following treatment with DMSO or SB939. Gel images were cropped and mirrored. Original blots are shown in Supplementary Fig. 6.
c) ChIP-seq occupancy plots showing the average level of DNA methylation (grey), H3K9me3 (red), H3K4me3 (orange), H3K9ac (green), H3K27ac (blue), H3K14ac (brown), and H2BK5ac (purple) 5 kb up- and downstream of all identified TINATs. Colored areas indicate the 95% confidence interval and numbers indicate the normalized read counts.
d) DNA methylation, H3K9me3 (red), H3K4me3 (orange), H3K9ac (green), H3K27ac (blue), H3K14ac (brown), and H2BK5ac (purple) levels around TINATs after DMSO (green bar), DAC (blue bar), SB939 (orange bar), or DAC+SB (red bar) treatment. Color intensity of the histone modifications represents Z-scores. Variance stabilized TINAT expression48 is shown to the right. TINATs were categorized into three groups using k-means clustering on the Z-scores of DNA methylation and histone modification levels relative to DMSO.
In untreated NCI-H1299 reporter cells, TINATs were silenced in association with DNA methylation and H3K9me3 around their TSS (Fig. 4c) but not with H3K27me3 (Supplementary Fig. 7a). Following treatment, DAC or DAC+SB ubiquitously decreased DNA methylation which was partially compensated by increased levels of H3K9me3 (Fig. 4c). Loss in DNA methylation was accompanied with an active promoter signature around TINATs as suggested by the presence of various active histone modifications (Fig. 4c and supplementary Fig. 7a). In stark contrast to DAC or DAC+SB treatment, SB939 or SAHA alone did not induce a classical promoter signature around TINATs as concluded by the lack of demethylation and H3K4me3, H3K9ac, or H3K27ac histone marks (Fig. 4c and Supplementary Fig. 7b). Rather, SB939 induced TINATs in association with H3K14ac, H2AK9ac, and H3K23ac (Fig. 4c and supplementary Fig. 7a). Although these marks were centered on TINATs, their signal intensity was low, potentially reflecting that only a small fraction of cells/alleles in the population responded to HDACi. Of note, most activating histone modifications displayed the strongest mean signal around TINATs after DAC+SB treatment and some modifications, such as H2BK5ac and H3K4ac, were exclusively found after combinatorial inhibition, thereby providing a potential foundation for the synergistic effects of DAC+SB treatment on TINAT expression.
To gain further insights into the interplay of chromatin modulation and TINAT expression, we clustered TINATs based on their surrounding DNA methylation and histone modification profiles. We identified three distinct clusters (Supplementary Fig. 7c) that differed in their respective epigenetic makeup (Fig. 4d and Supplementary Figure 7d). Cluster 1 was devoid of any of the investigated chromatin modifications in the vicinity of TINATs and was characterized by relatively low TINAT expression. TINATs of the second cluster harbored the most focal chromatin modifications that were strongly enriched in proximity to the TSS. Cluster 3 TINATs had lower levels of repressive DNA methylation and H3K9me3 than TINATs of cluster 2 and more widely distributed active chromatin marks after treatment. This chromatin signature was associated with the highest TINAT expression among the clusters. Together, these findings suggest that DNMTi and HDACi drive TINAT expression via distinct mechanisms and that TINATs are associated with a heterogeneous group of TSS classes.
TINATs arise from LTRs of the LTR12 family
Despite DNMTi and HDACi target different epigenetic pathways and are observed to employ different mechanism of TINAT activation, both inhibitor classes converged on activating identical TINATs. We therefore hypothesized that these regions harbor some universal sequence commonality. Since it has been shown that transposable elements (TEs) play significant roles in regulating gene networks through novel promoter, enhancer and splicing mechanisms15,16, we explored whether sequence-specific features of TEs explain TINAT activation.
Indeed, more than 80% of TINATs overlapped with TEs (Fig. 5a) and specifically the LTR class was more frequently associated with TINATs than expected by chance (Fig. 5b, top). For combination treatment, expression levels from LTR-derived TINATs were higher than from other TINATs (Supplementary Fig. 8a). Intriguingly, LTRs belonging to the LTR12 family, whose members are preferentially localized around the promoter region of genes (Fisher′s exact test, P < 2.2 × 10−16; Supplementary Fig. 8b), were strongly enriched for TINATs (Fig. 5b, bottom). Moreover, certain TE families were enriched for the different chromatin clusters identified previously (Supplementary Fig. 8c), suggesting that different epigenetic mechanisms are preferred for activation of certain TE families. Within the LTR12 family, LTR12C had the highest enrichment value (associated with ~50% of all TINATs, Supplementary Fig. 8d and Supplementary Table 4). Analysis of public RNA-seq data from cells treated with SAHA17 confirmed the selective transcriptional activation of LTR12C copies after HDACi (Supplementary Fig. 8e). Moreover, we observed increased LTR12C transcription after SAHA treatment in a neuroblastoma mouse xenograft model (Wilcoxon and Mann-Whitney two-sided test, P = 0.0079; Fig. 5c). Importantly, treatment with several chemotherapeutic agents did not affect LTR12C transcript levels (Supplementary Fig. 8f), suggesting that their induction is a specific effect of epigenetic modulation. Next, we anchored the start positions from TINATs to the LTR12C consensus sequence to identify if any sequence-specific context in LTR12C was contributing to the generation of TINATs (Supplementary Fig. 8g). This analysis uncovered two intriguing results: First, all TSS activity originated from the second half of the sense strand, suggesting that LTR12C encodes unidirectional transcriptional regulation similar to promoter function. Second, there was one major summit around position 1165bp that was activated synergistically by combination treatment which corresponded to the previously identified ERV9 provirus TSS18-20. The broad range of CAGE peaks in LTR12C corresponds to the promoter region of the solitary LTR thus supporting the notion that TINATs derive from cryptic silenced promoters21. This observation is in line with the previously described cases of LTR12C copies that harbor promoter activity (Supplementary Table 5). We therefore predicted that promoter-specific histone modifications increase around expressed LTR12C copies when treated with epigenetic drugs. As expected, we only observed a significant difference in H3K4me3 and H3K9ac around expressed LTR12C but not for LTR12C copies without TINATs after DAC+SB treatment (Supplementary Fig. 8h).
Figure 5. TINATs arise from long-terminal repeats especially of the LTR12 family.
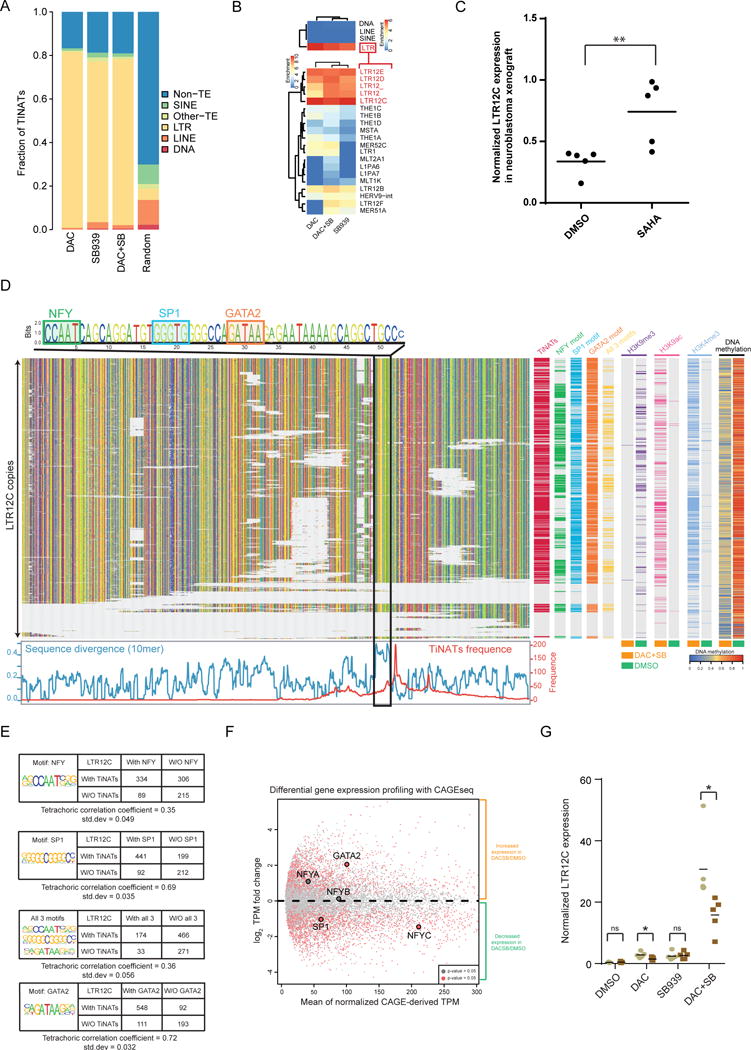
a) TINATs overlapping with transposable element (TE) classes.
b) Cluster analysis of enrichment scores for TINATs across TE classes (top panel) and LTR families (bottom).
c) qRT-PCR expression analysis of LTR12C copies relative to housekeepers in BE(2)-C neuroblastoma cells xenotransplanted into mice treated with DMSO or SAHA. Vertical lines represent the mean.
d) Sequence alignment of LTR12C copies. G, A, T, C, nucleotides are colored by yellow, green, red, and blue, respectively (top left). TF motifs are highlighted. TINAT frequency and sequence divergence between LTR12C copies with and without TINATs is shown below. Right panel displays the presence of TINATs, TF motifs, histone modifications, and DNA methylation.
e) Association between TINAT expression and the presence of NF-Y, SP1 and GATA2 motifs. Enrichment of SP1 and GATA2 sites in LTR12C with TINATs was significant (Pearson’s chi-squared test, P < 2.2e-16).
f) Differential gene expression between DAC+SB and DMSO using CAGE peaks. NF-Y, Sp1, and GATA2 are highlighted. NF-Y transcription factor is a trimeric complex of NYFA, NFYB and NFYC. Genes with significant expression differences are labeled in red (t-test p-value < 0.05).
g) Expression of LTR12C copies relative to housekeepers in the presence (brown) and absence (grey) of siRNAs targeting GATA2. Data from five independent experiments are shown. The mean from five independent experiments is shown.
Next, we asked whether LTR12C elements harbor TF binding sites that provide insight into master TFs that mediate de novo transcription, and whether there are sequence features that discriminate transcribed and non-transcribed LTR12C elements. Multiple-sequence alignment comparison of TINAT-producing and non-TINAT LTR12C copies identified that the most prominent sequence feature that segregated both LTR12C groups mapped immediately upstream of the 1165bp TINAT summit (Fig. 5d). The ERV9/LTR12 U3 enhancer and promoter region harbors several TF binding sites, such as NF-Y, Sp1 and GATA221. We explored whether the presence of these three TF motifs directly upstream of the LTR12C summit correlated with TINAT presence. TINAT expression correlated with the presence of a GATA2 motif (tetrachoric correlation coefficient (cc)= 0.72) and Sp1 motif (cc = 0.69) (Fig. 5e). Then, we checked expression levels of the LTR12C promoter TFs after DAC+SB treatment to explore the potential mechanism for TINAT activation. Using CAGE signal as a surrogate for gene expression, we identified that only GATA2 had significantly higher expression (t-test, P = 0.03) in DAC+SB relative to DMSO (Fig. 5f). These findings suggest GATA2 is likely the upstream TF responsible for TINAT activation and indeed, using siRNAs mediated knockdown, we validated the requirement of GATA2 for full TINAT activation (Fig. 5g and Supplementary Fig. 8i).
Discussion
We show that DNMTi and HDACi do not predominantly alter the expression of canonical genes, but induce the de novo transcription of LTRs of the LTR12 family. Previous efforts to understand the transcriptional response towards epigenetic therapy were largely based on gene expression microarrays and thus, were limited to the quantification of known transcripts and lacked information about their TSSs22,23. Our findings extend recent reports that demonstrated the presence of dsRNA molecules upon DNMTi4,5, originating from the bidirectional transcription of codogenic ERV envelope gene loci (e.g. Syncytin-1 and env-Fc2). While we confirmed Syncytin-1 expression and the subsequent induction of AIMs6 upon DNMTi, our data has important new implications. First, we show that HDAC inhibitors must exert their function independently. Second, the unidirectional transcription from up to thousands of solitary LTRs is an additional effect to the bidirectional transcription from full-length ERV copies following treatment. Therefore, we provide a novel mechanism for the action of different classes of epigenetic inhibitors.
With the exception of Syncytin-1 and few other codogenic ERVs that produce functional proteins24, most ERV genes became non-functional by different evolutionary forces25. Most of the ~700,000 ERV copies within the human genome exist as solitary LTRs26. Unlike other ERVs, the ~5500 LTRs of the ERV9 family (LTR12s) carry several tandem repeats containing multiple TF binding sites21,27. LTRs of this family have been shown to shape the transcriptomic landscape through enhancer-like and promoter-like mechanisms28,29, which have been adopted for tissue-specific functions18,19. Our data suggest that either the loss of DNA methylation or HDAC inhibition is sufficient to drive faint expression of especially LTR12C elements, but combinatorial inhibition is required for full activation. The loss in DNA methylation upon DNMTi is global and also occurs at LTR12Cs and other subfamilies that do not show a transcriptional response. We therefore propose that the selectivity of LTR expression is conferred by the disruption of repressive chromatin structure followed by binding of TFs to the regulatory sequence of exposed LTR elements. In line with the reported recruitment of GATA2 to LTR12 elements30, we show that GATA2 is required for full LTR12C expression. The selectivity of HDACi towards the activation of LTR12 family elements was also reported for multiple other cancer types based on candidate gene approaches31,32, indicating that this is a universal mechanism. However, non-epigenetic treatment examples of EVR9/LTR12 reactivation have been discovered in viral-induced tumors20 and in primary T cells infected with HIV33.
Splicing of ERV-derived transcripts into their genomic vicinity has been observed during normal development16 and in tumors20,34. There are reports from studies in mice or human cancer cells where a LTR element gives rise to a chimeric protein by virtue of being spliced to a protein-coding gene34,35. In line with these reports, we show that the treatment-induced expression of LTRs generates numerous fusion-transcripts that encode novel protein isoforms, often lacking N-terminal peptide sequences important for proper protein function. Given that truncated protein isoforms affect cellular function and contribute to human disease36,37, one expects that the simultaneous expression of aberrant peptides partially accounts to the clinical efficacy of these drugs.
So far, there are two major limitations to epigenetic therapy that could potentially be overcome by combining it with immunotherapy38. First, efficacy of DNMTi in different tumor entities is still quite limited and second, despite promising initial results in lung cancer39, no phase III randomized trial has yet demonstrated therapeutic synergism between DNMTi and HDACi. The combination of epigenetic inhibitors with immunotherapy raises the hope that epigenetic therapy will demonstrate an antineoplastic effect also in frequent cancer entities. Indeed, in preclinical cancer models, treatment with DNMTi or HDACi sensitizes tumors to the effects of immune checkpoint inhibition4,40. Moreover, combining DNMTi with allogeneic T-cell infusions in the treatment of relapsed AML patients41 indicates a curative potential42. Our data provide an elegant explanation for this priming effect, as epigenetic therapy may induce the expression of LTR-derived immunogenic antigens presented on MHC class I molecules for recognition by cytolytic T-cells. This would be of upmost importance for those cancer types with low mutational burden that respond poorly to immune therapy43. The here described mechanism likely synergizes with other effects of epigenetic therapy, including the inhibition of NMD44, transcription of viral defense genes4, increased antigen processing and presentation45, re-expression of epigenetically silenced inflammatory chemokines46, and up-regulation of CTAs47. Future proteomic approaches combined with T-cell cytotoxicity assays will further shed light on the interaction between epigenetic and immune therapy and the role of ERV-derived antigen presentation.
Methods
Methods, including statements of data availability and any associated accession codes and references, are available in the online version of the paper.
Online Methods
Engineering of the DAPK1 reporter cell line, 5′RACE, and the epigenetic compound screen are described in detail in Supplementary Note 1 and Supplementary Tables 6-9.
Cell culture and treatment
RAJI (ACC-319, DSMZ), MEC1 (ACC-497, DSMZ), HL60 (ACC-3, DSMZ), K562 (ACC-10, DSMZ), NCI-H1299 (CRL-5803, ATCC) cells were grown in RPMI 1640 supplemented with 10% FCS. T89G human glioblastoma cells (CRL-1690, ATCC) were kept in DMEM containing 10% FCS. Cell line authenticity and purity was confirmed using the Multiplex Cell Authentication and Cell Contamination Test by Multiplexion. Cells were treated with 500 nM (250 nM for HL60) DAC, 500 nM SB939, 1500 nM SAHA, or 500 nM (250 nM for HL60) DAC + 500 nM SB939 for 72, 18, 18, or 72+18 h, respectively, and compound-containing media was refreshed every 24 h.
Cap analysis of gene expression (CAGE) sequencing
CAGE was performed in two independent experiments on normal and treated NCI-H1299 cells using the CAGE™ Preparation Kit from DNAFORM.jp according to the manufacturer’s instructions. Enrichment of capped RNAs versus uncapped ribosomal transcripts was used to assess sample quality. Samples with a minimum of 400-fold enrichment over ribosomal RNA were subjected to sequencing on the Illumina Hi-Seq 2000 system in 50 bp single-end (replicate 1) and 100 bp paired-end (replicate 2) mode by the DKFZ Genomics and Proteomics Core facility. Resulting raw sequencing data was processed as follows: Multiplexed samples were separated by barcode, trimmed at the first position to remove non-specific guanines53 as well as to 50 bps in the case of the 100 bp paired-end reads, and aligned against the reference genome (hg19) using HISAT54 version 0.1.6-beta. Only uniquely mapped reads were retained and in the case of SB939 and DAC+SB939, files were down-sampled to 25×106 aligned reads. The resulting BAM files were loaded into CAGEr version 1.10.055 and CTSS were called using the following parameters (sequencingQualityThreshold = 20, mappingQualityThreshold = 20). After simple tpm normalization, clusterCTSS were generated using the paraclu method (threshold = 0.1, nrPassThreshold=2, thresholdIsTpm = TRUE, removeSingletons = TRUE, keepSingletonsAbove = 0.2, minStability=2, maxLength=100, reduceToNonoverlapping = TRUE). Finally, consensus TSSs across all conditions and replicates were created using the aggregateTagClusters function (tpmThreshold = 0.3, qLow = NULL, qUp = NULL, maxDist = 100, excludeSignalBelowThreshold=FALSE). Importantly, no confounding effects of the underlying sequencing protocol on TSS expression were observed (Supplementary Fig. 3a). Distance to the nearest Gencode GRCh37.p13 annotated TSS was calculated using HOMER50 software and statistical analysis was performed in DESEQ version (1.18.0)48. Size factors were calculated for the normalization of TSS expression and dispersion estimates for each gene were obtained using the estimateDispersions function with the following parameters (method=”per-condition”, sharingMode=”maximum”). Differential expression between control and DAC, SB939, SAHA, and DAC+SB treated cells was assessed by testing the differences between the base means of two conditions (nbinomTest). Benjamini-Hochberg adjusted q-values below 0.05 were considered as significantly differentially expressed.
RNA sequencing analysis
RNA-seq data was obtained from the Gene Expression Omnibus under accession GSE54912 and from the European Nucleotide Archive under accession PRJEB5049. Illumina and ABI_SOLID reads were aligned against the human hg19 reference genome using HISAT version 0.1.6.-beta with default parameters and bowtie version 1.0.0 with the following parameters (-C, –best), respectively. Overlap of aligned reads with TE subfamilies was counted using the summarizeOverlaps function of the GenomicAlignments R/Bioconductor package56 with default parameters. Read counts were normalized in edgeR57 using the total number of uniquely mapped reads as library size. After estimation of the dispersion, statistical significance was assessed by genewise exact tests for differences in the means between two groups of negative-binomially distributed counts.
Chromatin-immunoprecipitation (ChIP)
About 2×107 NCI-H1299 cells were cross-linked for 10 min using FCS-free RPMI 1640 containing 1.1 % formaldehyde. After cross-linking, 1/20th volume of 2.5 M glycine was added and incubated for 10 min to quench the cross-linking reaction. Cells were then washed three times with ice-cold PBS and scraped into a pre-chilled 15 ml polystyrene tube for subsequent centrifugation at 1000 g at 4 °C. Cell pellets were carefully resuspended in 1 ml Lysisbuffer 1 (LB1: 50 mM HEPES-KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 10 % glycerol, 0.5 % NP-40, 0.25 % Triton X-100) supplemented with protease inhibitors (1 tablet for 50 ml). Next, resuspended cells were incubated for 10 min at 4 °C on a rocker to permeabilize the cell membrane. After incubation, nuclei were centrifuged at 1000 g at 4° C and the supernatant was discarded. Hereafter, cells were washed in 1 ml cold Lysisbuffer 2 (LB2: 10 mM HEPES-KOH pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA). Finally, nuclei were washed twice in cold Lysisbuffer 3 (LB3: 10 mM HEPES-KOH pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1 % sodium deoxycholate, 0.5 % sodium lauroyl sarcosine) and then resuspended in 500-1000 μl LB3. Sonication of chromatin was performed at 4 °C in 12×24 mm glass tubes using the Covaris S220 Focused-ultrasonicator with the following settings: 30 min shearing, Duty Cycle 20%, Intensity 5, 200 Cycles per burst. Typically, this program resulted in fragment sizes between 150 bp and 450 bp. After shearing, cellular debris was removed by centrifugation at 16,000 g for 5 min and the supernatant was aliquoted and stored at −80 °C. To assure sufficient shearing efficiency, a small fraction of each sample was digested with Proteinase K at 65 °C for 16 h and thereafter RNase-treated and QIAquick Gel column-purified. The concentration of purified DNA was assessed by Nanodrop measurement and gel-analyzed to analyze fragment size distribution. Only samples with average fragment sizes of 150-300 bp were subjected to further chromatin immunoprecipitation.
ChIP-Assays were performed using the SX-8G IP-Star® Automated System in combination with the Auto ChIP kit according the manufacturers protocol (both Diagenode). IP reaction was carried out for 11 h using DiaMag protein A-coated magnetic beads (Diagenode) and the following antibodies: H3K4me3 (pAb-003-050, Diagenode); H3K27me3 (pAB-069-050, Diagenode); H3K9ac (17-658, Merck Millipore); H3K9me3 (ab8898, Abcam); H3K27ac (ab4729, Abcam); H3K4me1 (ab8895, Abcam); H3K36me3 (ab9050, Abcam); H3K23ac (39131, Active Motif); H4K8ac (61103, Active Motif); H3K14ac (ab52946, Abcam); H3K18ac (ab1191, Abcam); H4K12ac (ab46983, Abcam); H3K4ac (39381, Active Motif); H2AK9ac (ab177312, Abcam); H2BK5ac (ab40886, Abcam). After ChIP, DNA was isolated by Proteinase K digest at 65°C for 4 h and subsequent purification using Agencourt AMPure XP beads. For Chip-Seq analysis, size-selected libraries were prepared with the NEB Next Ultra DNA Library Kit. Sequencing was performed on the Illumina Hi-Seq 2000 system in 50 bp single-end mode by the DKFZ Genomics and Proteomics Core facility.
Reads were aligned against the human reference genome (hg19) using BWA version 0.5.9-r1658 with default parameters and reads with a mapping quality less than 1 or putative PCR duplicates were removed. MACS259 was used with default parameters to call peaks at a 1% FDR. Input-subtracted, whole-genome coverage tracks (bigWig files) of aligned reads were generated with a window size of 50 bps. To account for global differences in activating histone modifications after treatment (Fig. 4b), H3K4me3, H3K9ac, and H3K27ac tracks were multiplied by the multiplicative inverse of the mean signal intensity within 1.25 kp up- and downstream of the 100 TSSs with the lowest variance across treatments. The signal intensities of the other histone modifications were normalized to all aligned reads (RPM, reads per million) and used for down-stream analyses.
Whole-genome bisulfite sequencing (WGBS)
Whole-genome bisulfite sequencing of treated and untreated NCI-H1299 cells was performed as previously described60. Libraries were sequenced on the Hi-Seq 2000 system in 100 bp paired-end mode. CpG methylation was calculated as previously described61 and the BSmooth algorithm62 was employed to estimate the sample-wise methylation levels using the bsseq R/Bioconductor package with default parameters.
siRNA transfection and shRNA transduction
siRNA transfection of cultured cell lines was carried out using DharmaFECT 1 (Thermo Scientific) according to the manufacturers recommendations. In brief, cells were transfected using 1 μl transfection reagent per 0.02 pmol siRNA and all siRNAs (Dharmacon, siGenome series; DNMT1 – D-004605_1, _2, _4, _5; GATA2 – MU-009024-00 siRNA) were used as a pool of four individual sequences at a combined final concentration of 20 nM for DNMT1 and 10 nM for GATA2 knockdown. As a control, Dharmacon ON-Targetplus non-targeting siRNA #1 was used. In parallel to the siRNA-mediated GATA2 knockdown, epigenetic drug treatment was done as described above. Cells were harvested 96h post transfection and used for downstream analyses. DNMT1 and non-targeting (shLuciferase) shRNAs were cloned into the pRSI9 vector system (Cellecta) and lentiviral particles were produced in HEK293T cells using psPAX2 and pMD2.G packaging vectors. Sequences used for shRNA cloning can be found in Supplementary Table 10. 24h after transduction, transduced cells were enriched by treatment with 2μg/ml puromycin for 48h.
Comparison to FANTOM5 data
FANTOM5 CAGE-TSS expression across 625 tissues and primary cells was obtained using the hg19.cage_peak_phase1and2combined_tpm_ann.osc.txt file provided in the FANTOM5 website (http://fantom.gsc.riken.jp/5/datafiles/latest/extra/CAGE_peaks/). Cell lines, universal references, and cancer samples were excluded for this analysis. A TINAT was considered expressed in a given cell type if the sample contained an active TSS (tags per million > 0) with a distance of less than 150 bps to the nearest TINAT.
HLA-binding prediction
Immunogenic peptides were predicted for DAC+SB induced chimeric and out-of-frame protein isoforms by defining all novel amino acid 8 to 11mers and modeling the binding affinity to various high-frequency HLA alleles using NetMHCpan (v2.8)52. For each novel protein, the kmer with the strongest binding affinity for a given HLA allele was selected.
In vitro transcription and translation
TINATs were in vitro translated by using the TNT Coupled Reticulocyte Lysate System (Promega) with T7 Polymerase according to the manufacturer’s instructions. In brief, cDNA of DAC+SB treated NCI-H1299 cells was used as a template in PCRs with primers amplifying full-length mRNA- or TINAT-sequences (see Supplementary Table 11). A T7-promoter sequence was introduced by re-amplification of the purified PCR products with the same reverse primers and forward primers harboring an extended T7-promoter sequence at the 5′-end. PCR fragments were incubated with the Quick Coupled Transcription/Translation System (Promega) in the presence of [35S]-methionine.
Effect of TINATs ORFs on cell viability
Selected TINAT ORFs based on their potential capability to encode novel, so far not described proteins were synthesized and cloned in vector pMK-RQ (Thermo Fisher Scientific) (see Supplementary Table 3 for further information). The ORFs were then Gateway-shuttled (Thermo Fisher Scientific) into the lentiviral vector rwpTRIPZ, a Gateway compatible derivative of pTRIPZ (GE Healthcare) which allows doxycycline induction of the cloned gene. Lentiviral particles containing the diverse recombinant rwpTRIPZ constructs were generated in HEK293T cells using a second-generation packaging system. Particles were then transfected into H1299 reporter cells followed by puromycin selection. The expression of TINAT ORFs was induced in stable transfectants by addition of doxycycline (1 μg/ml final concentration) into the growth medium (RPMI 1640, Pan Biotech). Proper induction was monitored by qRT-PCR.
For in vitro proliferation assays, stably ORFs-overexpressing H1299 cells were plated into 96 well plates in technical triplicates at a number of 5 × 103 cells per well in a final volume of 100 μl complete RPMI with/without Doxycyclin. Cell proliferation was analyzed in technical triplicates 24, 48, and 72 h after induction with the Cell Titer-Blue Cell Viability Assay (Promega, cat. no. G8081) as described in the manual using Spectramax M5e (Molecular Devices) for the read-out.
Mouse xenograft studies with HDAC inhibitor
2 × 106 BE(2)-C viable neuroblastoma cells were resuspended in 100 μl Matrigel and 20 U/ml heparin and implanted into the subcutaneous tissue of right flank of 5- to 6-week old female athymic nude mice (HsdCpb: NMRI-Foxn1nu). Mice were randomly assigned to groups of five individuals bearing similarly sized tumors without blinding. Group size was estimated by the DKFZ biometry core facility. HDAC inhibitor SAHA was dissolved in 100% DMSO and given by intraperitoneal injection at a concentration of 150 mg/kg per day for 2 × 5 days. At explantation, tumor material used for isolation of total RNA was shock frozen in liquid nitrogen immediately after removal and stored at −80 °C. Total RNA was isolated with the RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. All animal studies were approved by the German Cancer Research Center (DKFZ) institutional animal care and use committee and the Regional Administrative Council Karlsruhe, Germany. All experiments were in accordance with the relevant regulatory standards.
qRT-PCR expression analysis
RNA was transcribed to cDNA using random hexamers and the Superscript™ III Reverse Transcriptase (Invitrogen) according to the manufacturer’s instructions. Unless stated otherwise, expression analysis was performed on the Roche Lightcycler® 480 system and target gene expression was normalized to the housekeeping genes GAPDH, β-Actin, and HPRT1 (for primer sequences see Supplementary Table 11).
Western blot
Total protein or histone extracts were isolated followed by electrophoretic separation and transfered to a polyvinylidene fluoride membrane. Antibodies against the following antigens were applied: Pan-Ac H3 (06-599, Millipore), DNMT1 (D63A6, Cell Signaling Technology), β-Actin-HRP conjugated (sc-47778, Santa Cruz Biotechnology), H3K9ac (17-658, Merck Millipore); H3K23ac (39131, Active Motif); H2BK20ac (ab52988, Abcam); H4K16ac (39167, Active Motif); H2BK12ac (ab40883, Abcam), H4K20ac (61531, Active Motif).
Analysis of transposable elements
The TINAT TE enrichment was computed based on Xie et al.15. Briefly, the enrichment score is the ratio between the observed and the expected number of transposable elements overlapping TINATs assuming a genome-wide random distribution model. TINAT start positions in each LTR12C copy were aligned to relative locations on the LTR12C consensus sequence and the LTR12C TSS frequency was defined as the accumulated density. De novo motif analysis was performed using HOMER50 on 640 LTR12Cs that fulfilled the following two criteria: 1) No CAGE signal (CTSS tags) in DMSO control and 2) TINAT expression in both CAGE-seq replicates after DAC+SB treatment. 304 LTR12C copies without any CAGE-seq signal (CTSS tags) before and after treatment were used a background. EMBOSS Needle tool63 was used to calculate the pairwise alignment between each LTR12C copy and the consensus sequence. The frequency of conserved 10mer DNA sequence in both LTR12C groups was calculated and the sequence divergence was defined as the difference of 10mer sequence frequency between both groups.
EpiTYPER MassARRAY quantitative DNA methylation analysis
MassARRAY was used for high-resolution DNA methylation analysis as previously reported64. For PCR amplification of target regions, tagged primers specific for bisulfite converted DNA were designed with the EpiDesigner Software (http://www.epidesigner.com/) and are listed in the Supplementary Table 11.
Transcript assembly and in silico translation
For TINAT transcript assembly, only properly-paired mates where the first in pair read originated from a TINAT were used as input for Stringtie version 1.0.1 (-g 150)9. Only the longest isoform per TINAT that overlapped with at least one exon of an annotated gene (Gencode v19) was used for subsequent in silico translation. In case a TINAT gave rise to multiple isoforms with the same length, the isoform with the highest coverage was used (Supplementary Table 2). To discriminate the main protein-coding from upstream open reading frames (ORFs) that are present in about 50 % of all human mRNAs65, only the first ORF that initiates from a strong ATG start codon (>80% sequence similarity to the Kozak consensus sequence66) and encodes >30 codons67 was considered. Prior to this, ATGs with a cap-to-ORF distance greater than 721 bps (95th percentile of the length of all human 5′ untranslated regions)68 were removed. The resulting translation products were aligned against the RefSeq (GRCh37.75) protein sequence of the corresponding splicing-acceptor gene using the Smith-Waterman algorithm69. Transcripts with no alignment for any isoform were classified as out-of-frame while transcripts with alignment for at least one isoform were denoted as in-frame. In-frame peptides were further classified into normal, chimeric-normal, truncated, or chimeric-truncated based on the following criteria: Normal; ORF peptide and RefSeq align perfectly. Chimeric-normal; the ORF encodes novel in-frame N-terminal amino acids followed by the full-length canonical RefSeq protein sequence. Truncated; the ORF lacks parts of the canonical N-terminal protein sequence. Chimeric-truncated; the ORF encodes novel in place of canonical N-terminal amino acids followed by the native peptide (Fig 3e). If the classification was ambiguous for different protein isoforms of the same gene, the hierarchically highest state (in the order: normal > truncated > chimeric-normal > chimeric-truncated) was used to assign a final state for the affected protein.
Polysome fractionation
Sucrose density gradients were produced by consecutively adding layers (790 μl/layer) of decreasing sucrose concentrations (50%, 41.9%, 33.8%, 25.6% and 17.5% in polysome buffer) into a Beckman Centrifuge Tube (11 × 60 mm). After each step, the tubes were frozen at −80°C. On the day before the experiment, tubes were slowly thawed overnight at 4°C. Harringtonine (10 μg/ml) was added to DAC+SB treated cells for 15 min at 37°C to deplete elongating ribosomes from mRNA molecules. Cells were washed in ice-cold PBS containing 100 μg/ml cycloheximide and lysed in 200 μl Polysome lysis buffer (15 mM Tris-HCl pH 7.4, 15 mM MgCl2, 300 mM NaCl, 100 μg/ml cycloheximide, 1% Triton-X-100, 0.1% β-mercaptoethanol, 200 U/ml RNAsin (Promega), 1 complete Mini Protease Inhibitor Tablet (Roche) per 10 ml). Nuclei were removed by centrifugation (9300 × g, 4°C, 10 min) and the cytoplasmic lysate was loaded onto a sucrose density gradient (17.5–50% in 15 mM Tris-HCl pH 7.4, 15 mM MgCl2, 300 mM NaCl). After ultracentrifugation (2.5 h, 35 000 rpm at 4°C in a SW60Ti rotor), gradients were eluted with a Teledyne Isco Foxy Jr. system into 14 fractions of similar volume. A rabbit HBB2 in vitro transcript was added to each fraction as a spike-in control (25 fmol/fraction) (Supplementary Table 11) and RNA was purified by phenol chloroform extraction and analyzed via qPCR. To assess RNA quality and equal purification efficiency across all fractions, the HBB2 in vitro transcript and endogenous Ncl mRNA were detected by Northern blotting.
Transcriptional directionality
Transcriptional directionality was calculated as previously described8 with modifications. The sum of CAGE tags mapping to the forward (Expf) or reverse (Expr) strand within ± 700 bps from the center position of TINAT or enhancer coordinates was used to calculate the directionality score (Expf − Expr)/(Expf + Expr). Ubiquitous cell line enhancer coordinates8 were used as a reference.
Statistical analysis
All statistical analyses were performed using the R statistical environment. Box plot center lines indicate data medians, box limits indicate the 25th and 75th percentiles, whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles, and outliers are shown by individual points. For group-wise comparison of two distributions from different samples/treatments, the two-tailed non-parametric Wilcoxon and Mann-Whitney test was used. For experimental settings with replicates of paired treatments/samples, a two-tailed student’s t test was applied. P values < 0.05 were considered statistically significant and significance levels are depicted as follows: * = p<0.05; ** = p<0.01; *** = p<0.001.
Code availability
Scripts are available upon request.
Data availability
CAGE, ChIP, and WGB-sequencing data generated in this study have been deposited in the NCBI Gene Expression Omnibus (GEO) under accession GSE81322.
Supplementary Material
Acknowledgments
We thank Christoph Weigel, Reka Toth, and Kathrin Chiappinelli for helpful feedback and discussions and Amos Tanay for critical proof-reading of the manuscript. We would also like to thank Karin Bauer, Kathrin Klimo, Marion Bähr and the Genomics and Proteomics Core Facility at the German Cancer Research Center for their excellent technical support and expertise. D.B. is supported by the German-Israeli Helmholtz Research School in Cancer Biology. D.L., J.L., H.S.J., B.Z., and T.W. are supported by the American Cancer Society grant RSG-14-049-01-DMC, and NIH grants R01HG007354, R01HG007175, R01ES024992, U01CA200060, and U24ES 026699. This work was supported in part by the Helmholtz Association, the DFG funded Priority Program SPP1463, and BMBF funded CancerEpiSys program and the “ICGC Data-Mining” project.
Footnotes
Author Contributions
C.S., M.D., D.B., D.B.L., S.L., M.S.I., H.B., S.H., M.H., A.L., A.R., G.S., J.S., R.W., J.P.M., K.R., D.W., C.C.O., and C.P. designed the experiments and performed experimental work. D.B., C.S., M.D., D.L., J.L., H.S.J., N.M.S., Y.H., B.Z., Y.A., C.D.I., B.B., and T.W. performed data analysis. I.O., O.W., and M.L. provided clinical expertise and data. D.B., M.D., T.W., and C.P. prepared the manuscript and figures. T.W., C.G., B.B., M.E., C.C.O., and C.P. provided project leadership. All authors contributed to the final manuscript.
Competing Financial Interest
The authors declare no competing financial interests.
References
- 1.Navada SC, Steinmann J, Lubbert M, Silverman LR. Clinical development of demethylating agents in hematology. The Journal of clinical investigation. 2014;124:40–46. doi: 10.1172/JCI69739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. The Journal of clinical investigation. 2014;124:30–39. doi: 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones PA. At the tipping point for epigenetic therapies in cancer. The Journal of clinical investigation. 2014;124:14–16. doi: 10.1172/JCI74145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chiappinelli KB, et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell. 2015;162:974–986. doi: 10.1016/j.cell.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roulois D, et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell. 2015;162:961–973. doi: 10.1016/j.cell.2015.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li H, et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget. 2014;5:587–598. doi: 10.18632/oncotarget.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nature genetics. 1999;21:103–107. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- 8.Andersson R, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507:455–461. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pertea M, et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature biotechnology. 2015;33:290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iyer MK, et al. The landscape of long noncoding RNAs in the human transcriptome. Nature genetics. 2015;47:199–208. doi: 10.1038/ng.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prensner JR, et al. The long noncoding RNA SChLAP1 promotes aggressive prostate cancer and antagonizes the SWI/SNF complex. Nature genetics. 2013;45:1392–1398. doi: 10.1038/ng.2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Almeida LG, et al. CTdatabase: a knowledge-base of high-throughput and curated data on cancer-testis antigens. Nucleic acids research. 2009;37:D816–819. doi: 10.1093/nar/gkn673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Heesch S, et al. Extensive localization of long noncoding RNAs to the cytosol and mono-and polyribosomal complexes. Genome biology. 2014;15:R6. doi: 10.1186/gb-2014-15-1-r6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie M, et al. DNA hypomethylation within specific transposable element families associates with tissue-specific enhancer landscape. Nature genetics. 2013;45:836–841. doi: 10.1038/ng.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goke J, et al. Dynamic transcription of distinct classes of endogenous retroviral elements marks specific populations of early human embryonic cells. Cell stem cell. 2015;16:135–141. doi: 10.1016/j.stem.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 17.Rafehi H, et al. Vascular histone deacetylation by pharmacological HDAC inhibition. Genome research. 2014;24:1271–1284. doi: 10.1101/gr.168781.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cohen CJ, Lock WM, Mager DL. Endogenous retroviral LTRs as promoters for human genes: a critical assessment. Gene. 2009;448:105–114. doi: 10.1016/j.gene.2009.06.020. [DOI] [PubMed] [Google Scholar]
- 19.Sokol M, Jessen KM, Pedersen FS. Human endogenous retroviruses sustain complex and cooperative regulation of gene-containing loci and unannotated megabase-sized regions. Retrovirology. 2015;12:32. doi: 10.1186/s12977-015-0161-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hashimoto K, et al. CAGE profiling of ncRNAs in hepatocellular carcinoma reveals widespread activation of retroviral LTR promoters in virus-induced tumors. Genome research. 2015 doi: 10.1101/gr.191031.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu XP, et al. The long terminal repeat (LTR) of ERV-9 human endogenous retrovirus binds to NF-Y in the assembly of an active LTR enhancer complex NF-Y/MZF1/GATA-2. J Biol Chem. 2005;280:35184–35194. doi: 10.1074/jbc.M508138200. [DOI] [PubMed] [Google Scholar]
- 22.New M, Olzscha H, La Thangue NB. HDAC inhibitor-based therapies: can we interpret the code? Molecular oncology. 2012;6:637–656. doi: 10.1016/j.molonc.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klco JM, et al. Genomic impact of transient low-dose decitabine treatment on primary AML cells. Blood. 2013;121:1633–1643. doi: 10.1182/blood-2012-09-459313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Parseval N, Lazar V, Casella JF, Benit L, Heidmann T. Survey of human genes of retroviral origin: identification and transcriptome of the genes with coding capacity for complete envelope proteins. Journal of virology. 2003;77:10414–10422. doi: 10.1128/JVI.77.19.10414-10422.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Katoh I, Kurata S. Association of endogenous retroviruses and long terminal repeats with human disorders. Frontiers in oncology. 2013;3:234. doi: 10.3389/fonc.2013.00234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 27.La Mantia G, et al. Identification of regulatory elements within the minimal promoter region of the human endogenous ERV9 proviruses: accurate transcription initiation is controlled by an Inr-like element. Nucleic acids research. 1992;20:4129–4136. doi: 10.1093/nar/20.16.4129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lania L, et al. Structural and functional organization of the human endogenous retroviral ERV9 sequences. Virology. 1992;191:464–468. doi: 10.1016/0042-6822(92)90211-7. [DOI] [PubMed] [Google Scholar]
- 29.Ling J, et al. The solitary long terminal repeats of ERV-9 endogenous retrovirus are conserved during primate evolution and possess enhancer activities in embryonic and hematopoietic cells. Journal of virology. 2002;76:2410–2423. doi: 10.1128/jvi.76.5.2410-2423.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pi W, et al. Long-range function of an intergenic retrotransposon. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:12992–12997. doi: 10.1073/pnas.1004139107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kronung SK, et al. LTR12 promoter activation in a broad range of human tumor cells by HDAC inhibition. Oncotarget. 2016;7:33484–33497. doi: 10.18632/oncotarget.9255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beyer U, Kronung SK, Leha A, Walter L, Dobbelstein M. Comprehensive identification of genes driven by ERV9-LTRs reveals TNFRSF10B as a re-activatable mediator of testicular cancer cell death. Cell death and differentiation. 2016;23:64–75. doi: 10.1038/cdd.2015.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sherrill-Mix S, Ocwieja KE, Bushman FD. Gene activity in primary T cells infected with HIV89.6: intron retention and induction of genomic repeats. Retrovirology. 2015;12:79. doi: 10.1186/s12977-015-0205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lock FE, et al. Distinct isoform of FABP7 revealed by screening for retroelement-activated genes in diffuse large B-cell lymphoma. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E3534–3543. doi: 10.1073/pnas.1405507111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mak KS, et al. Repression of chimeric transcripts emanating from endogenous retrotransposons by a sequence-specific transcription factor. Genome biology. 2014;15:R58. doi: 10.1186/gb-2014-15-4-r58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wiesner T, et al. Alternative transcription initiation leads to expression of a novel ALK isoform in cancer. Nature. 2015;526:453–457. doi: 10.1038/nature15258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vizoso M, et al. Epigenetic activation of a cryptic TBC1D16 transcript enhances melanoma progression by targeting EGFR. Nature medicine. 2015;21:741–750. doi: 10.1038/nm.3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chiappinelli KB, Zahnow CA, Ahuja N, Baylin SB. Combining Epigenetic and Immunotherapy to Combat Cancer. Cancer research. 2016 doi: 10.1158/0008-5472.CAN-15-2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Juergens RA, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer discovery. 2011;1:598–607. doi: 10.1158/2159-8290.CD-11-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim K, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:11774–11779. doi: 10.1073/pnas.1410626111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schroeder T, et al. Azacitidine and donor lymphocyte infusions as first salvage therapy for relapse of AML or MDS after allogeneic stem cell transplantation. Leukemia. 2013;27:1229–1235. doi: 10.1038/leu.2013.7. [DOI] [PubMed] [Google Scholar]
- 42.Steinmann J, et al. 5-Azacytidine and DLI can induce long-term remissions in AML patients relapsed after allograft. Bone marrow transplantation. 2015;50:690–695. doi: 10.1038/bmt.2015.10. [DOI] [PubMed] [Google Scholar]
- 43.Rizvi NA, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bhuvanagiri M, et al. 5-azacytidine inhibits nonsense-mediated decay in a MYC-dependent fashion. EMBO molecular medicine. 2014;6:1593–1609. doi: 10.15252/emmm.201404461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Setiadi AF, et al. Epigenetic enhancement of antigen processing and presentation promotes immune recognition of tumors. Cancer research. 2008;68:9601–9607. doi: 10.1158/0008-5472.CAN-07-5270. [DOI] [PubMed] [Google Scholar]
- 46.Peng D, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527:249–253. doi: 10.1038/nature15520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Almstedt M, et al. The DNA demethylating agent 5-aza-2′-deoxycytidine induces expression of NY-ESO-1 and other cancer/testis antigens in myeloid leukemia cells. Leukemia research. 2010;34:899–905. doi: 10.1016/j.leukres.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 48.Anders S, Huber W. Differential expression analysis for sequence count data. Genome biology. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao M, Kim P, Mitra R, Zhao J, Zhao Z. TSGene 2.0: an updated literature-based knowledgebase for tumor suppressor genes. Nucleic acids research. 2015 doi: 10.1093/nar/gkv1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kong L, et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic acids research. 2007;35:W345–349. doi: 10.1093/nar/gkm391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nielsen M, et al. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PloS one. 2007;2:e796. doi: 10.1371/journal.pone.0000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao X, Valen E, Parker BJ, Sandelin A. Systematic clustering of transcription start site landscapes. PloS one. 2011;6:e23409. doi: 10.1371/journal.pone.0023409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nature methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haberle V, Forrest AR, Hayashizaki Y, Carninci P, Lenhard B. CAGEr: precise TSS data retrieval and high-resolution promoterome mining for integrative analyses. Nucleic acids research. 2015;43:e51. doi: 10.1093/nar/gkv054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lawrence M, et al. Software for computing and annotating genomic ranges. PLoS computational biology. 2013;9:e1003118. doi: 10.1371/journal.pcbi.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Y, et al. Model-based analysis of ChIP-Seq (MACS) Genome biology. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lister R, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang Q, et al. Tagmentation-based whole-genome bisulfite sequencing. Nature protocols. 2013;8:2022–2032. doi: 10.1038/nprot.2013.118. [DOI] [PubMed] [Google Scholar]
- 62.Hansen KD, Langmead B, Irizarry RA. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome biology. 2012;13:R83. doi: 10.1186/gb-2012-13-10-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends in genetics: TIG. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 64.Claus R, et al. Quantitative analyses of DAPK1 methylation in AML and MDS. International journal of cancer Journal international du cancer. 2012;131:E138–142. doi: 10.1002/ijc.26429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Calvo SE, Pagliarini DJ, Mootha VK. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:7507–7512. doi: 10.1073/pnas.0810916106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kozak M. An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic acids research. 1987;15:8125–8148. doi: 10.1093/nar/15.20.8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nature reviews Molecular cell biology. 2010;11:113–127. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Grillo G, et al. UTRdb and UTRsite (RELEASE 2010): a collection of sequences and regulatory motifs of the untranslated regions of eukaryotic mRNAs. Nucleic acids research. 2010;38:D75–80. doi: 10.1093/nar/gkp902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Smith TF, Waterman MS. Identification of common molecular subsequences. Journal of molecular biology. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
CAGE, ChIP, and WGB-sequencing data generated in this study have been deposited in the NCBI Gene Expression Omnibus (GEO) under accession GSE81322.