

Abstract
Over the past few years, the advances in technology and methods that have revolutionized cryo-EM are allowing for key insights in a variety of areas in biology, and microbiology is no exception. A wide range of important macromolecular assemblies in prokaryotic and eukaryotic cells, as well as intact viruses, have now become accessible to investigation by new methods in 3D electron microscopy. We focus here on selected examples that illustrate this breadth, and review the application of methods in single particle cryo-EM and cryo-electron tomography to progress in the structural biology of CRISPR systems, visualization of small molecule drugs in membrane proteins, in situ visualization of bacterial nanomachines, and the analysis of antigen-antibody interactions to drive vaccine design.
Introduction
More than 2000 structures determined by single particle cryo-EM, cryo-electron tomography or from helical reconstruction have been deposited in the Electron Microscopy Data Bank in the past two years alone (EMDB, https://www.ebi.ac.uk/pdbe/emdb/). A comparison with a decade ago, when ~ 100 maps were deposited per year, provides a measure of the dramatic world-wide growth of the cryo-EM field. There is evidence of major progress in all sub-disciplines of cryo-EM including applications that use single particle and helical reconstruction, as well as those that use cryo-electron tomography, with or without sub-tomogram averaging [1]. And while the numbers of structures posting resolutions typical of those obtained by X-ray crystallography has increased substantially, much of the excitement with the emergence of cryo-EM, especially in microbiology, derives from determination of lower resolution structures of complexes both in vitro and in situ that are not tractable using conventional crystallographic methods.
In this review, we highlight four areas where cryo-EM has been, and likely will continue to be, of great use in the structural analysis of complexes of interest to microbiology. We begin with the example of CRISPR complexes, where cryo-EM methods reveal mechanisms for target recognition and inhibition. We next discuss the use of cryo-EM to visualize membrane proteins, such as drug transporters, in complex with small molecules, followed by an example of the use of cryo-electron tomography to analyze bacterial nanomachines in situ. Finally, we review recent highlights in the use of both single particle cryo-EM and sub-tomogram averaging to visualize antigen-antibody complexes on viruses and viral surface proteins.
Structural Biology of CRISPR Complexes
While CRISPR-Cas systems have garnered significant attention for their gene-editing capacity, these systems are found throughout a broad spectrum of prokaryotic organisms, providing an “adaptive” immune defense against invading genetic material. These systems, although highly diverse in both sequence and structure, are generally divided into 2 broad classes, which are further divided into subtypes: In class I (which includes types I, III, and IV, and encompasses the majority of CRISPR-Cas systems), a multi-subunit effector complex recognizes, unwinds, and degrades target RNA or DNA. Class II (which includes types II, V, and VI), uses a single-protein, multi-domain Cas9 (or similar) complex to target nucleic acids; because of this simplified structure, class II systems are most often used for gene editing applications [2]. While many key insights about the structure and function of these complexes have been derived from X-ray crystallography, CRISPR complexes are highly dynamic, making crystallization challenging. As such, cryo-EM has become a useful tool for understanding critical elements of CRISPR biology.
The class I effector complex forms an open coil shape, with a single-stranded RNA template (the guide RNA) curled through the center of the complex. Upon recognition of a target strand by the “protospacer adjacent motif” (PAM) region at the base of the complex, the target nucleic acid strand pairs with the matching sequence on the guide RNA. For type I complexes, which recognize double-stranded DNA (dsDNA), the dsDNA is unwound to generate an “R-loop”, with the target strand complementing the guide RNA (Figure 1A). A number of notable studies in the past several years have used cryo-EM to interrogate the structure and mechanisms of target recognition by type I effector complexes (Figure 1).
Figure 1.

Cryo-EM reveals mechanistic details for CRISPR-Cas systems. (A) Schematic of P. aeruginosa Type I-F CRISPR effector complex Csy, showing the guide RNA (cyan) within the backbone of the complex, with the target DNA strand (orange). Adapted from [6]. (B–F) Cryo-EM maps of CRISPR complexes. (B) P. aeruginosa Csy with DNA target strand (EMD 7048) [6]. Backbone Cas7f subunits (gray), Cas8f (purple), Cas5f (yellow), guide RNA (cyan) and dsDNA target (orange) are shown. (C) P. aeruginosa Csy with AcrF1 and AcrF2 inhibitors (EMD 8624) [5]. Cas7f subunits (blue), Cas8f (light purple), Cas5f (dark purple), guide RNA (yellow) AcrF1 (red), and AcrF2 (green) are shown. (D) D. vulgaris Type I-C with dsDNA (EMD 8296) [3]. Cas7 subunits (gray and pale blue), Cas8c (purple), Cas5c (green), and dsDNA (orange) are shown. Guide RNA is buried within the complex. (E) T. fusca Type I-E complex with full R-loop (EMD 8478) [4]. Cas7 subunits (gray), Cse1 (green and cyan), Cse2 (yellow), Cas5e (salmon), Cas6e (purple) and dsDNA (target strand, light orange, and non-target strand, dark orange) are shown. Guide RNA is buried within the complex. (F) T. thermophilus Type III Cmr complex (EMD 2900) [7]. Cmr4 subunits (gray and light blue), Cmr5 subunits (red and pink), Cmr6 (gold), Cmr1 (yellow), Cmr3 (green), Cmr2 (purple), guide RNA (dark blue), and target ssRNA (orange) are shown.
In 2016, Hochstrasser et al. determined the architecture of type I-C cascade from Desulfovibrio vulgaris, showing the path of not only the guide RNA and target DNA strand, but also the path of the non-target strand of the dsDNA (Figure 1D) [3]. In this complex, there are several fewer subunits than for the prototypical type I-E complex from Escherichia coli; whereas the non-target strand is organized by interactions with several different subunits in the type I-E complex, for the type I-C, the large Cas8c subunit not only recognizes the target dsDNA, but also organizes the entire R-loop. The dynamic nature of the formation of the R-loop (the unwinding of the dsDNA target) has made studying this process challenging. In 2017, cryo-EM was used to visualize multiple intermediates of the formation of the R-loop for the type I-E complex from thermophilic Thermobifida fusca (Figure 1E) [4]; this study suggested that an extra “pinch” of the non-target strand (which in the type I-E complex has a shorter path than the target strand) might serve as a recruitment signal for a nuclease to degrade the target dsDNA.
Also in 2017, two separate studies of the Pseudomonas aeruginosa type I-F complex used cryo-EM to visualize the process of target dsDNA recognition and binding, as well as the mechanism whereby phage-derived inhibitors can block CRISPR mediated degradation. Chowdhury et al. visualized for the first time the complete organization of all the type I-F effector complex subunits and guide RNA; they also determined the structures of the inhibitors AcrF1 and AcrF2, and showed that these inhibitors concealed moieties required for target dsDNA binding (Figure 1C) [5]. Guo et al. extended these studies by demonstrating the conformational changes that occur with dsDNA binding, and additionally visualized the binding site of a third inhibitor, AcrF10 (Figure 1B) [6].
Cryo-EM has also been used for non-type I structures: in 2015, cryo-EM structural analysis revealed the structure the type III CMR complex from Thermus thermophilus along with its single stranded RNA target (Figure 1F) [7]. And this past year, Shin et al. used cryo-EM to reveal how a different phage-derived inhibitor, AcrIIA4, targets the type II Cas9 complex from Streptococcus pyrogenes [8]. While none of these studies reached atomic resolution throughout the entire complex, the use of computational sorting to handle conformational heterogeneity was critical for visualizing each of these complexes in their entirety.
Small Molecule Interactions Mapped by Cryo-EM
Several recent publications have demonstrated that cryo-EM methods can be used to map the locations and potential interactions of small molecules bound to protein complexes, including membrane proteins [9,10]. And while X-ray crystallography remains the primary source of structural data used for drug design, cryo-EM will likely soon be increasing useful for this purpose because of its ability to visualize dynamic, heterogeneous complexes to high resolution, [1,11].
Noteworthy advances have been made especially in the studies of membrane proteins that function as transporters and channels, which are found in both prokaryotes and eukaryotes. Studies of the mammalian potassium channel KCNQ1 in complex with calmodulin have resulted in direct visualization of the interaction between the channel and ligand [12], while studies of the glutamate receptor have been able to visualize features such as bound ligands [13] and N-linked carbohydrates on the protein surface [14]. A particularly interesting area where rapid progress is being made is in the structural analysis of ATP-binding cassette (ABC) transporters, proteins that use ATPase activity to transport a diverse array of small molecules across bilayer membranes. The MacAB-TolC drug efflux pump, which is found in E. coli and other gram-negative bacteria, uses ATPase activity to drive transport of molecules out of the cell. On the inner membrane, the homodimeric ATPase MacB transporter (similar to eukaryotic transporters) transfers small molecules from the intracellular space to the hexameric MacA complex, which spans the periplasmic space. In turn, MacA connects to the TolC trimer on the outer membrane. While MacA and TolC have separately been crystallized, a multi-resolution cryo-EM map of an engineered, stable assembly of the three subsections was recently determined with resolutions ranging from ~ 3 Å for the highly stable periplasmic section, to ~8 Å for the dynamic MacB transporter (Figure 2A) [15]. Intriguingly, despite its relatively low resolution, this map revealed the presence of unexplained density between two helices that make up a periplasmic extension of MacB, which the authors suggest may occupy a substrate binding pocket (Figure 2A).
Figure 2.
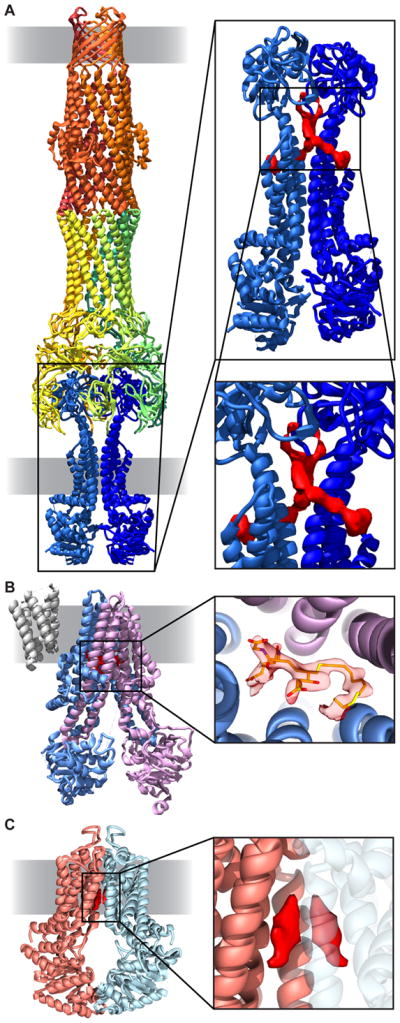
Visualization of small molecules bound to membrane proteins. (A) Structure of the MacAB-TolC drug efflux pump (left). TolC (orange), MacA (yellow and green), and MacB (blue) are shown in ribbon form. Extra density (red) is visible in potential substrate binding pocket (right, EMD 3653, PDB 5NIK) [15]. (B) Multidrug resistance protein (MRP1) with density for bound leukotriene C4. MRP1 subunits (purple, blue) are shown in ribbon (left); density for leukotriene C4 (orange) is visible in the structure (right, EMD 8560, PDB 5UJ9) [16]. (C) ABCG2 transporter (ribbon, orange and blue) with density (red) for cholesterol (EMD 3654, PDB 5NJ3) [17].
Cryo-EM structures of several additional ABC transporters are now at sufficient resolution to visualize density for bound ligands in the structure, as illustrated by a recent report of the structure of the mammalian multidrug resistance protein (MRP1) in complex with the ligand leukotriene C4 (Figure 2B). The structure reveals the mechanism by which MRP1 recognizes its ligand [16], as well as the overall change in conformation after ligand binding. A similar study of the structure of the mammalian ABC transporter ABCG2 to ~3.8 Å resolution (Figure 2C) enabled localization of two cholesterol molecules within the substrate binding pocket [17]. Interestingly, the latter structure was determined in nanodiscs, thus preserving native lipid interactions, which may be important to preserve a near-native environment for dynamic membrane proteins such as transporters. Several additional high resolution structures of ABC transporters and related proteins have been reported over the last two years; however, given the relatively high flexibility and structural heterogeneity of transporters, the direct visualization of small molecule substrates within the binding pocket remains a challenge for many of these complexes [18–22].
Bacterial Nanomachines in situ
As with the MacAB-TolC complex, many large, multi-subunit “nanomachines”, such as secretion systems, flagella, and chemosensory complexes, are challenging to study in their native conformations. For large, dynamic, multi-part nanomachines in prokaryotes, cryo-EM offers the unique possibility of determining the structure of these macromolecular complexes in situ, without needing to express or purify any components, while maintaining the integrity of the cellular architecture. This can be accomplished using cryo-electron tomography, where a frozen specimen – either a very thin whole bacterium, a thick frozen section of a cell, or a “mini-cell” (Figure 3A–B, reviewed in [23]) – is imaged under the microscope over a range of angles to generate a 3D volume. Since the individual 3D volumes generated by tomography are at low signal-to-noise ratios, averaging subvolumes that contain the complexes of interest can be used to obtain a higher level of structural detail.
Figure 3.

The Type III Secretion System in situ. (A) Slice through a tomogram of a Salmonella Typhimurium minicell, showing multiple secretion systems (blue arrows) crossing the inner (IM) and outer (OM) membranes. (B) Segmentation of the tomogram in (A). (C) Cross-section through 3D average of the T3SS, showing the outer membrane (OM) and inner membrane (IM), as well as the cytoplasmic complex and needle portions of the T3SS. From [32]. (D) 3D cryo-EM reconstruction of the T3SS shown in (C) (EMD 8544).
Although the resolutions achieved by tomography combined with sub-volume averaging are still significantly lower than what is possible at present with single particle cryo-EM, they allow the visualization of structures at medium resolution, good enough to reveal the overall organization of large macromolecular machines in the context of the cell [24]. In combination with X-ray crystallography, and cryo-EM methods, in situ analysis by cryo-electron tomography has produced important biological information on several complex systems, such as secretion and flagellar systems in prokaryotes.
One example where in situ structural analysis has been informative is the Type III secretion system (T3SS, Figure 3). The T3SS nanomachine is composed of more than 20 subunits, spanning the periplasmic space. These systems can be divided into two broad classes: the virulence T3SS (vT3SS), which engages with, and delivers export substrates to host cells to modulate host cell behavior, and the flagellar T3SS (fT3SS), which uses homologous intracellular and periplasmic machinery to drive flagellar motion. An ATPase (which drives export of material through the secretion system or flagellar rotation), a cytoplasmic (C-) ring, and an export apparatus are located intracellularly. These subunits connect to the basal body, which spans the inner membrane and connects to either the needle, pilus, or flagellum. The needle or flagellum then extends out through the outer membrane into the extracellular space (reviewed in [25,26]).
While many of the pieces of the T3SS had previously been studied structurally in vitro, cryo-electron tomography has been used more recently to visualize the complete T3SS within cells at progressively higher resolutions [27,28]. In one example, mini cells of Shigella flexneri were used to visualize the complete vT3SS prior to host cell engagement, and in complex with host red blood cells. Using sub-tomogram averaging of > 4000 individual vT3SS complexes, Hu et al. were able to achieve ~27 Å resolution for the pre-engaged structure, sufficient to fit existing models for a variety of components, and to show the basic structure of the cytoplasmic sorting platform [29]. This latter discovery was confirmed in a lower resolution study comparing S. flexneri vT3SS with Salmonella enterica serovar Typhimurium vT3SS and fT3SS, verifying the existence of 6-fold symmetry in cytoplasmic region, which the authors considered likely to be the ATPase structure [30]. Similar studies with Chlamydia trachomatis vT3SS prior to and after contact with host red blood cells demonstrated that the engagement with the host cell led to a significant compaction of both the basal body and the distance between the inner and outer membranes [31]. In situ structural analysis of the S. Typhimurium vT3SS to 17 Å resolution (Figure 3C–D) has also led to the surprising discovery that the structure of the isolated needle, as determined previously from purified samples, is significantly thinner than the needle in situ. This suggests that interactions with the complete T3SS apparatus may have an effect on the structure and organization of the needle complex [32], underscoring the importance of in situ analysis for studies of nanomachines.
Cryo-EM and Vaccine Design
For more than a decade, cryo-EM has been used to visualize antibody/antigen interactions; as techniques for sample preparation and data processing improve, single-particle and tomography-based techniques are becoming a method of choice to interrogate antibody binding and structural epitopes. For icosahedral viruses, where high symmetry can in some cases improve the probability of achieving high resolution, cryo-EM has proved especially useful for understanding virus-antibody interactions. For enveloped and non-symmetric viruses, the use of tomography combined with sub-volume averaging has provided a new window into virus architecture and footprints for the binding locations of therapeutically important antibodies, as initially described for studies on the HIV envelope glycoprotein [33].
High resolution cryo-EM structures for members of the flavivirus family (including the Dengue (DENV) and Zika (ZIKV) viruses, among others) have recently been published [34–36]. A critical challenge – for Dengue and Zika viruses in particular – is antibody-dependent enhancement, where antibodies specific for one previously acquired strain can lead to exacerbation of infection by a second strain. Over the past several years, several groups have used cryo-EM to understand the mechanism of binding for highly neutralizing antibodies. One common theme appears to be the ability of these antibodies to cross-link or simultaneously bind multiple E subunits on the surface of the virus, as has been shown for DENV3 [37], DENV2 [38], and ZIKV [39,40]. For example, Hasan et al. showed in their 6.2 Å structure that the therapeutic, neutralizing ZIKV-117 antibody crosslinks E proteins both within and across dimers, while not cross-reacting with other flaviviruses (Figure 4A) [39].
Figure 4.

Neutralizing antibodies bound to virus. (A) Zika virus (background) bound with neutralizing antibody ZIKV-117 (orange and gray ribbon, foreground), which crosslinks two E-protein dimers (blue and purple). (EMD 8548, PDB 5UHY) [39]. (B) Monoclonal antibody mAb100 (purple) bound to soluble Ebola glycoprotein (red, foreground). (EMD 3110) [46]. Schematic rendering of an Ebola VLP with envelope glycoprotein is shown in the background. (C) Influenza H1 spike (blue), determined on the surface of intact influenza virus, bound with 7B2 antibody (purple) (EMD 6612) [49]. Schematic rendering of influenza virion with HA (light blue) and NA (dark blue) spikes on the surface (background).
While many members of the enterovirus family (which includes the coxsackie viruses, enteroviruses, rhinoviruses, and polio) have been studied extensively by X-ray crystallography, many emerging strains of this family of viruses are challenging to culture and purify, requiring structural studies of small quantities of heterogeneous samples. Also of interest for vaccine studies is the visualization of empty, or procapsid, virus shells, which retain the external icosahedral shell but lack genomic material. Cryo-EM has been used to visualize such procapsid shells, sometimes mixed into a single sample with mature virus, for a variety of enterovirus family members, including for rhinovirus C (RV-15a) [41], coxsackie virus 16A [42], and poliovirus [43]. Additionally, the structures of enterovirus family members have been determined in complex with antibodies. For example, Ye et al. used cryo-EM to visualize the enterovirus 71 at high resolution with the antibody D5, which binds to a “propeller”-like structure on the surface of the virus in a bivalent fashion [44]. And in a study that made use of the ability of cryo-EM to determine asymmetric structures from heterogeneous samples, the coxsackie virus B3 entry intermediate was captured with a single nanodisc-bound receptor attached. This structure [45], which required asymmetric reconstruction, was determined to ~7.8 Å resolution, and revealed the asymmetric nature of structural changes on the virus after receptor engagement.
Although somewhat more challenging to study due to the lack of symmetry, non-icosahedral viruses have also been studied extensively by cryo-electron tomography and sub-volume averaging. In some instances, the tomography has been complemented by cryo-EM analysis of isolated viral protein assemblies to achieve higher resolution, as for example in the case of the surface glycoprotein of Ebola virus. Ebola is a filamentous, membrane-enclosed virus with trimers of an envelope glycoprotein that is responsible for both receptor binding and viral entry. In 2016, Misasi et al. used a soluble, purified version of the envelope glycoprotein to visualize the binding of the monoclonal antibody mAb100 to the base of the spike, and the antibody mAb114 to the head (Figure 4B) [46]. Pallesen et al. used a similar strategy to obtain high resolution structures of the Ebola glycoprotein bound to the three antibodies in the ZMAPP therapeutic cocktail [47], two of which are neutralizing in cell culture (c2G4 and c4G7), and one of which is not (c13C6). In a complementary approach, Tran et al. visualized the ZMAPP antibodies c2G4 and c4G7 on the surface of virus. Despite the low (~ 25 Å) resolution of the study, these structures nevertheless showed the location and orientation of antibodies on the native spike, and suggested that crosslinking between closely neighboring spikes may be a mechanism of neutralization [48].
Similar cryo-electron tomography-based strategies have also been useful for studies influenza virions, which display both trimeric hemagglutinin (HA) and tetrameric neuraminidase (NA) on the viral surface. Because it has been proposed that antibodies directed towards the conserved stalk region of the HA molecule may be more broadly neutralizing than those bound to the highly variable head region, Tran et al. compared native H1 (Figure 4C) and H5 spikes, each bound to specific antibodies, to spikes made of the H5 head domain and H1 stalk domain. Although the chimeric HA protein had a somewhat more open structure than the native H1 and H5 spikes, stalk H1 and head H5 antibodies were still bound, suggesting this chimeric protein might be useful as a vaccine candidate [49].
Conclusion
The recent advances in electron microscopy that have allowed atomic resolution for single particle analysis of select proteins have also increased the flexibility and power of a variety of electron-microscopy based techniques, enabling new discoveries in microbiology. In this review, we have discussed CRISPR complexes, membrane proteins and dynamic complexes visualized with small molecules, structures of molecular machines determined in situ, and the visualization of vaccine epitopes on viruses and viral proteins, but these areas are just a few select examples of areas in microbiology where cryo-EM is beginning to have an impact. The versatility of these cryo-EM based techniques will likely continue to yield critical biological insights in the years to come.
Highlights.
Recent developments in cryo-EM are producing new insights in microbiology
Dynamic complexes and membrane proteins can be visualized at high resolution
Nanomachines visualized in situ in bacterial cells using cryo-electron tomography
Virus/antibody complexes reveal mechanisms of neutralization and aid vaccine design
Acknowledgments
This work was supported by the Intramural program of the National Cancer Institute, NIH, Bethesda, MD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Subramaniam S, Earl LA, Falconieri V, Milne JL, Egelman EH. Resolution advances in cryo-EM enable application to drug discovery. Curr Opin Struct Biol. 2016;41:194–202. doi: 10.1016/j.sbi.2016.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mohanraju P, Makarova KS, Zetsche B, Zhang F, Koonin EV, van der Oost J. Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science. 2016;353:aad5147. doi: 10.1126/science.aad5147. [DOI] [PubMed] [Google Scholar]
- 3.Hochstrasser ML, Taylor DW, Kornfeld JE, Nogales E, Doudna JA. DNA Targeting by a Minimal CRISPR RNA-Guided Cascade. Mol Cell. 2016;63:840–851. doi: 10.1016/j.molcel.2016.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiao Y, Luo M, Hayes RP, Kim J, Ng S, Ding F, Liao M, Ke A. Structure Basis for Directional R-loop Formation and Substrate Handover Mechanisms in Type I CRISPR-Cas System. Cell. 2017;170:48–60 e11. doi: 10.1016/j.cell.2017.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5**.Chowdhury S, Carter J, Rollins MF, Golden SM, Jackson RN, Hoffmann C, Nosaka L, Bondy-Denomy J, Maxwell KL, Davidson AR, et al. Structure Reveals Mechanisms of Viral Suppressors that Intercept a CRISPR RNA-Guided Surveillance Complex. Cell. 2017;169:47–57. e11. doi: 10.1016/j.cell.2017.03.012. This study presented the first high resolution structure of the P. aeruginosa CRISPR complex in complex with the guide RNA, visualizing all subunits and the structure of the RNA, as well as that of bound inhibitors AcrF1 and AcrF2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6**.Guo TW, Bartesaghi A, Yang H, Falconieri V, Rao P, Merk A, Eng ET, Raczkowski AM, Fox T, Earl LA, et al. Cryo-EM Structures Reveal Mechanism and Inhibition of DNA Targeting by a CRISPR-Cas Surveillance Complex. Cell. 2017;171:414–426. e412. doi: 10.1016/j.cell.2017.09.006. High resolution structures of the P. aeruginosa CRISPR complex with and without target dsDNA revealed large tertiary and quaternary structural changes upon target binding. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taylor DW, Zhu Y, Staals RH, Kornfeld JE, Shinkai A, van der Oost J, Nogales E, Doudna JA. Structures of the CRISPR-Cmr complex reveal mode of RNA target positioning. Science. 2015;348:581–585. doi: 10.1126/science.aaa4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shin J, Jiang F, Liu JJ, Bray NL, Rauch BJ, Baik SH, Nogales E, Bondy-Denomy J, Corn JE, Doudna JA. Disabling Cas9 by an anti-CRISPR DNA mimic. Sci Adv. 2017;3:e1701620. doi: 10.1126/sciadv.1701620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Merk A, Bartesaghi A, Banerjee S, Falconieri V, Rao P, Davis MI, Pragani R, Boxer MB, Earl LA, Milne JLS, et al. Breaking Cryo-EM Resolution Barriers to Facilitate Drug Discovery. Cell. 2016;165:1698–1707. doi: 10.1016/j.cell.2016.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bartesaghi A, Merk A, Banerjee S, Matthies D, Wu X, Milne JL, Subramaniam S. 2.2 A resolution cryo-EM structure of beta-galactosidase in complex with a cell-permeant inhibitor. Science. 2015;348:1147–1151. doi: 10.1126/science.aab1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boland A, Chang L, Barford D. The potential of cryo-electron microscopy for structure-based drug design. Essays Biochem. 2017;61:543–560. doi: 10.1042/EBC20170032. [DOI] [PubMed] [Google Scholar]
- 12.Sun J, MacKinnon R. Cryo-EM Structure of a KCNQ1/CaM Complex Reveals Insights into Congenital Long QT Syndrome. Cell. 2017;169:1042–1050 e1049. doi: 10.1016/j.cell.2017.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Twomey EC, Yelshanskaya MV, Grassucci RA, Frank J, Sobolevsky AI. Channel opening and gating mechanism in AMPA-subtype glutamate receptors. Nature. 2017;549:60–65. doi: 10.1038/nature23479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14*.Meyerson JR, Chittori S, Merk A, Rao P, Han TH, Serpe M, Mayer ML, Subramaniam S. Structural basis of kainate subtype glutamate receptor desensitization. Nature. 2016;537:567–571. doi: 10.1038/nature19352. Structures of the kainate glutamate receptor GluK2, which include features for glycosylation and other high-resolution features, revealed mechanism for desensitization. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15**.Fitzpatrick AWP, Llabres S, Neuberger A, Blaza JN, Bai XC, Okada U, Murakami S, van Veen HW, Zachariae U, Scheres SHW, et al. Structure of the MacAB-TolC ABC-type tripartite multidrug efflux pump. Nat Microbiol. 2017;2:17070. doi: 10.1038/nmicrobiol.2017.70. Multi-resolution cryo-EM map enabled definition of the structure of TolC and MacA to high resolution, and the structure of the flexible MacB transporter to sub-nanometer resolution. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16*.Johnson ZL, Chen J. Structural Basis of Substrate Recognition by the Multidrug Resistance Protein MRP1. Cell. 2017;168:1075–1085 e1079. doi: 10.1016/j.cell.2017.01.041. Near-atomic resolution structures of MRP1 in apo- and substrate-bound forms showed density for the bound substrate, and revealed structural changes upon ATP hydrolysis. [DOI] [PubMed] [Google Scholar]
- 17.Taylor NMI, Manolaridis I, Jackson SM, Kowal J, Stahlberg H, Locher KP. Structure of the human multidrug transporter ABCG2. Nature. 2017;546:504–509. doi: 10.1038/nature22345. [DOI] [PubMed] [Google Scholar]
- 18.Oldham ML, Grigorieff N, Chen J. Structure of the transporter associated with antigen processing trapped by herpes simplex virus. Elife. 2016;5:e21829. doi: 10.7554/eLife.21829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qian H, Zhao X, Cao P, Lei J, Yan N, Gong X. Structure of the Human Lipid Exporter ABCA1. Cell. 2017;169:1228–1239 e1210. doi: 10.1016/j.cell.2017.05.020. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Z, Chen J. Atomic Structure of the Cystic Fibrosis Transmembrane Conductance Regulator. Cell. 2016;167:1586–1597 e1589. doi: 10.1016/j.cell.2016.11.014. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Z, Liu F, Chen J. Conformational Changes of CFTR upon Phosphorylation and ATP Binding. Cell. 2017;170:483–491 e488. doi: 10.1016/j.cell.2017.06.041. [DOI] [PubMed] [Google Scholar]
- 22.Kim Y, Chen J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science. 2018;359:915–919. doi: 10.1126/science.aar7389. [DOI] [PubMed] [Google Scholar]
- 23.Farley MM, Hu B, Margolin W, Liu J. Minicells, Back in Fashion. J Bacteriol. 2016;198:1186–1195. doi: 10.1128/JB.00901-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oikonomou CM, Jensen GJ. Cellular Electron Cryotomography: Toward Structural Biology In Situ. Annu Rev Biochem. 2017;86:873–896. doi: 10.1146/annurev-biochem-061516-044741. [DOI] [PubMed] [Google Scholar]
- 25.Deng W, Marshall NC, Rowland JL, McCoy JM, Worrall LJ, Santos AS, Strynadka NCJ, Finlay BB. Assembly, structure, function and regulation of type III secretion systems. Nat Rev Microbiol. 2017;15:323–337. doi: 10.1038/nrmicro.2017.20. [DOI] [PubMed] [Google Scholar]
- 26.Gold V, Kudryashev M. Recent progress in structure and dynamics of dual-membrane-spanning bacterial nanomachines. Curr Opin Struct Biol. 2016;39:1–7. doi: 10.1016/j.sbi.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 27.Kawamoto A, Morimoto YV, Miyata T, Minamino T, Hughes KT, Kato T, Namba K. Common and distinct structural features of Salmonella injectisome and flagellar basal body. Sci Rep. 2013;3:3369. doi: 10.1038/srep03369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kudryashev M, Stenta M, Schmelz S, Amstutz M, Wiesand U, Castano-Diez D, Degiacomi MT, Munnich S, Bleck CK, Kowal J, et al. In situ structural analysis of the Yersinia enterocolitica injectisome. Elife. 2013;2:e00792. doi: 10.7554/eLife.00792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu B, Morado DR, Margolin W, Rohde JR, Arizmendi O, Picking WL, Picking WD, Liu J. Visualization of the type III secretion sorting platform of Shigella flexneri. Proc Natl Acad Sci U S A. 2015;112:1047–1052. doi: 10.1073/pnas.1411610112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Makino F, Shen D, Kajimura N, Kawamoto A, Pissaridou P, Oswin H, Pain M, Murillo I, Namba K, Blocker AJ. The Architecture of the Cytoplasmic Region of Type III Secretion Systems. Sci Rep. 2016;6:33341. doi: 10.1038/srep33341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nans A, Kudryashev M, Saibil HR, Hayward RD. Structure of a bacterial type III secretion system in contact with a host membrane in situ. Nat Commun. 2015;6:10114. doi: 10.1038/ncomms10114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32**.Hu B, Lara-Tejero M, Kong Q, Galan JE, Liu J. In Situ Molecular Architecture of the Salmonella Type III Secretion Machine. Cell. 2017;168:1065–1074 e1010. doi: 10.1016/j.cell.2017.02.022. 17 Å resolution reconstruction of Type III secretion systems in situ in Salmonella Typhimurium minicells using cryo-electron tomography and subtomogram averaging. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu J, Bartesaghi A, Borgnia MJ, Sapiro G, Subramaniam S. Molecular architecture of native HIV-1 gp120 trimers. Nature. 2008;455:109–113. doi: 10.1038/nature07159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34*.Zhang X, Ge P, Yu X, Brannan JM, Bi G, Zhang Q, Schein S, Zhou ZH. Cryo-EM structure of the mature dengue virus at 3.5-A resolution. Nat Struct Mol Biol. 2013;20:105–110. doi: 10.1038/nsmb.2463. Cryo-EM structure of Zika virus, showing organization of the capsid shell with high resolution features such as glycosylation, revealing similarities and differences to the closely related Dengue viruses. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sirohi D, Chen Z, Sun L, Klose T, Pierson TC, Rossmann MG, Kuhn RJ. The 3.8 A resolution cryo-EM structure of Zika virus. Science. 2016;352:467–470. doi: 10.1126/science.aaf5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kostyuchenko VA, Lim EX, Zhang S, Fibriansah G, Ng TS, Ooi JS, Shi J, Lok SM. Structure of the thermally stable Zika virus. Nature. 2016;533:425–428. doi: 10.1038/nature17994. [DOI] [PubMed] [Google Scholar]
- 37.Fibriansah G, Tan JL, Smith SA, de Alwis R, Ng TS, Kostyuchenko VA, Jadi RS, Kukkaro P, de Silva AM, Crowe JE, et al. A highly potent human antibody neutralizes dengue virus serotype 3 by binding across three surface proteins. Nat Commun. 2015;6:6341. doi: 10.1038/ncomms7341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fibriansah G, Ibarra KD, Ng TS, Smith SA, Tan JL, Lim XN, Ooi JS, Kostyuchenko VA, Wang J, de Silva AM, et al. DENGUE VIRUS. Cryo-EM structure of an antibody that neutralizes dengue virus type 2 by locking E protein dimers. Science. 2015;349:88–91. doi: 10.1126/science.aaa8651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hasan SS, Miller A, Sapparapu G, Fernandez E, Klose T, Long F, Fokine A, Porta JC, Jiang W, Diamond MS, et al. A human antibody against Zika virus crosslinks the E protein to prevent infection. Nat Commun. 2017;8:14722. doi: 10.1038/ncomms14722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang J, Bardelli M, Espinosa DA, Pedotti M, Ng TS, Bianchi S, Simonelli L, Lim EXY, Foglierini M, Zatta F, et al. A Human Bi-specific Antibody against Zika Virus with High Therapeutic Potential. Cell. 2017;171:229–241. e215. doi: 10.1016/j.cell.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Y, Hill MG, Klose T, Chen Z, Watters K, Bochkov YA, Jiang W, Palmenberg AC, Rossmann MG. Atomic structure of a rhinovirus C, a virus species linked to severe childhood asthma. Proc Natl Acad Sci U S A. 2016;113:8997–9002. doi: 10.1073/pnas.1606595113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fan C, Ye X, Ku Z, Kong L, Liu Q, Xu C, Cong Y, Huang Z. Beta-Propiolactone Inactivation of Coxsackievirus A16 Induces Structural Alteration and Surface Modification of Viral Capsids. J Virol. 2017;91:e00038–00017. doi: 10.1128/JVI.00038-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marsian J, Fox H, Bahar MW, Kotecha A, Fry EE, Stuart DI, Macadam AJ, Rowlands DJ, Lomonossoff GP. Plant-made polio type 3 stabilized VLPs-a candidate synthetic polio vaccine. Nat Commun. 2017;8:245. doi: 10.1038/s41467-017-00090-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44*.Ye X, Fan C, Ku Z, Zuo T, Kong L, Zhang C, Shi J, Liu Q, Chen T, Zhang Y, et al. Structural Basis for Recognition of Human Enterovirus 71 by a Bivalent Broadly Neutralizing Monoclonal Antibody. PLoS Pathog. 2016;12:e1005454. doi: 10.1371/journal.ppat.1005454. Cryo-EM analysis enterovirus 71 and related Virus-Like-Particle (VLP) structures to high resolution, also revealing reveal potential mechanisms for neutralization by the D5 antibody. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee H, Shingler KL, Organtini LJ, Ashley RE, Makhov AM, Conway JF, Hafenstein S. The novel asymmetric entry intermediate of a picornavirus captured with nanodiscs. Sci Adv. 2016;2:e1501929. doi: 10.1126/sciadv.1501929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46**.Misasi J, Gilman MS, Kanekiyo M, Gui M, Cagigi A, Mulangu S, Corti D, Ledgerwood JE, Lanzavecchia A, Cunningham J, et al. Structural and molecular basis for Ebola virus neutralization by protective human antibodies. Science. 2016;351:1343–1346. doi: 10.1126/science.aad6117. Mechanisms by which two diferent antibodies bind and neutralize the Ebola glycoprotein were determined using a combination of X-ray crystallography and cryo-EM of soluble glycoprotein trimers. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pallesen J, Murin CD, de Val N, Cottrell CA, Hastie KM, Turner HL, Fusco ML, Flyak AI, Zeitlin L, Crowe JE, Jr, et al. Structures of Ebola virus GP and sGP in complex with therapeutic antibodies. Nat Microbiol. 2016;1:16128. doi: 10.1038/nmicrobiol.2016.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tran EE, Nelson EA, Bonagiri P, Simmons JA, Shoemaker CJ, Schmaljohn CS, Kobinger GP, Zeitlin L, Subramaniam S, White JM. Mapping of Ebolavirus Neutralization by Monoclonal Antibodies in the ZMapp Cocktail Using Cryo-Electron Tomography and Studies of Cellular Entry. J Virol. 2016;90:7618–7627. doi: 10.1128/JVI.00406-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49*.Tran EE, Podolsky KA, Bartesaghi A, Kuybeda O, Grandinetti G, Wohlbold TJ, Tan GS, Nachbagauer R, Palese P, Krammer F, et al. Cryo-electron Microscopy Structures of Chimeric Hemagglutinin Displayed on a Universal Influenza Vaccine Candidate. MBio. 2016;7:e00257. doi: 10.1128/mBio.00257-16. Cryo-electron tomography and sub-tomogram averaging of native influenza HA spikes (including H1, H5, and a chimeric H5/1 HA) in complex with antibodies on the surface of virus-like-proteins to define immunogenic epitopes. [DOI] [PMC free article] [PubMed] [Google Scholar]