

Abstract
The core binding factor (CBF) gene RUNX1 is a target of chromosomal translocations in leukemia, including the t(8;21) in acute myeloid leukemia (AML). Normal CBF function is essential for the activity of AML1-ETO, product of the t(8;21), and for the survival of several leukemias lacking RUNX1 mutations. Using virtual screening and further optimization, we developed Runt domain inhibitors (RDIs) which bind directly to the RD and disrupt its interaction with CBFβ On target activity was demonstrated by the RDIs’ ability to depress hematopoietic cell formation in zebrafish embryos, reduce growth and induce apoptosis of t(8;21) acute myeloid leukemia (AML) cell lines, and reduce progenitor activity of mouse and human leukemia cells harboring the t(8;21), but not normal bone marrow cells. RDIs had similar effects on murine and human T cell acute lymphocytic leukemia (T-ALL) cell lines. Our results confirmed that RDIs might prove efficacious in various AMLs and in T-ALL.
Keywords: leukemia, RUNX, CBFB, AML1-ETO, TEL-AML1, protein-protein interaction inhibitor, PPI
Introduction
Acute myeloid leukemia (AML) often harbors non-random clonal chromosomal aberrations. Among them, the 8;21 translocation t(8;21)(q22;q22) is one of the most common in de novo AML, occurring in 10% of adult and 12% of pediatric AMLs (1, 2). The translocation fuses the N-terminal 177 amino acids of Runx1 (also known as AML1, encoded by RUNX1) to ETO (eight twenty-one, encoded by RUNX1T1), generating the chimeric protein AML1-ETO, an essential causative factor of t(8;21)-positive AML (2). Runx1 is a sequence-specific DNA binding transcription factor and member of heterodimeric core binding factors (CBFs) that play important roles in hematopoiesis (1, 2). Loss of Runx1 during embryonic development results in a failure of hematopoietic stem cell (HSC) emergence, whereas loss in adult HSCs leads to a pre-leukemic state (3–8). ETO has no known role in normal hematopoiesis.
Clinically, t(8;21)-positive leukemia is associated with favorable prognosis, with 70% of patients achieving complete remission following standard therapy (9, 10). However, many patients retain AML1-ETO expressing cells in their bone marrow due to incomplete eradication of the leukemic cells (11, 12). As a result 35–40% of these patients relapse within five years and have poor long-term survival (9, 10). We hypothesize that direct therapeutic targeting of the chimeric protein AML1-ETO may reduce the rate of relapse and improve long-term survival.
AML1-ETO has five domains conserved with its Drosophila homologues: the Runt domain (RD) from Runx1, and four from ETO (eTAFH, HHR, Nervy, MYND) (2). We and others solved the structures of all five domains and their interacting partners, and introduced amino acid substitutions to assess their contribution to AML1-ETO’s transforming ability (13–23). We determined that the interaction between the Runt domain of AML1-ETO and CBFβ, the non-DNA binding partner of all three RUNX proteins, is important for AML1-ETO mediated leukemogenesis (17). In addition, the interaction of CBFβ with the Runt domain is essential for the ability of TEL-AML1 (ETV6-RUNX1), frequently found in B-ALL, to promote the serial replating of B cell progenitors in vitro (17).
Recent studies provided evidence that normal CBF functions are required for the maintenance and survival of certain leukemia cells. Specifically, knock down of wild type Runx1 reduced growth and induced apoptosis in t(8;21) cell lines, and also in AML1-ETO transformed human CD34 positive cells (24, 25). Knock down of Runx1 also induced apoptosis of MLL-AF9 transformed cells, and deletion of Runx1 and CBFβ extended the disease latency in a mouse MLL-AF9 model (25), suggesting that a subset of AMLs that do not harbor mutations in the CBF genes may rely on continuous CBF function. T cell acute lymphocytic leukemia (T-ALL) also appears to rely on sustained Runx1 activity. A small molecule targeting cyclin-dependent kinase 7 displayed activity on a subset of cancer cell lines, including T-ALL, and its mechanism of action appeared to involve the down regulation of Runx1 expression (26).
Herein we describe the development of a tool compound that binds to the Runt domain of RUNX proteins and inhibits their interaction with CBFβ. These Runt domain inhibitors (RDIs) inhibit growth in culture as well as clonogenic potential of AML1-ETO and T-ALL leukemia cell lines. The tool compounds show clear effects on the expression of well-characterized RUNX1 target genes, and decrease the runx1-dependent process of hematopoietic cell formation from the dorsal aorta in zebrafish embryos. Analysis of genome-wide changes in gene expression identified lipid and sterol biosynthesis and ribosome biogenesis pathways, which are RUNX regulated (27, 28), as significantly affected by inhibitor treatment.
Materials and Methods
Virtual screening
Details of the virtual screen are described in the Supplementary Information.
Fluorescence resonance energy transfer (FRET) assays
Cerulean-Runt domain and Venus-CBFβ were expressed, purified and used in FRET assays as described previously (29–31). Cerulean-Runt domain and Venus-CBFβ proteins were used at a concentration of 100 nM for all assays.
Saturation transfer difference NMR
Details of the saturation transfer difference NMR experiments are described in the Supplementary Information.
Chemical synthesis
Details of the chemical synthesis including relevant NMR and mass spec data are provided in the Supplementary Information.
Mice
All mouse procedures were approved by the University of Pennsylvania University Animals Resource Center (ULAR) and Institutional Animal Care and Use Committees (IACUC) of the University of Pennsylvania. C57BL/6 mice were used in all studies.
Zebrafish experiments
Zebrafish were maintained according to IACUC-approved protocols. Tg(−6.0itga2b(CD41):eGFP (32) and Tg(cmyb:egfp) (33) lines were described previously. Whole mount in situ hybridization WISH was performed on PFA (4%) fixed embryos at the timepoints indicated using previously published probes (34, 35) and established methods (http://zfin.org/ZFIN/Methods/ThisseProtocol.html). Qualitative phenotypes (≥2 replicate clutches, n ≥ 20 embryos/per condition) were scored using a dissecting microscope (Zeiss) as compared to stage-matched sibling controls.
Human samples
Patient AML specimen pheresis and bone marrow mononuclear cells were obtained from the University of Pennsylvania Stem Cell and Xenograft Core, under the approval from the University of Pennsylvania Institutional Review Board (IRB).
Mouse and human cell lines
Details for the mouse and human cell lines are provided in the Supplementary Information.
MTT Cell Proliferation Assay
Details for proliferation assays are provided in the Supplementary Information.
CFU-C Assay
Details for CFU-C assays are provided in the Supplementary Information.
Western blotting
Details for Western blotting are provided in the Supplementary Information.
Flow cytometry
Details for flow cytometry of mouse and human cells as well as zebrafish embryos are provided in the Supplementary Information.
Gene expression analysis (quantitative real-time PCR and microarrays)
Details of the gene expression analysis are provided in the Supplementary Information. The accession number for the microarray data reported in this paper is GEO: GSE70495.
Statistical analyses
Statistical analyses were performed using the Graph Pad Prism 6.0 software package.
Results
Virtual screening to identify lead compounds targeting the Runt domain
We conducted a virtual screen utilizing the computer program LUDI (36) to identify initial lead compounds that bind to the Runt domain at the CBFβ interface (20, 21) and inhibit CBFβ binding. We employed the LUDI/CAP (Chemicals Available for Purchase) library containing 78,000 commercially available drug-like compounds, i.e. compounds that meet Lipinski’s criteria (37) in terms of solubility, number of hydrogen bond donors and acceptors, and molecular weight. These compounds were docked to the CBFβ binding surface on the Runt domain using LUDI and ranked by the scoring function implemented in the program (38) for theoretical binding efficacy estimation. Based on the LUDI results and a visual inspection of the predicted interactions with the protein for the 500 top scoring compounds, we selected 100 compounds with diverse molecular scaffolds for experimental screening.
Evaluation of compound efficacy using a fluorescence resonance energy transfer (FRET) assay
We used Cerulean-Runt domain and Venus-CBFβ fusion proteins at a 100 nM concentration for compound screening by fluorescence resonance energy transfer (FRET) (29–31). Compounds were screened at 200 µM in a competition experiment using the FRET assay. Five compounds showed 20% – 60% inhibition of the Runt domain - CBFβ interaction at 200 µM. Four of these compounds were discontinued due to toxicity, covalent binding to the protein, or promiscuous inhibition. The fifth compound, the aminotriazole AI-9–76 (Figure 1, Scheme 1) was identified as an initial hit. As the hydrazine moiety in AI-9–76 is a potential source of toxicity, we made O (oxadiazole2-thione) and S (thiadiazole2-thione) substitutions (Figure 1, Scheme 1). While the O substitution (AI-9–75) resulted in a loss of activity, the compound with a S substitution (AI-7–54) retained activity in the FRET assay.
Figure 1. Library of analogs synthesized.

Schemes 1–5 illustrate the synthetic routes used for library generation and refer to sets of compounds described in the main text.
Verification of ligand binding to the Runt domain by Nuclear Magnetic Resonance (NMR) spectroscopy
The FRET assay detects inhibition of CBFβ – Runt domain binding by small molecules, but cannot determine to which protein the compound binds. In order to establish that the lead compound does indeed bind to the Runt domain, we employed Nuclear Magnetic Resonance (NMR) spectroscopy. Two-dimensional 15N-1H HSQC spectra have been used very effectively to detect binding to proteins, however 15N-1H HSQC spectra of the Runt domain are very poor and missing almost half of the peaks due to exchange broadening (39), thus we were not able to detect any chemical shift changes upon addition of AI-7–54 (not shown). Spectra of the Runt domain bound to DNA are of good quality but the addition of our inhibitors resulted in precipitation. To overcome these obstacles, we utilized saturation transfer difference (STD) experiments (40, 41) that rely on the transfer of saturation from the protein to bound ligand to detect binding. To enhance the sensitivity of the experiment, we increased the size of the proteins by using Cerulean-RD and Venus-CBFβ, the same protein constructs that were used in the FRET assay. A saturation transfer effect was observed only for the AI-7–54 plus Cerulean-Runt domain mixture (Figure 2A), indicating that AI-7–54 binds to the Runt domain. Importantly, the absence of signals on the Venus-CBFβ STD spectrum confirms that AI-7–54 does not interact with either CBFβ or Venus (nor by analogy to Cerulean which has a very similar primary sequence). To confirm this, we collected high quality 15N-1H HSQC spectra for CBFβ in the presence of AI-7–54 and observed no chemical shift changes (not shown). Taken together, these results confirm that the Runt domain is the protein to which AI-7–54 binds.
Figure 2. NMR STD and FRET data for Runt domain inhibitors.

A. Results of NMR saturation transfer (STD) analysis for AI-7–54. The structure of AI-7–54 is shown with a 1D 1H NMR spectrum of the compound below. Arrows indicate resonance assignments. The middle spectrum shows 1D difference spectrum for Cerulean-Runt domain + AI-7–54. Bottom spectrum shows 1D difference spectrum for Venus-CBFβ, demonstrating a lack of binding.
B. Results of STD analysis for AI-8–45, as in panel A.
C,D. Results of FRET analysis for the initial lead AI-7–54 (C) and optimized compounds AI-8–45 and AI-9–54 (D). Calculated IC50 values (average of two measurements ± standard deviation) are shown.
Development of inhibitors with increased potency
The STD NMR and FRET data confirmed that the AI-7–54 scaffold is a valid initial lead. Indeed, the 1,3,4-thiadiazole heterocyclic ring system has been reported to have a wide range of pharmacological activities in the literature (42). To further develop this class of compounds, we performed a structure activity relationship (SAR) analysis, first by replacing the furan ring in AI-7–54 with thiophene, phenyl, 6-methyl-2-pyridyl, and pyrazine (Figure 1, Scheme 2). The phenyl substitution (4b, yielding 5-phenyl-1,3,4-thiadiazole-2(3H)-thione) improved the activity (IC50 = 11 ± 1.3 µM), whereas other substitutions yielded compounds that were inactive or weaker than AI-7–54. The addition of several substitutions into the phenyl ring based on the 4b scaffold did not improve activity (Figure 1, Scheme 3). We further elaborated the molecule as shown in Figure 1, Scheme 4. Methylation of the thioamide in compound 5a yielding 2-(2-fluorophenyl)-5-(methylthio)-1,3,4-thiadiazole (Figure 1, Scheme 4, 6a) resulted in a complete loss of activity, clearly defining the importance of the thioamide moiety. We then introduced a series of substitutions for the phenyl ring (Figure 1, Scheme 4). Substitution with benzofuran (6b), and replacement of the furan ring with an oxadiazole (6d) resulted in a complete loss of activity. On the other hand, compound 6c in which a benzene ring was added to the furan of AI-7–54 was active (IC50 = 13 ± 1.9 µM). We explored the SAR around compound 6c by introducing a series of substitutions into the phenyl ring (Figure 1, Scheme 5). Introduction of a 3-Cl yielding the compound 5-(5–3-chlorophenyl)furan-2-yl)-1,3,4-thiadiazole-2(3H)-thione (AI-8–45) (7f) improved the activity, however AI-8–45 has somewhat limited solubility. STD NMR analysis of AI-8–45 showed that protons in the furan ring as well as protons on the phenyl ring are in close contact with the protein (Figure 2B), consistent with the SAR. To improve the solubility we introduced 3-Cl pus 4-methoxyethyl ether yielding AI-9–54 (7k) that had similar activity as AI-8–45 in the FRET assay (Figure 2D). The SAR for this library of analogs was somewhat flat with no dramatic improvements in activity, so we selected two of the most active compounds (Table 1), AI-8–45 (IC50 = 2.0 ± 1.1 µM) and AI-9–54 (IC50 = 1.8 ± 0.6 µM) for use as tool compounds for biological studies. We used the starting scaffold AI-7–54 that has a 12-fold higher IC50 (24 ± 7µM, Figure 2C) as a negative control.
Table 1.
| Compound name |
Compound Structure | FRET IC50 (µM) |
|---|---|---|
| AI-7–54 (3) |
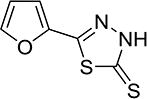
|
24 ± 7.1 |
| AI-8–153 (6c) |
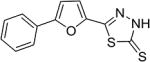
|
13 ± 1.9 |
| AI-8–45 (7f) |
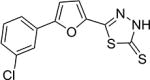
|
2.0 ± 1.1 |
| AI-9–24 (7a) |
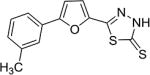
|
3.7 ± 1.6 |
| AI-9–13 (7b) |
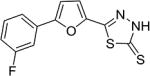
|
6.2 ± 0.5 |
| AI-9–23 (7c) |
|
17 ± 3.9 |
| AI-8–117 (7d) |
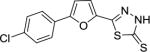
|
3.0 ± 0.3 |
| AI-9–27 (7g) |
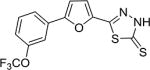
|
9.5 ± 1.4 |
| AI-8–103 (7i) |
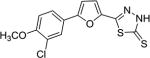
|
2.6 ± 0.6 |
| AI-9–54 (7k) |
|
1.8 ± 0.6 |
Runt domain inhibitors (RDIs) inhibit hematopoietic stem and progenitor cell (HSPC) formation in zebrafish embryos
Zebrafish embryos have been used successfully to directly screen for or to validate inhibitors based on their ability to augment or repress markers of HSPC formation in vivo (33, 43, 44). HSPCs differentiate from hemogenic endothelium of the dorsal aorta in all vertebrate species. This process is strictly dependent on Runx1, and highly sensitive to Runx1 dosage (45–47). In zebrafish embryos, HSPCs form de novo in the ventral dorsal aorta (VDA), then migrate through the blood stream to colonize the caudal hematopoietic tissue (CHT) to expand and differentiate (48); hence a consequence of decreased HSPC production in the VDA is fewer HSPCs in the CHT at later time points. To determine whether the predicted RDIs had on target activity in vivo, we analyzed their ability to modify HSPC numbers in the VDA and CHT of zebrafish embryos. Transgenic zebrafish embryos expressing fluorescent proteins marking HSPCs and/or adjacent vasculature were incubated with the RDIs AI-8–45 and AI-9–45, and as a control either vehicle (DMSO) or AI-7–54, which is structurally similar but exhibits significantly weaker potency. The number of HSPCs in the dorsal aorta and caudal hematopoietic tissue was then measured by fluorescence microscopy and/or flow cytometry. Incubation of zebrafish embryos with RDIs AI-8–45 and AI-9–45 from 12 to 36 hours post fertilization (hpf), during the window of HSPC specification, decreased Flk1:dsRed+Myb:Gfp+ HSPCs in the VDA at 36 hpf, as determined by flow cytometric analysis of whole Tg(kdrl:dsred;cmyb:egfp) embryos (Figure 3A). Likewise, treatment of Tg(−6.0itga2b:egfp) embryos from 18 to 48 hpf with either AI-8–45 or AI-9–54 decreased the number of CD41:Gfp+ HSPCs in the CHT at 48 hpf compared to pairwise-stage matched AI-7–54 treated controls (Figure 3B,C). Importantly, none of the RDIs had an effect on the viability or growth of the embryos, or on the formation of Flk1:dsRed+ endothelial cells (not shown) at the doses examined. Therefore, the RDIs appear to have on-target in vivo activity based on their ability to depress a process that is highly dependent on Runx1 dosage.
Figure 3. The RDIs inhibit hematopoietic stem and progenitor cell (HSPC) formation in zebrafish embryos.

A. Transgenic Tg(kdrl:dsred;cmyb:egfp) zebrafish HSPC-reporter embryos were incubated with the RDIs (2.5 µM) from 12–36 hpf, and analyzed at 36 hpf by flow cytometry. Shown is the percentage of Flk1:dsRed+Myb:Gfp+ HSPCs per whole embryo. Data are compiled from 3 clutches (n = 20 embryos/condition embryos). Significance determined by Student’s t-test.
B. Cd41:Gfp+ HSPCs in the caudal hematopoietic tissue (CHT) of representative 48 hpf Tg(−6.0itga2b:egfp) zebrafish embryos following incubation with RDIs from 18–48 hpf. Comparisons of AI-8–45 to AI-7–54, and AI-9–54 to A7–54 were performed in separate experiments.
C. Number of Cd41:Gfp+cells in the CHT as determined from fluorescence microscopy analysis, averaged from 20 embryos/condition in replicate experiments. Shown are pair-wise comparative data from two representative clutches.
Runt domain inhibitors (RDIs) induce growth arrest
Recent studies showed that Runx1 is required for the maintenance of leukemia cells expressing AML1-ETO, MLL-AF9, and T-ALL cells (24–26). We used an MTT cell viability assay to determine whether the RDIs inhibited the growth of leukemia cells that are either positive or negative for AML1-ETO expression. The RDIs AI-8–45 and AI-9–45 significantly reduced the growth of Kasumi-1 cells, a human AML cell line that harbors t(8;21), after 48 hours in culture (Figure 4A). In contrast, AI-7–54, the structurally similar but weaker potency inhibitor had no effect on Kasumi-1 cells, indicating that the effects are due to specific targeting of the RD-CBFβ interaction. We also tested the efficacy of AI-8–45 and AI-9–54 in murine and human T-ALL cell lines including 720 (derived from a Tcf12+/− mouse expressing a Tal1 transgene under the control of the Lck promoter (49)), Jurkat, and 8946 (murine T-ALL induced with doxycycline-repressible MYC transgene (50)). AI-8–45 significantly reduced the growth of all three T-ALL cell lines, and to an even greater extent of the 720 T-ALL line than of Kasumi-1 cells, indicating that the RDIs have activity in non-t(8;21) leukemia (Figure 4B–D). AI-9–54 reduced the growth of two of the three T-ALL cell lines (720 and 8946, but not Jurkat). In contrast, the growth of K562, a human CML cell line positive for BCR-ABL, was not inhibited by RDIs at 48 hours (Figure 4E).
Figure 4. RDIs reduce cell growth and induce apoptosis in leukemia cells.
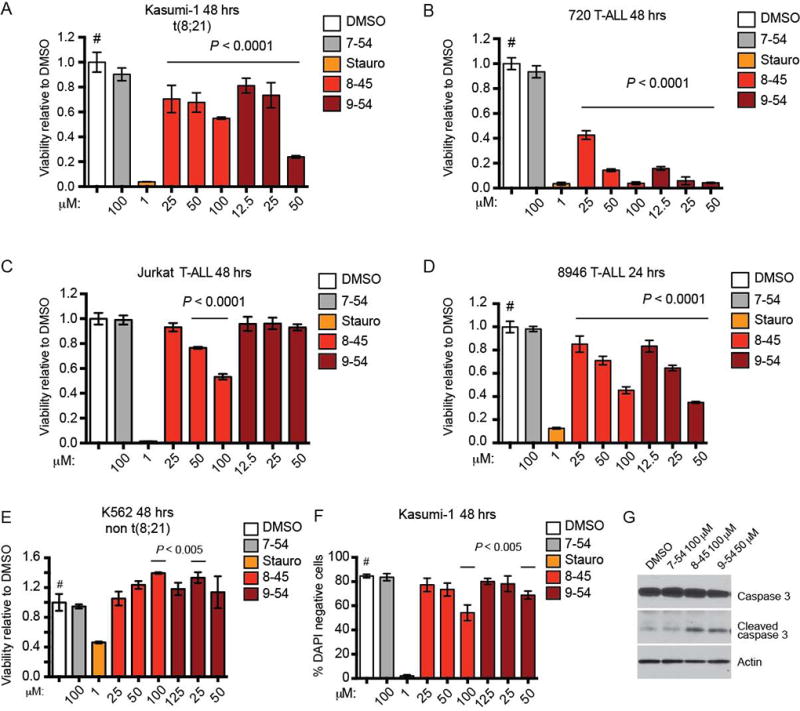
A–E. RDIs AI-8–45 (8–45) and AI-9–54 (9–54) inhibit proliferation of the AML cell line Kasumi-1 and the T-ALL lines 720, Jurkat (8–45 only), and 8946, but not K562 as detected by MTT cell viability assay. AI-7–54 (7–54) is the negative control, and staurosporine (Stauro) is a positive control. Data represent mean values for triplicates ± standard deviation (SD) (two independent experiments). P values were calculated by one-way ANOVA (staurosporine-treated cells were not included in the ANOVA analysis). Dunnett’s Multiple Comparison test was performed using DMSO treated cells as the comparator (#); horizontal lines above columns indicate significant differences from DMSO treated cells (P ≤ 0.05).
F. RDIs reduce the percentage of live (DAPI negative) Kasumi-1 cells as measured by flow cytometry. Data represents mean values of two independent experiments; statistical analysis as in A–E.
G. RDI treatment results in increased caspase-3 cleavage in 720 T-ALL cells (48 hrs).
We assessed whether the reduction in cell growth was accompanied by increased apoptosis. After 48 hours in culture, Kasumi-1 cells treated with AI-8–45 and AI-9-54, but not the less active compound AI-7–54, showed a statistically significant and dose-dependent increase in the percentage of DAPI-positive dead cells (Figure 4F). This was accompanied by an increase in the level of cleaved caspase-3 (Figure 4G). Taken together, these results showed that RDIs cause growth arrest, leading to caspase-3 mediated apoptosis in leukemia cells in vitro.
RDI effect on clonogenic potential
We assessed the activity of the RDIs on the clonogenic potential of murine AML cells. The AML cells were isolated from a secondary recipient of spleen cells transplanted from a primary recipient that had received fetal liver cells transduced with retroviruses expressing AML1-ETO9a and NRasG12D (51). AML1-ETO9a is a shortened form of AML1-ETO lacking the C-terminal MYND domain, and is more potent at inducing leukemia than full length AML1-ETO (52). The active compounds AI-8–45 and AI-9–54, but not the control compound AI-7–54, significantly reduced colony formation of the AML1-ETO9a + NRasG12D AML cells (Figure 5A). Neither AI-8–45 nor AI-9–54 had any effect on colony formation by normal mouse bone marrow cells (Figure 5A). These data demonstrate that the RDIs reduce the clonogenic potential of AMLs transformed with the AML1-ETO fusion protein, but do not affect normal bone marrow progenitors at the same concentrations, indicating that a therapeutic window is available.
Figure 5. Effect of RDIs on colony formation by normal and leukemic mouse bone marrow cells, and in human AML samples.
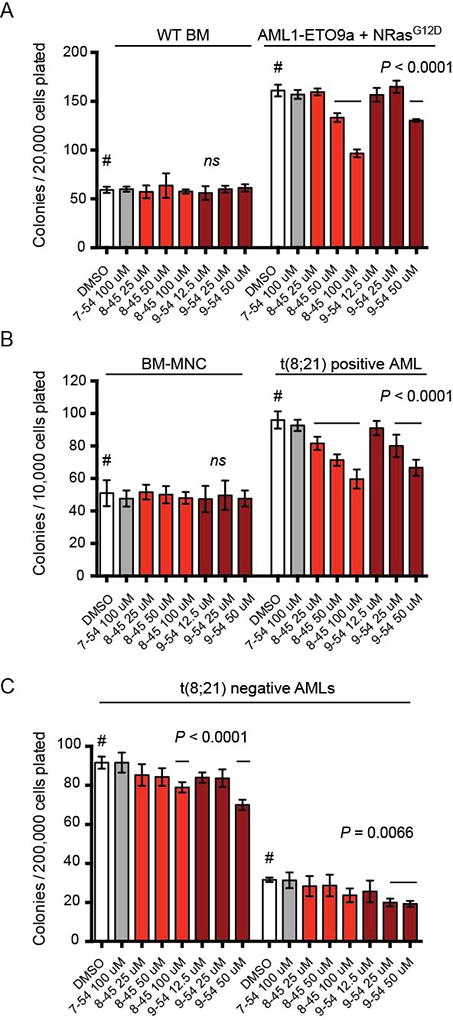
A. Various concentrations of compound were added to methylcellulose cultures containing 20,000 wild type bone marrow cells or 20,000 leukemic mouse cells transformed with AE9a and NRasG12D. All compounds were dissolved in DMSO (final concentration 0.2%). Colonies were counted 7 days after plating. Shown is a representative experiment (n=3 per compound concentration, two experiments). Error bars represent SD. Significance relative to DMSO was determined by one-way ANOVA and Dunnett’s multiple-comparison test as in Figure 4.
B–C. Various concentrations of compound were added to methylcellulose cultures containing bone marrow mononuclear cells (MNC) or primary AML samples. Colonies were counted 14 days after plating. Shown is a representative experiment (n=3 per compound concentration, two experiments). Significance relative to DMSO treatment was determined by one-way ANOVA and Dunnett’s multiple-comparison test as in Figure 4.
We also determined the effect of RDIs on the clonogenic potential of primary human t(8;21) AML. AI-8–45 and AI-9–54 significantly reduced the colony numbers of five t(8;21) positive human AML samples, but not that of normal bone marrow mononuclear cells (BM-MNCs) (Figure 5B). The control compound AI-7–54 had no effect on colony numbers in either the primary AML samples or normal bone marrow cells. We also examined the effect of AI-8–45 and AI-9–54 on colony formation by AML cells that do not harbor the t(8;21). The RDIs reduced the progenitor activity of some but not all human AML cell samples, indicating that they may have efficacy in a subset of AML not associated with Runx1 mutations (Figure 5C).
Molecular pathways affected by Runt domain inhibitors (RDIs)
To gain a better understanding of the mechanism of action of the RDIs we performed global gene expression analysis of the highly responsive 720 T-ALL cells following treatment. 720 T-ALL cells were treated with RDIs and harvested 8 hours later, before they underwent apoptosis, and expression was analyzed using microarrays. Overall, we detected only modest changes in expression levels after short-term inhibitor treatment (Figure 6A and Table S1). Pathway analysis revealed that genes involved in lipid and sterol biosynthesis, and in ribosome biogenesis to be among the most significantly downregulated (Figure 6B). Both of these processes are regulated by Runx proteins, and therefore are likely to reflect on-target effects (27, 53, 54). We confirmed the microarray data for several genes, including Deptor, which encodes an mTOR inhibitor; Cdkn1a, encoding the cell cycle inhibitor p21; Dhcr24 (encoding for 24-dehydrocholesterol reductase); as well as known Runx1 targets, Cebpa and Csf1r (encoding a transcription factor and cytokine receptor, respectively) (55, 56) by qPCR. The expression of all four genes was affected by treatment with AI-8–45 and AI-9–45, but not by the control compound AI-7–54 (Figure 6C,D), validating the microarray data for these genes.
Figure 6. Microarray analysis of gene expression changes induced by RDIs.
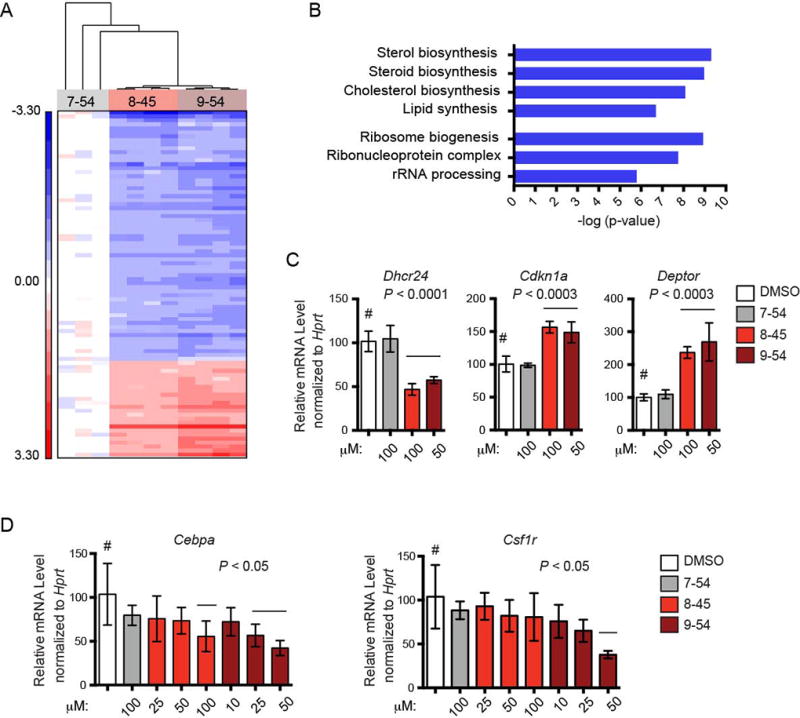
A. Hierarchical clustering of 87 transcript IDs (78 genes) differentially expressed (False Discovery Rate 5%) among 720 T-ALL cells treated for 8 hrs with AI-7–54 (100 µM), AI-8–45 (100 µM), or AI-9–54 (50 µM). Red represents genes up-regulated relative to mean expression level in AI-7–54 treated cells; blue represents genes down-regulated relative to mean expression level in AI-7–54 treated cells.
B. KEGG pathways downregulated following RDI treatment from functional annotation clustering.
C. Relative expression of genes in 720 T-ALL cells treated with RDIs for 16 hours, measured by qPCR. Data represents mean values for triplicates ± SD, n= two experiments. Significance relative to DMSO treatment was determined by one-way ANOVA. Dunnett’s Multiple Comparison test was performed using DMSO treated cells as the comparator (#); horizontal lines above columns indicate significant differences from DMSO treated cells (P ≤ 0.05).
D. Relative expression of Runx1-regulated differentiation genes, as in panel C.
Discussion
Based on an initial hit from a virtual screening approach, we developed tool compounds that bind to the Runt domain and are low µM inhibitors of the CBFβ-Runt domain interaction. The RDIs meet all the criteria outlined by Frye as the key properties of a high quality chemical probe (57), including a clear molecular profile of activity, mechanism of action, identity of active species, and proven utility. Furthermore, the inclusion of a weakly active control compound in all the biological evaluations provides high confidence that the observed activities derive from on-target activity. As the Runt domain is highly conserved and CBFβ binds all three RUNX proteins, it is important to keep in mind that the effect of the RDIs likely reflects inhibition of CBFβ binding to all RUNX proteins in a particular cell type. Recently, it has been reported that the benzodiazepine Ro5-3335 is also an inhibitor of CBFβ-Runx (44), with in vivo activity in zebrafish embryos consistent with predicted on-target effects. However, direct binding of Ro5-3335 to RUNX or CBFβ was not well documented, thus the biochemical direct target of Ro5-3335 remains to be established.
Based on our finding that the Runt domain’s interaction with CBFβ is critical for t(8;21) positive leukemia (17) as well as several recent reports that normal Runx1 function is required for the maintenance of some leukemias (24–26), we hypothesized that molecules targeting the RD:CBFβ interaction might achieve therapeutic efficacy against multiple leukemias. Indeed, we showed that the RDIs induced growth arrest and apoptosis not only in t(8;21) leukemia cells but also in a subset of non t(8;21) leukemia cells. Interestingly, the RDIs showed marked efficacy against the T-ALL cell lines that overexpress Tal1. RUNX1 was shown to form an auto-regulatory loop with TAL1 and GATA3 in TAL1 overexpressing T-ALL cells, and knocking down RUNX1 inhibited cell growth and induced apoptosis (58). In addition, RUNX1 was shown to be part of a complex that forms at a super enhancer created by somatic mutations in T-ALL (59). Thus RUNX1 appears to serve as a key member of an interconnected auto-regulatory loop involved in reinforcing and stabilizing the malignant cell state. In contrast, BCR-ABL positive K562 cells were minimally affected by the RDIs, as were multiple human primary AML cells.
RUNX proteins have now been implicated in numerous developmental pathways. Their role in leukemia is well established. In addition, there is emerging evidence for roles in various epithelial cancers. The tool compounds we have developed provide reagents to study the function of RUNX proteins in various settings. The advantage of such small molecule tools, unlike genetic approaches, is the rapid inhibition of the target, the ability to monitor time-dependent effects, and the opportunity to withdraw inhibitor and monitor the return to the previous state. For all these reasons, the RDIs provide a unique tool for studying RUNX protein function.
Supplementary Material
Supplemental Table 1. A list of 87 unique transcript IDs that are differentially expressed as shown in Figure 6a.
Acknowledgments
NIH R01 CA108056 (JHB and NAS), R01 CA149976 (NAS), and R01 DK098241 (TEN) and a Scholar Award from the Leukemia and Lymphoma Society (TEN) supported this work. The authors thank Martin Carroll and the University of Pennsylvania Stem Cell and Xenograft Core Facility for the AML cells, and for advice on culturing them.
Footnotes
Conflict of Interest
The authors declare they have no competing financial interests.
References
- 1.Mangan JK, Speck NA. RUNX1 mutations in clonal myeloid disorders: from conventional cytogenetics to next generation sequencing, a story 40 years in the making. Critical reviews in oncogenesis. 2011;16(1–2):77–91. doi: 10.1615/critrevoncog.v16.i1-2.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lam K, Zhang DE. RUNX1 and RUNX1-ETO: roles in hematopoiesis and leukemogenesis. Frontiers in bioscience. 2012;17:1120–39. doi: 10.2741/3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cai Z, de Bruijn MFTR, Ma X, Dortland B, Luteijn T, Downing JR, et al. Haploinsufficiency of AML1/CBFA2 affects the embryonic generation of mouse hematopoietic stem cells. Immunity. 2000;13:423–31. doi: 10.1016/s1074-7613(00)00042-x. [DOI] [PubMed] [Google Scholar]
- 4.Cai X, Gaudet JJ, Mangan JK, Chen MJ, De Obaldia ME, Oo Z, et al. Runx1 loss minimally impacts long-term hematopoietic stem cells. PloS one. 2011;6(12):e28430. doi: 10.1371/journal.pone.0028430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Motoda L, Osato M, Yamashita N, Jacob B, Chen LQ, Yanagida M, et al. Runx1 protects hematopoietic stem/progenitor cells from oncogenic insult. Stem cells. 2007 Dec;25(12):2976–86. doi: 10.1634/stemcells.2007-0061. [DOI] [PubMed] [Google Scholar]
- 6.Jacob B, Osato M, Yamashita N, Wang CQ, Taniuchi I, Littman DR, et al. Stem cell exhaustion due to Runx1 deficiency is prevented by Evi5 activation in leukemogenesis. Blood. 2009 Dec 14;:1610–1620. doi: 10.1182/blood-2009-07-232249. Epub 2009/12/17. Eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Growney JD, Shigematsu H, Li Z, Lee BH, Adelsperger J, Rowan R, et al. Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood. 2005;106:494–504. doi: 10.1182/blood-2004-08-3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Putz G, Rosner A, Nuesslein I, Schmitz N, Buchholz F. AML1 deletion in adult mice causes splenomegaly and lymphomas. Oncogene. 2006;25:929–39. doi: 10.1038/sj.onc.1209136. [DOI] [PubMed] [Google Scholar]
- 9.Grimwade D, Walker H, Oliver F, Wheatley K, Harrison C, Harrison G, et al. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children's Leukaemia Working Parties. Blood. 1998 Oct 1;92(7):2322–33. Epub 1998/09/25. eng. [PubMed] [Google Scholar]
- 10.Grimwade D, Walker H, Harrison G, Oliver F, Chatters S, Harrison CJ, et al. The predictive value of hierarchical cytogenetic classification in older adults with acute myeloid leukemia (AML): analysis of 1065 patients entered into the United Kingdom Medical Research Council AML11 trial. Blood. 2001 Sep 1;98(5):1312–20. doi: 10.1182/blood.v98.5.1312. [DOI] [PubMed] [Google Scholar]
- 11.Gulley ML, Shea TC, Fedoriw Y. Genetic tests to evaluate prognosis and predict therapeutic response in acute myeloid leukemia. The Journal of molecular diagnostics : JMD. 2010 Jan;12(1):3–16. doi: 10.2353/jmoldx.2010.090054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duployez N, Willekens C, Marceau-Renaut A, Boudry-Labis E, Preudhomme C. Prognosis and monitoring of core-binding factor acute myeloid leukemia: current and emerging factors. Expert review of hematology. 2015 Feb;8(1):43–56. doi: 10.1586/17474086.2014.976551. [DOI] [PubMed] [Google Scholar]
- 13.Liu Y, Cheney MD, Gaudet JJ, Chruszcz M, Lukasik SM, Sugiyama D, et al. The tetramer structure of the Nervy homology two domain, NHR2, is critical for AML1/ETO's activity. Cancer Cell. 2006 Apr;9(4):249–60. doi: 10.1016/j.ccr.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 14.Liu Y, Chen W, Gaudet J, Cheney MD, Roudaia L, Cierpicki T, et al. Structural Basis for Recognition of SMRT/N-CoR by the MYND Domain and Its Contribution to AML1/ETO's Activity. Cancer Cell. 2007 Jun;11(6):483–97. doi: 10.1016/j.ccr.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corpora T, Roudaia L, Oo ZM, Chen W, Manuylova E, Cai X, et al. Structure of the AML1-ETO NHR3-PKA(RIIalpha) complex and its contribution to AML1-ETO activity. J Mol Biol. 2010 Sep 24;402(3):560–77. doi: 10.1016/j.jmb.2010.08.007. Epub 2010/08/17. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park S, Chen W, Cierpicki T, Tonelli M, Cai X, Speck NA, et al. Structure of the AML1-ETO eTAFH domain-HEB peptide complex and its contribution to AML1-ETO activity. Blood. 2009 Apr 9;113(15):3558–67. doi: 10.1182/blood-2008-06-161307. Epub 2009/02/11. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roudaia L, Cheney MD, Manuylova E, Chen W, Morrow M, Park S, et al. CBFbeta is critical for AML1-ETO and TEL-AML1 activity. Blood. 2009 Mar 26;113(13):3070–9. doi: 10.1182/blood-2008-03-147207. Epub 2009/01/31. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun XJ, Wang Z, Wang L, Jiang Y, Kost N, Soong TD, et al. A stable transcription factor complex nucleated by oligomeric AML1-ETO controls leukaemogenesis. Nature. 2013 Aug 1;500(7460):93–7. doi: 10.1038/nature12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei Y, Liu S, Lausen J, Woodrell C, Cho S, Biris N, et al. A TAF4-homology domain from the corepressor ETO is a docking platform for positive and negative regulators of transcription. Nat Struct Mol Biol. 2007 Jul;14(7):653–61. doi: 10.1038/nsmb1258. [DOI] [PubMed] [Google Scholar]
- 20.Bravo J, Li Z, Speck NA, Warren AJ. The leukaemia-associated AML1 (Runx1)-CBFb complex functions as a DNA-induced molecular clamp. Nat Struct Biol. 2001;8:371–7. doi: 10.1038/86264. [DOI] [PubMed] [Google Scholar]
- 21.Tahirov TH, Inoue-Bungo T, Morii H, Fujikawa A, Sasaki M, Kimura K, et al. Structural analyses of DNA recognition by the AML1/Runx-1 Runt domain and its allosteric control by CBFb. Cell. 2001;104:755–67. doi: 10.1016/s0092-8674(01)00271-9. [DOI] [PubMed] [Google Scholar]
- 22.DeKelver RC, Yan M, Ahn EY, Shia WJ, Speck NA, Zhang DE. Attenuation of AML1-ETO cellular dysregulation correlates with increased leukemogenic potential. Blood. 2013 May 2;121(18):3714–7. doi: 10.1182/blood-2012-11-465641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kwok C, Zeisig BB, Qiu J, Dong S, So CW. Transforming activity of AML1-ETO is independent of CBFbeta and ETO interaction but requires formation of homo-oligomeric complexes. Proc Natl Acad Sci U S A. 2009 Feb 24;106(8):2853–8. doi: 10.1073/pnas.0810558106. Epub 2009/02/10. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ben-Ami O, Friedman D, Leshkowitz D, Goldenberg D, Orlovsky K, Pencovich N, et al. Addiction of t(8;21) and inv(16) acute myeloid leukemia to native RUNX1. Cell reports. 2013 Sep 26;4(6):1131–43. doi: 10.1016/j.celrep.2013.08.020. [DOI] [PubMed] [Google Scholar]
- 25.Goyama S, Schibler J, Cunningham L, Zhang Y, Rao Y, Nishimoto N, et al. Transcription factor RUNX1 promotes survival of acute myeloid leukemia cells. J Clin Invest. 2013 Sep 3;123(9):3876–88. doi: 10.1172/JCI68557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014 Jul 31;511(7511):616–20. doi: 10.1038/nature13393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Young DW, Hassan MQ, Pratap J, Galindo M, Zaidi SK, Lee SH, et al. Mitotic occupancy and lineage-specific transcriptional control of rRNA genes by Runx2. Nature. 2007 Jan 25;445(7126):442–6. doi: 10.1038/nature05473. [DOI] [PubMed] [Google Scholar]
- 28.Teplyuk NM, Zhang Y, Lou Y, Hawse JR, Hassan MQ, Teplyuk VI, et al. The osteogenic transcription factor runx2 controls genes involved in sterol/steroid metabolism, including CYP11A1 in osteoblasts. Molecular endocrinology. 2009 Jun;23(6):849–61. doi: 10.1210/me.2008-0270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gorczynski MJ, Grembecka J, Zhou Y, Kong Y, Roudaia L, Douvas MG, et al. Allosteric inhibition of the protein-protein interaction between the leukemia-associated proteins Runx1 and CBFbeta. Chem Biol. 2007 Oct;14(10):1186–97. doi: 10.1016/j.chembiol.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 30.Illendula A, Pulikkan JA, Zong H, Grembecka J, Xue L, Sen S, et al. Chemical biology. A small-molecule inhibitor of the aberrant transcription factor CBFbeta-SMMHC delays leukemia in mice. Science. 2015 Feb 13;347(6223):779–84. doi: 10.1126/science.aaa0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Illendula A, Gilmour J, Grembecka J, Tirumala VS, Boulton A, Kuntimaddi A, et al. Small Molecule Inhibitor of CBFbeta-RUNX Binding for RUNX Transcription Factor Driven Cancers. EBioMedicine. 2016 Jun;8:117–31. doi: 10.1016/j.ebiom.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin HF, Traver D, Zhu H, Dooley K, Paw BH, Zon LI, et al. Analysis of thrombocyte development in CD41-GFP transgenic zebrafish. Blood. 2005 Dec 1;106(12):3803–10. doi: 10.1182/blood-2005-01-0179. Epub 2005/08/16. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.North TE, Goessling W, Walkley CR, Lengerke C, Kopani KR, Lord AM, et al. Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature. 2007 Jun 21;447(7147):1007–11. doi: 10.1038/nature05883. Epub 2007/06/22. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carroll KJ, Esain V, Garnaas MK, Cortes M, Dovey MC, Nissim S, et al. Estrogen defines the dorsal-ventral limit of VEGF regulation to specify the location of the hemogenic endothelial niche. Developmental cell. 2014 May 27;29(4):437–53. doi: 10.1016/j.devcel.2014.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li L, Jin H, Xu J, Shi Y, Wen Z. Irf8 regulates macrophage versus neutrophil fate during zebrafish primitive myelopoiesis. Blood. 2011 Jan 27;117(4):1359–69. doi: 10.1182/blood-2010-06-290700. [DOI] [PubMed] [Google Scholar]
- 36.Bohm HJ. The computer program LUDI: a new method for the de novo design of enzyme inhibitors. J Comput Aided Mol Des. 1992;6(1):61–78. doi: 10.1007/BF00124387. [DOI] [PubMed] [Google Scholar]
- 37.Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods. 2000 Jul-Aug;44(1):235–49. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 38.Bohm HJ. On the use of LUDI to search the Fine Chemicals Directory for ligands of proteins of known three-dimensional structure. Journal of computer-aided molecular design. 1994 Oct;8(5):623–32. doi: 10.1007/BF00123669. [DOI] [PubMed] [Google Scholar]
- 39.Perez-Alvarado GC, Munnerlyn A, Dyson HJ, Grosschedl R, Wright PE. Identification of the regions involved in DNA binding by the mouse PEBP2a protein. FEBS L. 2000;470:125–30. doi: 10.1016/s0014-5793(00)01296-5. [DOI] [PubMed] [Google Scholar]
- 40.Mayer M, Meyer B. Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. Journal of the American Chemical Society. 2001 Jun 27;123(25):6108–17. doi: 10.1021/ja0100120. [DOI] [PubMed] [Google Scholar]
- 41.Mayer M, Mayer B. Characterization of ligand binding by saturaton transfer difference NMR spectroscopy. Angewandte. 1999;38(12):1784–8. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1784::AID-ANIE1784>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 42.Li P, Shi L, Gao MN, Yang X, Xue W, Jin LH, et al. Antibacterial activities against rice bacterial leaf blight and tomato bacterial wilt of 2-mercapto-5-substituted-1,3,4-oxadiazole/thiadiazole derivatives. Bioorganic & medicinal chemistry letters. 2015 Feb 1;25(3):481–4. doi: 10.1016/j.bmcl.2014.12.038. [DOI] [PubMed] [Google Scholar]
- 43.North TE, Goessling W, Peeters M, Li P, Ceol C, Lord AM, et al. Hematopoietic stem cell development is dependent on blood flow. Cell. 2009 May 15;137(4):736–48. doi: 10.1016/j.cell.2009.04.023. Epub 2009/05/20. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cunningham L, Finckbeiner S, Hyde RK, Southall N, Marugan J, Yedavalli VR, et al. Identification of benzodiazepine Ro5-3335 as an inhibitor of CBF leukemia through quantitative high throughput screen against RUNX1-CBFbeta interaction. Proc Natl Acad Sci U S A. 2012 Sep 4;109(36):14592–7. doi: 10.1073/pnas.1200037109. Epub 2012/08/23. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kalev-Zhylinska M, Horsfield JA, Flores MVC, Postlethwait JH, Vitas MR, Baas AM, et al. Runx1 is required for zebrafish blood and vessel development and expression of a human RUNX1-CBF2T1 transgene advances a model for studies of leukemogenesis. Development. 2002;129:2015–30. doi: 10.1242/dev.129.8.2015. [DOI] [PubMed] [Google Scholar]
- 46.Matheny CJ, Speck ME, Cushing PR, Zhou Y, Corpora T, Regan M, et al. Disease mutations in RUNX1 and RUNX2 create nonfunctional, dominant-negative, or hypomorphic alleles. Embo J. 2007 Feb 8;26:1163–75. doi: 10.1038/sj.emboj.7601568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.North TE, Gu T-L, Stacy T, Wang Q, Howard L, Binder M, et al. Cbfa2 is required for the formation of intra-aortic hematopoietic clusters. Development. 1999;126:2563–75. doi: 10.1242/dev.126.11.2563. [DOI] [PubMed] [Google Scholar]
- 48.Murayama E, Kissa K, Zapata A, Mordelet E, Briolat V, Lin HF, et al. Tracing hematopoietic precursor migration to successive hematopoietic organs during zebrafish development. Immunity. 2006 Dec;25(6):963–75. doi: 10.1016/j.immuni.2006.10.015. [DOI] [PubMed] [Google Scholar]
- 49.Cullion K, Draheim KM, Hermance N, Tammam J, Sharma VM, Ware C, et al. Targeting the Notch1 and mTOR pathways in a mouse T-ALL model. Blood. 2009 Jun 11;113(24):6172–81. doi: 10.1182/blood-2008-02-136762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999 Aug;4(2):199–207. doi: 10.1016/s1097-2765(00)80367-6. [DOI] [PubMed] [Google Scholar]
- 51.Zuber J, Radtke I, Pardee TS, Zhao Z, Rappaport AR, Luo W, et al. Mouse models of human AML accurately predict chemotherapy response. Genes Dev. 2009 Apr 1;23(7):877–89. doi: 10.1101/gad.1771409. Epub 2009/04/03. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yan M, Kanbe E, Peterson LF, Boyapati A, Miao Y, Wang Y, et al. A previously unidentified alternatively spliced isoform of t(8;21) transcript promotes leukemogenesis. Nat Med. 2006 Aug;12(8):945–9. doi: 10.1038/nm1443. [DOI] [PubMed] [Google Scholar]
- 53.Teplyuk NM, Galindo M, Teplyuk VI, Pratap J, Young DW, Lapointe D, et al. Runx2 regulates G protein-coupled signaling pathways to control growth of osteoblast progenitors. The Journal of biological chemistry. 2008 Oct 10;283(41):27585–97. doi: 10.1074/jbc.M802453200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cai X, Gao L, Teng L, Ge J, Oo Z, Kumar AR, et al. Runx1 deficiency decreases ribosome biogenesis and confers stress resistance to hematopoietic stem and progenitor cells. Cell Stem Cell. 2015 doi: 10.1016/j.stem.2015.06.002. [DOI] [PMC free article] [PubMed]
- 55.Wilson NK, Foster SD, Wang X, Knezevic K, Schutte J, Kaimakis P, et al. Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell. 2010 Oct 8;7(4):532–44. doi: 10.1016/j.stem.2010.07.016. Epub 2010/10/05. eng. [DOI] [PubMed] [Google Scholar]
- 56.Hoogenkamp M, Lichtinger M, Krysinska H, Lancrin C, Clarke D, Williamson A, et al. Early chromatin unfolding by RUNX1 - a molecular explanation for differential requirements during specification versus maintenance of the hematopoietic gene expression program. Blood. 2009 Apr 1; doi: 10.1182/blood-2008-11-191890. Epub 2009/04/03. Eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Frye SV. The art of the chemical probe. Nature chemical biology. 2010 Mar;6(3):159–61. doi: 10.1038/nchembio.296. [DOI] [PubMed] [Google Scholar]
- 58.Sanda T, Lawton LN, Barrasa MI, Fan ZP, Kohlhammer H, Gutierrez A, et al. Core transcriptional regulatory circuit controlled by the TAL1 complex in human T cell acute lymphoblastic leukemia. Cancer Cell. 2012 Aug 14;22(2):209–21. doi: 10.1016/j.ccr.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014 Dec 12;346(6215):1373–7. doi: 10.1126/science.1259037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. A list of 87 unique transcript IDs that are differentially expressed as shown in Figure 6a.