

Abstract
Ehrlichia chaffeensis, a tick-transmitted rickettsial bacterium, is the causative agent of human monocytic ehrlichiosis. Biochemical characterization of this and other related Rickettsiales remains a major challenge, as they require a host cell for their replication. We investigated the use of an axenic medium for E. chaffeensis growth, assessed by protein and DNA synthesis, in the absence of a host cell. E. chaffeensis organisms harvested from in vitro cultures grown in a vertebrate cell line were fractionated into infectious dense-core cells (DC) and the non-infectious replicating form, known as reticulate cells (RC) by renografin density gradient centrifugation and incubated in the axenic medium containing amino acids, nucleotides, and different energy sources. Bacterial protein and DNA synthesis were observed in RCs in response to glucose-6-phosphate, although adenosine triphosphate, alpha-ketoglutarate or sodium acetate supported protein synthesis. The biosynthetic activity could not be detected in DCs in the axenic medium. While the data demonstrate de novo protein and DNA synthesis under axenic conditions for E. chaffeensis RCs, additional modifications are required in order to establish conditions that support bacterial replication, and transition to DCs.
Introduction
Ehrlichia chaffeensis is an obligate intracellular, tick-transmitted bacterium that is maintained in nature in a cycle involving at least one and perhaps several vertebrate reservoir hosts1,2. Human infections with E. chaffeensis cause the disease human monocytic ehrlichiosis (HME) which is characterized by an acute onset of febrile illness that can progress to a fatal outcome, particularly in immune compromised individuals, elderly and children3,4. People undergoing blood transfusions and organ transplantations are also at high risk in acquiring E. chaffeensis infections and HME5,6. Knowledge of the biology and natural history of E. chaffeensis, and of the epidemiology, clinical features, and laboratory diagnosis has expanded considerably since its discovery7–10.
The life cycle of E. chaffeensis involves a tick vector and a mammalian host. In both mammalian and tick cells, E. chaffeensis transitions between two forms; the smaller dense-core cells (DCs) with dense nucleoid and the larger pleomorphic reticulate cells (RCs). RCs possess uniformly dispersed nucleoid filaments and ribosomes, sometimes forming long projections of the cell wall, protrusions of the cytoplasmic membrane into the periplasmic space, or budding protoplast fragments into the periplasmic space11–13. DCs are considered the infectious form of the bacterium, which enter naïve host cells by phagocytosis, then transform to non-infectious RCs within a phagosome and replicate prior to retransforming and releasing as DCs from the cells. We are yet to understand the detailed differences in proteins expressed in the two distinct forms of E. chaffeensis in vertebrate and tick cells and how the entire process is regulated.
Recent advances with Coxiella burnetii demonstrate that axenic media aids greatly in studies focused on biochemical and genetic analysis of the pathogen14–19. Similarly, the use of axenic media for protein biosynthesis has been reported for Chlamydia trachomatis, although bacterial replication is yet to be demonstrated20,21. These studies suggest that it is possible to take advantage of the cell-free growth medium for other important obligates belonging to Rickettsiales, such as E. chaffeensis. An axenic growth medium, called acidified citrate cysteine medium (ACCM), supported about three logs of growth of C. burnetii over 6 days in a microaerobic environment14. The ability to grow obligate intracellular bacteria under axenic conditions represents a major advancement, as it will enable new paths of investigation, such as aiding the manipulation of the pathogenic organisms in the absence of host cells, clonal purification of bacterial mutants, and detailed biochemical and genetic studies. Indeed, its application is well documented in greatly advancing the research on C. burnetii15. However, axenic growth methods require considerable optimization to adapt to each obligate pathogen of interest.
The purpose of this study is to evaluate the possibility of developing axenic culture methods for E. chaffeensis. In this study, we attempted to use the medium previously described for C. trachomatis, referred to as CIP-120. We describe the use of CIP-1 in supporting both protein and DNA biosynthesis in DC and RC forms of E. chaffeensis in the absence of host cells.
Results
Protein synthesis in cell-free E. chaffeensis assessed in axenic media
The axenic medium used for C. trachomatis is a complex mixture containing intracellular phosphate buffer (IPB) supplemented with 1% FBS, 25 μM of equimolar mix of all 20 amino acids (AA), 0.5 mM glucose 6-phosphate (G6P), 1.0 mM ATP, 0.5 mM DTT, and 50 μM each of GTP, UTP, and CTP as described in20, except when using alpha ketoglutarate (0.5 mM) or sodium acetate (0.5 mM) as the carbon source. When assessing 35S Cys-Met incorporation, concentrations of these two cold amino acids were reduced to 1 μM and then the two radioactive amino acids were supplemented in the form of 70 µCi of 35S-Cys-Met. Protein synthesis in the axenic media is verified with inclusion of chloramphenicol (CHL) or rifampin (RIF). In the current study, we prepared the medium with or without an energy source and used it to determine if it supports E. chaffeensis protein synthesis in the absence of a host cell. E. chaffeensis dense-core cells (DCs) and reticulate cells (RCs) were purified from the infected Vero cells or DH82 cells by renografin gradient centrifugation which fractionated at the interface between PBS/25% renografin (top layer) and between 25–35% renografin fraction (bottom layer), respectively. Incubation of E. chaffeensis purified from the bottom layer of the renografin fraction, where the RCs appeared to have concentrated, resulted in detectable protein synthesis. Protein synthesis by bacteria isolated from the top fraction, which is likely to contain DCs, was comparably much lower (Fig 1). Protein synthesis was abolished when CHL was included in the media and similarly with the inclusion of RIF (Fig. 1).
Figure 1.

Incorporation of 35S Cys-Met into E. chaffeensis organisms recovered from renografin fractionated bacteria in the axenic medium. (A) E. chaffeensis organisms isolated from Vero cells used in the axenic media assessment; autoradiography image assessing the incorporation of 35S Cys-Met in E. chaffeensis recovered from renografin purified top (TL) and bottom (BL) layered fractions incubation in the axenic media with or without chloramphenicol (CHL) and with G6P as an energy source and incubated for 24 h (G6P) (B). As in panel A, but quantitation of radiolabel incorporation by scintillation count analysis data (C). As in panel A except that the organisms were recovered from infected HL60 cells; this experiment also included a fraction of the purified organisms incubated in the axenic media with ATP as the energy source (ATP) (D). As in panel C, but the scintillation counting data were presented.
Defining DCs and RCs in the renografin fractions
The presence of DCs and RCs in the top and bottom layers of the renografin-purified fractions, respectively, was confirmed by Western blot analysis, in vitro infection assessment (Fig. 2) and transmission electron microscopy (Fig. 3). We previously reported the enhanced expression of E. chaffeensis ClpB in RCs compared to the infectious DCs22. In the current study, ClpB expression was assessed by Western blot analysis using the total proteins recovered from the E. chaffeensis organisms fractionated as the top and bottom layers on renografin gradient centrifugation. ClpB expression was significantly higher in the bottom layer compared to that found in the top layer (Fig. 2A). The protein expression data for ClpB suggest that the replicating form of E. chaffeensis is fractionated at the interface of the 25–35% renografin. To further confirm the presence of DCs and RCs in top and bottom layers, respectively, cell-free organisms recovered from each of the two fractions were used to re-infect naïve Vero cells (Fig. 2B). E. chaffeensis infection in Vero cells was primarily detected when fractionated organisms from the top layer were used, but not from the bottom layer. Infectivity of fractionated Ehrlichia organisms from the top layer was also confirmed by judging their ability to infect HL60 cells where we estimated the percent of infected host cells (Fig. 2C). Contrary to our assumption, the presence of RC fraction in the bottom layer and DC in the top layer was puzzling, as DCs are considered more dense organisms. Therefore to further confirm this observation, we performed transmission electron microscopy to detect the presence of DC and RC forms of E. chaffeensis in top and bottom layers, respectively (Fig. 3). Indeed, the pleomorphic and larger form of E. chaffeensis is evident primarily in the bottom layer, while the condensed form of the organisms was primarily observed in the top layer. Together, the experiments confirmed that the E. chaffeensis DC form is present primarily in the renografin fractionated top layer, whereas the RC form is present in the bottom layer.
Figure 2.
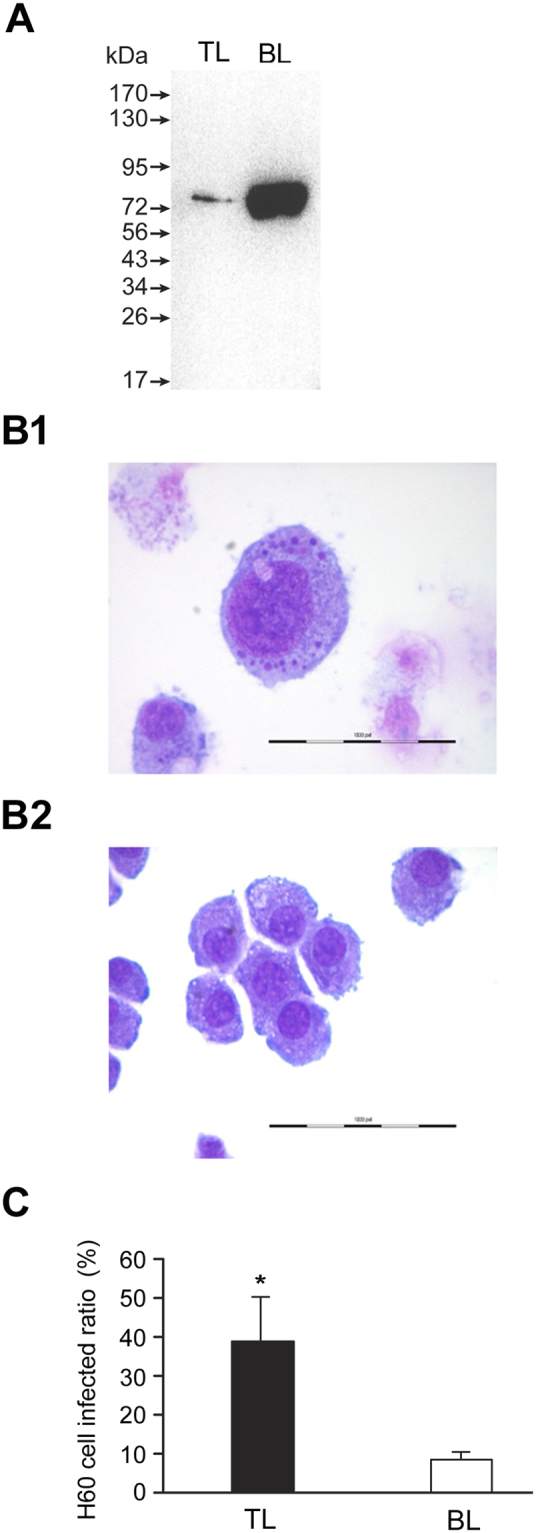
The presence of DCs and RCs assessed in the TL and BL of the renografin purified E. chaffeensis fractions. (A) Western blot analysis performed with ClpB polyclonal antibody which revealed higher levels of the protein expression in the BL derived bacterial fraction proteins; (B) cell-free E. chaffeensis recovered from TL and BL assessed for reinfection of naïve Vero cells; infection was detected only with the fraction derived from the TL (B1), but not in the BL (B2) (C). Infectivity of fractionated Ehrlichia organisms from the TL and BL was further confirmed by measuring the numbers of infected cells following incubation for three days following inoculation into naïve HL60 cultures. Infectivity with TL-derived Ehrlichia was significantly more than BL derived bacteria.
Figure 3.
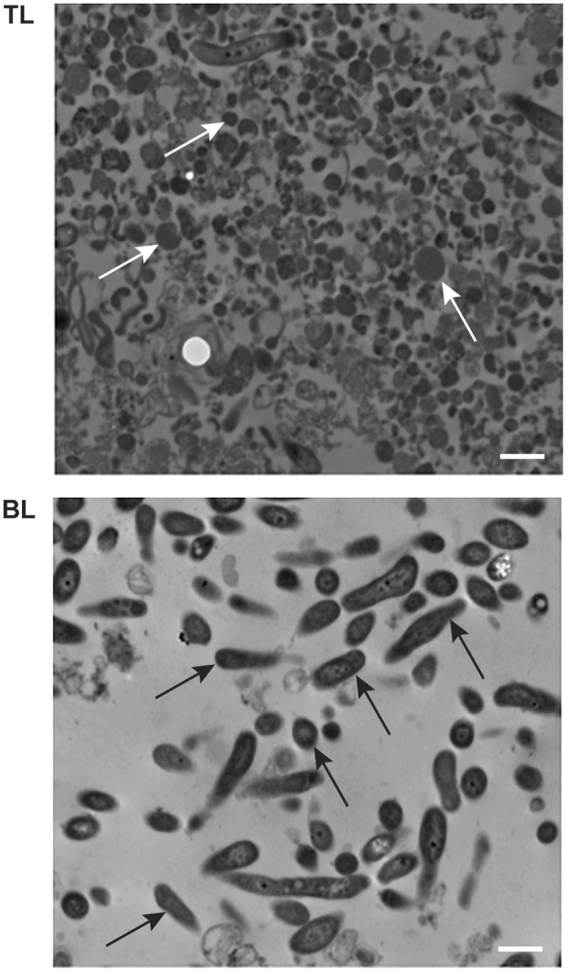
Transmission electron microscopy (TEM) analysis to define the E. chaffeensis organisms present in TL and BL. TEM images of the top (A) and bottom (B) layers where prototypical small DCs and pleomorphic RCs of E. chaffeensis were observed, respectively, throughout the images (a few DC and RC are identified with arrows). (Scale bars = 1 μm).
Impact of pH and alternate energy sources on protein biosynthesis in the axenic media
We investigated if protein biosynthesis by cell-free Ehrlichia in the axenic media has any pH preference, and secondly, if an altered pH may promote biosynthesis by the DC form (Fig. 4). As in the previous experiment, 35S-Cys-Met incorporation in the RC form was significantly higher compared to the control for all three different pHs, but there was no significant difference in the levels of incorporation among the three different pHs of the medium. No significant changes in 35S-Cys-Met incorporation were noted for the DC form of the bacterium compared to its respective antibiotic control. We then assessed the impact of different energy sources for the cell-free protein biosynthesis. The axenic medium supplemented with G6P, α-ketoglutarate or sodium acetate supported protein biosynthesis similarly for the RC form of E. chaffeensis recovered from DH82 cultures. G6P appeared to be more favored as the carbon source, but the difference was not significant compared to other carbon sources (Fig. 5). We also tested the impact of excluding the reducing agent (DTT) in the axenic medium for protein biosynthesis. The absence of DTT had only a marginal negative effect.
Figure 4.

Impact of pH variations assessed on the 35S Cys-Met incorporation into E. chaffeensis cell-free organisms. (A) Autoradiography imaging was performed to assess the impact of three different pH units; 5, 6 and 7 for DCs and RCs. In the first lane, we included rifampin (RIF) containing sample with the media at pH 5.0 to serve as a negative control (B). As in panel A, except that the scintillation counting data were presented. Significant change noted relative to RIF controls are identified with a *above each bar. (Note: Original images used in preparing Figure 4A were provided as the Supplementary information file.)
Figure 5.

Axenic media assessment using different carbon sources for the RC fraction. (A) Autoradiography imaging was assessed with the axenic media containing G6P with DTT (G6P); without an energy source (no G6P); with G6P in the absence of DTT (G6P, No DTT); with α-keto glutaric acid (α-KG) or sodium acetate (NaAce) as energy sources. The first lane included axenic media with G6P and CHL to serve as the negative control. (B) As in panel A, but scintillation counting data were presented. Significant change noted relative to CHL control in the experimental samples are identified with a *above each bar.
Limited protein biosynthesis confirmed by Western blot analysis
Based on the incorporation data in the above-described experiments, we reasoned that only limited protein biosysthesis has occurred in RCs. To verify this hypothesis, we compared protein profiles of total proteins resolved on an acrylamide gel before and after assessing by Western blot analysis using antisera against E. chaffeensis (Fig. 6). Despite the presence of increased abundance of a selected sub-group of proteins when RCs were incubated in the axenic medium containing different carbon sources, increase in total proteins was only moderate compared to that observed in the axenic medium containing CHL (Fig. 6A). Consistent with these data, Ehrlichia immunogenic proteins, assessed using a polyclonal sera, were also moderately increased when the RCs were incubated in the axenic media (Fig. 6B).
Figure 6.
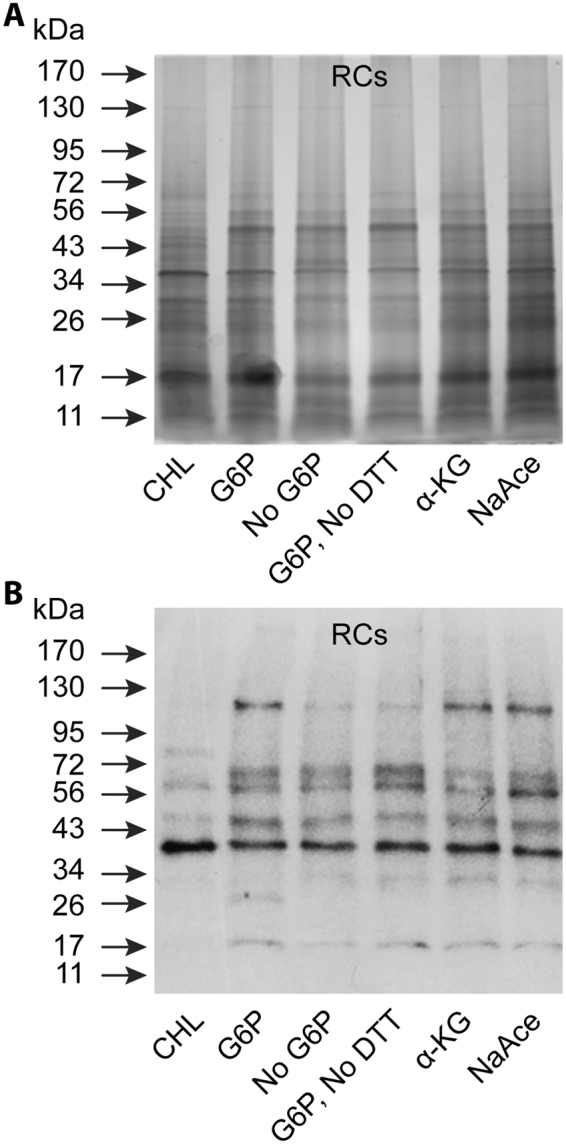
Protein biosynthesis assessed by protein fractionation and Western blot analysis. (A) Silver stained SDS containing polyacrylamide gel-resolved protein fractions were assessed for the protein abundance variations in RCs incubated in the axenic media with different carbon sources as in Fig. 5. (B) As in panel A, except that the resolved proteins were transferred to a nylon membrane and assessed by Western blot analysis using mouse polyclonal serum against E. chaffeensis.
DNA synthesis assessed in axenic media supports the limited protein biosynthesis data
We then tested if the axenic media also promoted DNA synthesis (Fig. 7). 3H thymidine incorporation was measured in the axenic media along with 35S Cys-Met incorporation to assess DNA and protein synthesis, respectively, for the RC form of E. chaffeensis. This assay was carried out at different pH conditions ranging from pH 5 to 9. There were no major differences in the incorporation of 3H thymidine or 35S Cys-Met when the RC form of E. chaffeensis was incubated at pH 6–9. Importantly, 3H thymidine incorporation in RC fraction was consistent with that of the 35S Cys-Met incorporation.
Figure 7.
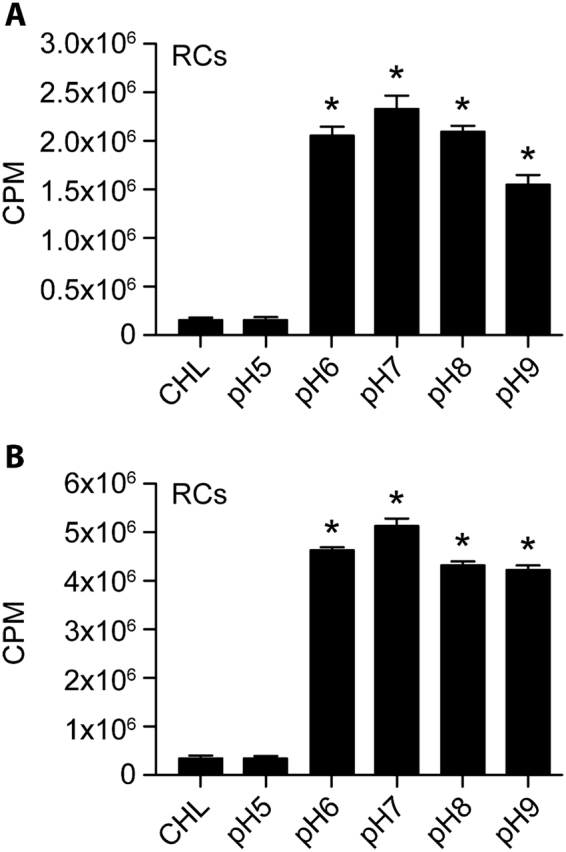
Protein biosynthesis and DNA synthesis assessed simultaneously by measuring the 35S Cys-Met and 3H thymidine incorporation, respectively in the axenic media at varying pHs of the media. (A) Scintillation counting data for the 35S Cys-Met assessed with G6P as the energy source. (B) As in panel A, except that the scintillation counting data for the 3H thymidine incorporation was assessed. Significant changes noted relative to CHL control are identified with a *above each bar.
RNA synthesis assessed in axenic media is consistent with DNA and protein biosynthesis
We performed a quantitative RT-PCR experiment targeting 16S rRNA using the cell-free RC fraction, as DNA and protein synthesis were observed primarily in this form of E. chaffeensis. The analysis was performed on RNA recovered from RCs incubated in the axenic media for 0 h, 2 h, 6 h, 12 h and 24 h. Triplicate RNA samples from each incubation time point were assessed by Taq-Man probe-based, quantitative real-time RT-PCR (Table 1). There was no significant change in the RNA expression level between 0 h and 2 h of incubation, while RNA copy numbers beyond 2 h of incubation resulted in a steady decline. These data suggest that the RC form of E. chaffeensis is viable only within the first two hours of incubation where protein biosynthesis and DNA synthesis occurred. The decline in RNA beyond 2 h of incubation may have resulted from continuous loss of RC viability in the axenic media. While RNA degradation is unexpected after 2 h incubation, the data are consistent with the observed moderate increase in protein and DNA biosynthesis in the RC form of E. chaffeensis.
Table 1.
E. chaffeensis 16S rRNA assessed for the RC form following different times points of incubation in the axenic media. (* refers to significant fold change at different time points compared to 0 h value.)
| Hours | Ct | SD | Fold change compared to 0 hour | *P-value |
|---|---|---|---|---|
| 0 | 18.24 | 0.7 | N/A | N/A |
| 2 | 18.64 | 0.31 | None | 0.47 |
| 6 | 20.01 | 0.5 | 3.3 | 0.02* |
| 12 | 22.87 | 0.15 | 24 | 0.0007* |
| 24 | 24.64 | 0.22 | 86 | 0.0003* |
Discussion
Two major limitations of carrying out research on obligate intracellular bacterial pathogens, including the study of Rickettsiales belonging to the Anaplasmataceae family, are the lack of fully established methods for targeted mutagenesis and the inability to grow the bacteria in the absence of a host cell. Targeted mutagenesis methods aid greatly in understanding the contributions of various genes involved in pathogenesis and in defining the genes critical for the pathogens’ vector and vertebrate host cell-specific growth. However, the ability to grow the pathogens in a cell-free medium can greatly facilitate studies focused on understanding functions of various bacterial proteins without the influence of a host cell. Further, growth in a host cell-free medium will aid in rapidly recovering mutant organisms and clonally purifying mutants9. Indeed, recent studies on C. burnetii demonstrated that significant progress could be made with the advent of fully established methods of mutagenesis and axenic growth14,16,23,24. To address these two major deficiencies for the field of research on Anaplasmataceae family pathogens, we recently described methods for creating stable targeted mutations to both disrupt and also restore the function of a disrupted gene in E. chaffeensis25. In the present study, we focused on the second major challenge for the field: the development of an axenic culture medium to grow E. chaffeensis. We believe that the data described here are critical for moving the field forward in various fronts, while requiring making improvements to the axenic media growth method to promote the transition of the replicating form to the infectious form of E. chaffeensis. Axenic media growth conditions will be valuable for studies focused on several Anaplasmataceae pathogens; 1) to aid in identifying and characterizing effector proteins involved in influencing the host; 2) in studying the potential interactions of the bacterial phagosome with mitochondria, host cytoplasmic proteins and nucleus; and 3) in facilitating the clonal purification of mutated organisms resulting from random and targeted mutagenesis methods.
Firstly, we presented a method for purification of E. chaffeensis DCs and RCs from host cells by employing renografin density gradient centrifugation. We discovered that the DC form of the bacterium fractionated at a lower concentration of renografin compared to the RC form. The presence of DCs and RCs within the gradient fractions was confirmed by three independent methods: the ability to infect naïve host cells, morphology, and by protein expression. Our studies demonstrate that axenic media supports protein and DNA synthesis only in the RCs of E. chaffeensis. Our study also provides definitive proof that the DC form is the infectious form, while the RC form is a pleomorphic replicating form that is actively involved in protein and DNA synthesis. Axenic media-specific protein synthesis was further confirmed by inclusion of the inhibitors, chloramphenicol or rifampin, in the cell-free media.
Axenic media-specific protein synthesis in E. chaffeensis is similar to a prior study demonstrating the cell-free protein biosynthesis for C. trachomatis20. In the current study we presented evidence that the axenic media also supported bacterial DNA synthesis in E. chaffeensis. Despite protein and DNA synthesis shown in the absence of host cells for RCs, our data suggest that the abundance is limited. We further investigated if variations in the pH of the media and altered energy sources may improve the protein biosynthesis. G6P appeared to be the best energy source for RC fraction. We did not note any conditions in the axenic media with the capacity to promote protein synthesis in DC fraction of E. chaffeensis.
Two important goals to improve the axenic media for E. chaffeensis are to modify the media conditions, such as adding thymidine in the media cocktail to 1) promote increased DNA and protein synthesis, resulting in the continued replication of the RC form; and 2) to transform the RCs to DCs under axenic media conditions. These improvements may be possible if the axenic media growth is assessed with purified, host cell-derived E. chaffeensis RC-containing phagosomes in place of purified RCs (Fig. 8). We reasoned that the phagosomal microenvironment might mimic in vivo conditions, although it may limit the number of bacterial replications. Axenic growth may also be further improved with the addition of purified mitochondria to the media containing the host cell-free RC fraction or RC-containing phagosomes (Fig. 8). Prior studies by us and other investigators focused on transmission electron microscopy11–13 demonstrated that mitochondria are closely associated with E. chaffeensis-containing phagosome vacuoles of infected host cells. E. chaffeensis and other Anaplasmataceae pathogens may benefit from mitochondria in multiple ways, including obtaining energy and metabolites.
Figure 8.
Proposed model to make improvements to the axenic media to promote E. chaffeensis replication in vitro in the absence of a host cell. RCCV and DCCV refer to RC containing and DC containing phagosome vacuoles, respectively.
In summary, the data presented here represent a significant step in advancing the goal of developing axenic media growth of E. chaffeensis. While the method needs improvement, we believe the strategy used in the current study can be adapted to other important pathogens belonging to Anaplasmataceae pathogens belonging to the genera Ehrlichia, Anaplasma and Neorickettsia.
Materials and Methods
Cultivation of E. chaffeensis
E. chaffeensis was cultivated in canine macrophage cell line, DH82 as described previously26. Similarly, E. chaffeensis in Vero cells (ATCC, Manassas, VA) was cultured in the complete MEM medium (Thermo Fisher Scientific, Waltham, MA) supplemented with 7% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA) and 2 mM L-glutamine (Mediatech, Manassas, VA). Cultivation of E. chaffeensis in HL60 cells (ATCC, Manassas, VA) was in complete RPMI 1640 medium (Thermo Fisher Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA) and 2 mM L-glutamine (Mediatech, Manassas, VA) by following the protocols described elsewhere for Anaplasma phagocytophilum27. To prepare cell-free Ehrlichia inocula, about 80–100% E. chaffeensis-infected DH82 cells in a T25 flask were harvested by centrifugation at 400 × g for 10 min at 4 °C. The pellets were resuspended in 5 ml of serum-free medium, and the cells were disrupted with glass beads by vortexing twice for 30 sec. The cell debris and unbroken cells were removed by centrifugation at 200 × g for 10 min at 4 °C. The supernatant was passed through a 2.7 µm pore-size syringe filter (Whatman, Pittsburgh, PA). HL60 cells were incubated with host cell-free E. chaffeensis (about 100 bacteria per host cell) for 120 min to allow for internalization. Non-ingested E. chaffeensis were removed by washing with PBS, and the cells were incubated for an additional 3 days in T150 flasks. Similar infection protocol is followed when infecting Vero cells or DH82 cells. When the infectivity reached to 80–90%, the infected host cell cultures were harvested by centrifugation at 15,000 × g for 5 min at 4 °C and used for purifying the host cell-free bacteria, as outlined below.
Purification of E. chaffeensis
E. chaffeensis organisms in the forms of dense-core cells (DCs) and reticulate cells (RCs) were purified by renografin density gradient centrifugation as described previously28,29 with some minor modifications. In brief, pellets of infected host cells were suspended in sterile PBS. The cells were then homogenized at 4 °C using a 10 ml syringe with a 231/2-G needle; typically 10–15 strokes were used to disrupt the cells. Homogenization was carried out until approximately 90% of cells were disrupted without major breakage of nuclei, as monitored by light microscopy. The disrupted cell suspension was centrifuged at 500 × g for 5 min at 4 °C. The supernatant was collected and filtered through 2.7 µm sterile syringe filter. The filtered supernatant was then centrifuged at 15,000 × g for 15 min at 4 °C. The pellet was resuspended into sterile PBS and 2 mL of the suspension was layered over discontinuous renografin gradients (3 mL 25%, 4 mL of 35% renografin in PBS, vol/vol). These gradients were centrifuged at 100,000 × g for 1 h at 4 °C using a Swinging Bucket rotor (S50-ST) in a Sorvall MTX150 ultracentrifuge (Waltham, MA). Fractions at the interfaces of PBS-25% and 25–35% renografin were collected using a sterile syringe, diluted with three volumes of PBS, and then centrifuged at 15,000 × g for 15 min at 4 °C. The pellets were washed with PBS to remove residual renografin by repeating the centrifugation step 15,000 × g for 15 min at 4 °C, and then the final purified pellets were resuspended in PBS for use in the cell free activity experiments.
Preparation of axenic medium
The axenic medium was prepared according to the previous study on C. trachomatis cultured in axenic medium as per the compositions and concentrations of each component20. Depending on the experiment carried out, the medium contained or excluded glucose 6-phoshate (G6P) or adenosine triphosphate (ATP) or alpha ketoglutarate or sodium acetate to serve as carbon sources. Similarly, pH of the media is modified as per the experimental need.
Protein synthesis by 35S-cysteine–methionine incorporation
Protein synthesis in cell-free purified fractions of E. chaffeensis was measured by incorporation of 35S-Cys-Met (Perkin Elmer, Waltham, MA) as described previously20. For normalization of bacterial total protein content, the suspensions of E. chaffeensis cell-free fractions were lysed in 1% SDS solution for 5 min at 100 °C and the total protein concentration was determined using Protein Assay kit (Bio-Rad, Hercules, CA). Subsequently, the suspensions of E. chaffeensis cell-free fractions were equally split into micro-centrifuge tubes at the amount of 30 µg total proteins. Partially opened micro-centrifuge tubes containing 500 μL of media supplemented with 70 µCi of 35S-Cys-Met were incubated at 37 °C for 24 h in a tri-gas humidified incubator set to maintain 2.5% O2. E. chaffeensis cell-free organisms were pelleted at the end of incubation by centrifugation at 15,000 × g for 15 min at 4 °C, washed with K-36 buffer (0.05 M K2HPO4, 0.05 M KH2PO4, 0.1 M KCl, 0.15 M NaCl, pH7.0) twice, and disrupted by adding 30 µL of 2 × SDS-PAGE sample buffer and by boiling for 5 min20. Ten µL of lysate each was then transferred to a tube containing 5 mL of biosafe liquid II and used for quantification of 35S-Cys-Met incorporation using the protocol 4 (35S) in a liquid scintillation counter (TRI-CARB 2100TR, PerkinElmer, Waltham, MA). For visualizing the radiolabel incorporation into bacterial proteins, equal volumes of sample lysates were also separated in an SDS/PAGE and the gel was dried and exposed to an X-ray film. Similarly, cell-free growth experiments were carried out in the absence of 35S-Cys-Met, resolved on an SDS-PAGE gel, and stained using silver nitrate staining kit (Thermo Fisher Scientific, Waltham, MA) as per the manufacturer’s recommendations.
DNA synthesis by 3H-thymidine incorporation
Purified E. chaffeensis cell-free fractions were also assessed for incorporation of 3H-thymidine (Perkin Elmer, Waltham, MA) into the bacterial DNA simultaneously with the incorporation 35S-Cys-Met into proteins30. Briefly, E. chaffeensis cell-free organisms were incubated for 48 h at 37 °C with 2.5% O2 in micro-centrifuge tubes containing 500 µL of medium supplemented with 20 μCi of 3H-thymidine and 70 μCi of 35S-Cys-Met. E. chaffeensis were pelleted at 15,000 × g for 15 min at 4 °C, washed with K-36 twice, lysed in 30 µL of 2 × SDS-PAGE sample buffer and then boiled for 5 min. 10 µL of lysate each was added into 5 mL of Biosafe liquid II (Grainger, Hartford, CT) and used for quantification of 3H-thymidine incorporation (the protocol 10, 3H) and 35S-Cys-Met incorporation (the protocol 4, 35S) using a liquid scintillation counting machine (TRI-CARB 2100TR, PerkinElmer, Waltham, MA), respectively.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
Five μL of NuPAGE SDS sample buffer and 2 μL of NuPAGE reducing agent (Invitrogen/Thermo Fisher Scientific, Waltham, MA) were added to each of 10 μL of sample solution following cell-free incubation experiments in the axenic medium, boiled for 5 min, and then loaded onto a Mini-PROTEAN Precast Bis-Tris 4% to 14% polyacrylamide gels (Bio-Rad, Hercules, CA) and subjected to electrophoresis (100 mA/gel for 60 minutes). The gels were then recovered from the gel assembly and stained using a Silver staining kit (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s recommendations.
Western blot analysis to assess protein synthesis
For the detection of ClpB protein of E. chaffeensis, the above-described electrophoresed proteins were transferred onto a nitrocellulose membrane (Thermo Fisher Scientific, Waltham, MA) by subjecting to electro-blotting using an electrophoretic transfer unit (Bio-Rad, Hercules, CA). Protein transfer buffer was prepared as per the manufacturer’s instructions and used in the protein transfer protocols. Subsequently, E. chaffeensis ClpB expression was assessed using polyclonal rabbit antisera raised against recombinant E. chaffeensis proteins for ClpB, respectively22. Secondary anti-rabbit antibody conjugated with horseradish peroxidase (Sigma-Aldrich, St. Louis, MO, USA) and Super Signal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, Waltham, MA) were used for the signal detection, respectively. We also performed similar blot analysis using polyclonal sera generated in the murine host in response to E. chaffeensis infection31.
Quantitative real-time RT-PCR to measure E. chaffeensis 16S rRNA expression
Cultures of E. chaffeensis grown in several T150 flasks were used in recovering cell-free RC form of the organisms fractionated on a renografin density gradient centrifugation as described above. E. chaffeensis RC organisms in triplicate microcentrifuge tubes were incubated for 0 h, 2 h, 6 h, 12 h and 24 h with 500 µL of axenic medium containing G6P and ATP at 37 °C with 2.5% O2. At the end of the specified incubation times, cells were recovered by centrifugation at 15,000 × g for 10 min at 4 °C. The bacterial pellets were then inactivated immediately in the TRI reagent solution, and then used to isolate total RNA by TRI reagent protocol (Sigma-Aldrich, St. Louis, MO). Final recovered RNA from each tube was resuspended in 25 μl of nuclease-free water, then DNase treated to remove residual genomic DNAs using RQ1 DNase (Thermo Fisher Scientific, Waltham, MA). RNA from each tube was diluted 1:1000 in nuclease-free water and 2 μl each was used in 25 μl reaction in performing Taq-Man probe-based real-time RT-PCR targeted to the E. chaffeensis 16S RNA as previously described32. The RNA levels in each sample were expressed by Ct values. Variation among triplicates for each time point was calculated and presented with the respective standard deviations observed. Fold changes were calculated relative to the Ct values observed for the RNA recovered before incubation (0 h) compared to different incubation times. The data were then assessed for statistical significance.
Preparation of E. chaffeensis cultures for use in transmission electron microscopy
Purified E. chaffeensis DCs and RCs were resuspended in PBS and used in transmission electron microscopy analysis by following the methods described previously12. Briefly, all TEM preparation steps were followed by repelleting samples by centrifugation at 4 °C for 5 min at 200 × g, unless otherwise specified. Pelleted DCs and RCs (in 10 μl volume) were fixed with 0.5 ml of Karnovsky’s fixative containing 2% paraformaldehyde, 2.5% gluteraldehyde in 0.1 M cacodylate buffer (pH7.4) overnight at 4 °C. The cell-free E. chaffeensis organisms were then washed three times with 1 ml of 0.1 M cacodylate buffer, post-fixed in 2% osmium tetroxide in the same cacodylate buffer, washed, enblock stained in 1% aqueous uranyl acetate for 1 h, washed, dehydrated in a 50–100% ascending series of acetone, infiltrated and embedded in Spurr’s resin. Ultrathin sections (<95 nm thick silver to gold) were cut using ultramicrotome, examined with a CM 100 TEM (FEI Company, Hillsboro, OR, USA now Thermo Fisher Scientific), and images captured with a Hamamatsu C8484 digital camera using a AMT digital image capture system (Chazy, NY).
Statistics analysis
Differences in quantitative protein, DNA synthesis and RNA expression between groups were assessed using Student’s t-test using the online software (http://www.socscistatistics.com/tests/studentttest/Default.aspx), with significance considered for p < 0.05.
Electronic supplementary material
Acknowledgements
This work was supported by the PHS grant # AI070908 from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, USA. This manuscript is contribution number from the Kansas Agricultural Experiment Station18–272-J. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication. We thank Ms. Mal Rocks Hoover for her help in formatting Fig. illustrations. We also thank Dr. Jodi McGill for critical reading of the manuscript.
Author Contributions
R.G. conceived and directed the research design and its execution. V.E., Y.Z., C.C., L.C., H.L., A.O. and D.B. contributed to discussions in designing the experiments. V.E. and Y.Z. performed the major portions of the experiments, while C.C., L.C. and H.L. assisted in executing some of the experiments. D.B. assisted in performing the TEM studies. All authors contributed to the manuscript’s drafting and editing to its final form.
Competing Interests
The authors declare no competing interests.
Footnotes
Vijay K. Eedunuri and Yuntao Zhang contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-27574-z.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Yabsley MJ. Natural history of Ehrlichia chaffeensis: vertebrate hosts and tick vectors from the United States and evidence for endemic transmission in other countries. Vet Parasitol. 2010;167:136–148. doi: 10.1016/j.vetpar.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 2.Lockhart JM, Davidson WR, Stallknecht DE, Dawson JE, Little SE. Natural history of Ehrlichia chaffeensis (Rickettsiales: Ehrlichieae) in the piedmont physiographic province of Georgia. J. Parasitol. 1997;83:887–894. doi: 10.2307/3284284. [DOI] [PubMed] [Google Scholar]
- 3.Paddock CD, Childs JE. Ehrlichia chaffeensis: a prototypical emerging pathogen. Clin Microbiol Rev. 2003;16:37–64. doi: 10.1128/CMR.16.1.37-64.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walker DH, Dumler JS. Emergence of the Ehrlichiosis as human health problems. Emerg Infect Dis. 1996;2:18–29. doi: 10.3201/eid0201.960102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McQuiston JH, Childs JE, Chamberland ME, Tabor E. Transmission of tick-borne agents of disease by blood transfusion: a review of known and potential risks in the United States. Transfusion. 2000;40:274–284. doi: 10.1046/j.1537-2995.2000.40030274.x. [DOI] [PubMed] [Google Scholar]
- 6.Sachdev SH, Joshi V, Cox ER, Amoroso A, Palekar S. Severe life-threatening Ehrlichia chaffeensis infections transmitted through solid organ transplantation. Transpl Infect Dis. 2014;16:119–124. doi: 10.1111/tid.12172. [DOI] [PubMed] [Google Scholar]
- 7.James E. Ehrlichia chaffeensis: a Prototypical Emerging Pathogen Christopher D. Paddock. Childs Clin Microbiol Rev. 2003;16:37–64. doi: 10.1128/CMR.16.1.37-64.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuczynska-Wisnik, D., Cheng, C., Ganta, R. R. & Zolkiewski, M. Protein aggregation in Ehrlichia chaffeensis during infection of mammalian cells. FEMS Microbiol Lett. 364, 10.1093/femsle/fnx059 (2017). [DOI] [PMC free article] [PubMed]
- 9.Cheng C, Nair AD, Jaworski DC, Ganta RR. Mutations in Ehrlichia chaffeensis Causing Polar Effects in Gene Expression and Differential Host Specificities. PLoS One. 2015;10:e0132657. doi: 10.1371/journal.pone.0132657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McClure EE, et al. Engineering of obligate intracellular bacteria: progress, challenges and paradigms. Nat Rev Microbiol. 2017;15:544–558. doi: 10.1038/nrmicro.2017.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Popov VL, Chen SM, Feng HM, Walker DH. Ultrastructural variation of cultured Ehrlichia chaffeensis. J Med Microbiol. 1995;43:411–421. doi: 10.1099/00222615-43-6-411. [DOI] [PubMed] [Google Scholar]
- 12.Dedonder SE, Cheng C, Willard LH, Boyle DL, Ganta RR. Transmission electron microscopy reveals distinct macrophage- and tick cell-specific morphological stages of Ehrlichia chaffeensis. PLoS One. 2012;7:e36749. doi: 10.1371/journal.pone.0036749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang JZ, Popov VL, Gao S, Walker DH, Yu XJ. The developmental cycle of Ehrlichia chaffeensis in vertebrate cells. Cell Microbiol. 2007;9:610–618. doi: 10.1111/j.1462-5822.2006.00812.x. [DOI] [PubMed] [Google Scholar]
- 14.Omsland A, et al. Host cell-free growth of the Q fever bacterium Coxiella burnetii. Proc Natl Acad Sci USA. 2009;106:4430–4434. doi: 10.1073/pnas.0812074106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Omsland A. Axenic growth of Coxiella burnetii. Adv Exp Med Biol. 2012;984:215–229. doi: 10.1007/978-94-007-4315-1_11. [DOI] [PubMed] [Google Scholar]
- 16.Omsland A, et al. Isolation from animal tissue and genetic transformation of Coxiella burnetii are facilitated by an improved axenic growth medium. Appl Environ Microbiol. 2011;77:3720–3725. doi: 10.1128/AEM.02826-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Omsland A, Cockrell DC, Fischer ER, Heinzen RA. Sustained axenic metabolic activity by the obligate intracellular bacterium Coxiella burnetii. J Bacteriol. 2008;190:3203–3212. doi: 10.1128/JB.01911-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vallejo Esquerra E, Yang H, Sanchez SE, Omsland A. Physicochemical and Nutritional Requirements for Axenic Replication Suggest Physiological Basis for Coxiella burnetii Niche Restriction. Front Cell Infect Microbiol. 2017;7:190. doi: 10.3389/fcimb.2017.00190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sandoz KM, Beare PA, Cockrell DC, Heinzen RA. Complementation of Arginine Auxotrophy for Genetic Transformation of Coxiella burnetii by Use of a Defined Axenic Medium. Appl Environ Microbiol. 2016;82:3042–3051. doi: 10.1128/AEM.00261-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Omsland A, Sager J, Nair V, Sturdevant DE, Hackstadt T. Developmental stage-specific metabolic and transcriptional activity of Chlamydia trachomatis in an axenic medium. Proc Natl Acad Sci USA. 2012;109:19781–19785. doi: 10.1073/pnas.1212831109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Omsland A, Sixt BS, Horn M, Hackstadt T. Chlamydial metabolism revisited: interspecies metabolic variability and developmental stage-specific physiologic activities. FEMS Microbiol Rev. 2014;38:779–801. doi: 10.1111/1574-6976.12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang T, et al. Aggregate-reactivation activity of the molecular chaperone ClpB from Ehrlichia chaffeensis. PLoS One. 2013;8:e62454. doi: 10.1371/journal.pone.0062454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Omsland A, Hackstadt T, Heinzen RA. Bringing culture to the uncultured: Coxiella burnetii and lessons for obligate intracellular bacterial pathogens. PLoS Pathog. 2013;9:e1003540. doi: 10.1371/journal.ppat.1003540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stead, C. M., Omsland, A., Beare, P. A., Sandoz, K. M. & Heinzen, R. A. Sec-mediated secretion by Coxiella burnetii. BMC Microbiol. 13, 10.1186/1471-2180-13-222 (2013). [DOI] [PMC free article] [PubMed]
- 25.Wang, Y., Wei, L., Liu, H., Cheng, C. & Ganta, R. R. Targeted gene knockout and restore function mutations in Ehrlichia chaffeensis. Scientific Reports. 10.1038/s41598-017-16023-y (2017). [DOI] [PMC free article] [PubMed]
- 26.Cheng C, Ganta RR. Laboratory maintenance of Ehrlichia chaffeensis and Ehrlichia canis and recovery of organisms for molecular biology and proteomics studies. Curr Protoc Microbiol. 2010;78:1864–1873. doi: 10.1002/9780471729259.mc03a01s9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang B, et al. Anaplasma phagocytophilum APH_1387 is expressed throughout bacterial intracellulardevelopment and localizes to the pathogen-occupied vacuolar membrane. Infect Immun. 2010;78:1864–1873. doi: 10.1128/IAI.01418-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caldwell HD, Kromhout J, Schachter J. Purification and partial characterization of the major outer membrane protein of Chlamydia trachomatis. Infect Immun. 1981;31:1161–1176. doi: 10.1128/iai.31.3.1161-1176.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cockrell DC, Beare P. A., Fischer, E. R., Howe, D. & Heinzen, R. A. A. method for purifying obligate intracellular Coxiella burnetii that employs digitonin lysis of host cells. J Microbiol Methods. 2008;72:321–325. doi: 10.1016/j.mimet.2007.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaplan LA, Bott TL, Bielicki JK. Assessment of [3H] thymidine incorporation into DNA as a method to determine bacterial productivity in stream bed sediments. Appl Environ Microbiol. 1992;58:3614–3621. doi: 10.1128/aem.58.11.3614-3621.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ganta RR, et al. Differential clearance and immune responses to tick cell-derived versus macrophage culture-derived Ehrlichia chaffeensis in mice. Infect Immun. 2007;75:135–145. doi: 10.1128/IAI.01127-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sirigireddy KR, Ganta RR. Multiplex detection of Ehrlichia and Anaplasma species pathogens in peripheral blood by real-time reverse transcriptase-polymerase chain reaction. J Mol Diagn. 2005;7:308–316. doi: 10.1016/S1525-1578(10)60559-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.