

Abstract
Background
With increasing emphasis on pragmatic trials, new randomized clinical trial designs are being proposed to enhance the “real world” nature of the data generated. We describe one such design, appropriate for unmasked pragmatic clinical trials in which the control arm receives usual care, called “Trials within Cohorts” that is increasingly used in various countries because of its efficiency in recruitment, advantages in reducing subject burden, and ability to better mimic real-world consent processes.
Methods
Descriptive, ethical, and US regulatory analysis of the Trials within Cohorts design.
Results
Trials within Cohorts design involves, after recruitment into a cohort, randomization of eligible subjects, followed by an asymmetric treatment of the two arms: those selected for the experimental arm provide informed consent for the intervention trial, while the data from the control arm are used based on prior broad permission. Thus, unlike the traditional Zelen post-randomization consent design, the cohort participants are informed about future research within the cohort; however, the extent of this disclosure currently varies among studies. Thus, ethical analysis is provided for two types of situations: when the pre-randomization disclosure and consent regarding the embedded trials are fairly explicit and detailed versus when they consist of only general statements about future data use. These differing ethical situations could have implications for how ethics review committees apply US research rules regarding waivers and alterations of informed consent.
Conclusion
Trials within Cohorts is a promising new pragmatic randomized controlled trial design that is being increasingly used in various countries. Although the asymmetric consent procedures for the experimental versus control arm subjects can initially raise ethical concerns, it is ethically superior to previous post-randomization consent designs and can have important advantages over traditional trial designs.
Keywords: Informed consent, pragmatic trial, pragmatic randomized controlled trial, ethics
There is increasing recognition of the value of pragmatic clinical trials, especially as it relates to the vision of a learning healthcare system that aims to closely integrate the delivery of medical services with clinical research. In such a system, the generation of knowledge would be “embedded into the core of the practice of medicine” leading to “continual improvement in care.”1 The advent of a modern electronic health record system makes it feasible and relatively inexpensive to conduct studies in the context of routine clinical practice.2 Such a vision provides an opportunity to think creatively about novel trial designs that can fulfill this pragmatic imperative.
In this article, we describe an emerging pragmatic trial paradigm called “Trials within Cohorts” (TwiCs) which involves longitudinal cohort studies that provide a platform for randomized clinical trials. To date, studies using the design have obtained research ethics committee approval in 10 countries (Australia, Canada, Finland, France, Germany, Mexico, the Netherlands, Spain, United Kingdom and the United States) with the most growth in the United Kingdom,3–7 Canada,8–11 and the Netherlands.12 The rare disease SPIN (Scleroderma Patient-centered Intervention Network) cohort has obtained institutional review board (IRB) approval to recruit to its cohort and conduct four intervention trials using the design in the United States, Canada, Mexico, France, and Spain.10
We first describe the features of TwiCs, and their strengths and limitations as a pragmatic randomized controlled trial (RCT) design. Because the TwiCs design is novel and unfamiliar to most research ethics committees/IRBs—and also because it involves an element of post-randomization consent which has a history of controversy13—we largely focus on the ethical issues in conducting TwiCs. We place TwiCs within a brief history of RCTs that obtain informed consent after randomization and then provide an ethical analysis of TwiCs, including a discussion of how it might be regulated by US IRBs.
Trials within Cohorts
RCTs remain the gold standard to prove effectiveness of interventions and this is no less true when the goal is to show the real-world effectiveness of the intervention in learning healthcare systems. However, the standard approach to RCTs is often complicated by slow recruitment rates, limited generalisability, limited long-term follow up, and high costs. The “Trials within Cohorts” design (formerly referred to as the “cohort multiple RCT design”14,15) was created to address these problems when unmasked studies are used to compare an intervention of interest with a usual care control arm.
In the TwiCs design, a cohort of participants with the condition(s) of interest is recruited for a longitudinal cohort study. At the time of recruitment into the cohort, the participants are given information about the process for their potential involvement in future intervention studies (i.e. TwiCs) and consent is obtained for potential future use of their data. A critical point, and one which varies from TwiCs to TwiCs, is whether this discussion also includes an explicit consent to be randomly assigned to control or intervention in unspecified future trials. Some implementations of TwiCs have given no specific information about future clinical trials (only general information about future use of their data in other studies) to the cohort participants,4,16 while others obtain consent (at initial recruitment into the cohort) regarding future randomization prior to TwiCs and use of data in future TwiCs12,17 as described further below.
After randomization to any given trial within the cohort, additional consent to receive the intervention is obtained from participants who have been randomly assigned to the new intervention. Those assigned to the treatment as usual control arm do not provide any additional consent after randomization.
The TwiCs design has several advantages over standard RCT design. First, difficulty with recruitment is a common concern in RCTs. The TwiCs design takes advantage of the fact that recruitment into observational cohort studies is often easier and less selective. Once a cohort is established, controls for multiple future clinical trials are available without further recruitment efforts. There is now preliminary evidence that recruitment for RCTs within such established cohorts can be highly efficient when compared to recruitment without such cohorts.18
Second, disclosure of information and informed consent can be tailored to the needs of the participants (e.g. those not offered the new intervention are not burdened with information about the risks and potential benefits of trial intervention). Thus, the informed consent process is “patient centered” and “real world” in its goals—replicating, as much as is ethically feasible, the real-world routine healthcare where clinicians provide patients with the information they need, at the time they need it. This may in fact increase the autonomous decision-making by patient-subjects by reducing some of the widely discussed challenges in the consent process, such as decisional burden, confusion, and information overload.19
Third, the design reduces some problems related to patient preferences in standard RCT designs. For instance, when a condition does not have highly effective interventions, the prospect of trying a new, if unproven, intervention is often an incentive for patients to enrol. In standard RCT designs, this often results in those randomized to the “treatment as usual” arm dropping out or experiencing disappointment. But this does not occur in the TwiCs design.
Another advantage of embedding RCTs within an established cohort is that periodic research data collection that is part of the longitudinal study can provide outcome data in addition to data from medical records.10
The TwiCs design does have limitations. It is only applicable to unmasked studies and requires (at least) one “usual care” control arm; however, it is not unusual for pragmatic studies emulating “real world” conditions to have unmasked, usual care control designs. TwiCs will also share the limitations of unmasked studies in general regarding potentially biased outcome reporting.
Another limitation of TwiCs design is the potential bias introduced by non-compliance in the intervention arm (i.e. patients who decline to enroll as well as those who enrol but do not adhere). This involves two issues. First, in the traditional approach to informed consent only those willing to try either arm are recruited. This approach will usually result in fewer dropouts in the intervention arm than in the TwiCs approach. However, this is because traditional designs will have excluded the “unwilling to enroll” at an earlier stage, and the actual number of persons complying with the intervention may be similar in TwiCs. Also, the traditional design is less pragmatic (less generalizable) because only those willing to enter the RCT are enrolled in either arm. Furthermore, in the TwiCs design, added information on the acceptability and adherence rates of new treatments in the real world is provided by the behaviour of those in the intervention arm.
Second, to reduce bias due to non-compliance in the intervention arm, TwiCs studies are typically analyzed as intention-to-treat. But if the dropouts in the intervention arm in TwiCs design are greater than in a traditional design RCT, there could be relative disadvantage in terms of loss of power. One mitigating factor is that because the dropouts in the control arm will be very rare in a TwiCs design, a TwiCs design has “room for more non-compliance” in the intervention arm in comparison with an RCT where non-compliance is expected in both arms.20 This relative power advantage in TwiCs may not apply, however, if the non-compliance in the intervention arm is very high.20
A brief background on RCT designs with consent following randomization
The TwiCs design is a descendent of a family of older proposals variously known as “Zelen design,” “randomized consent,” or “pre-randomization” designs.21 A brief history of these proposals and their implementation illustrates some of their strengths and weaknesses and also helps to clarify how TwiCs is different from these earlier proposals.
The original proposal for post-randomization con-sent, called a “Zelen single-consent design,” (named after the biostatistician who proposed it) was the simplest: patients were, without prior consent or knowledge, randomized between “best standard” care (usual care) and an intervention.21 Subjects assigned to the intervention were then asked for consent, while the others served as control subjects without their knowledge (thus the label “single-consent”). Several advantages were proposed for the single-consent design.21 It reduces the need for investigators to present, and patients to confront, stressful aspects of research participation, such as knowing that their treatment is going to be randomly chosen and being denied access to an experimental treatment. Furthermore, single-consent designs might increase the efficiency of accrual, in part because patients (assigned to the intervention arm) might be more inclined to enroll knowing that they were guaranteed to receive the intervention.
Zelen22 seems to have interpreted the US Federal research regulations to say that as long as research subjects received only “established and accepted methods necessary to meet [their] needs,” informed consent was not necessary. However, the Office of Protection from Research Risks (OPRR) eventually disagreed and reprimanded the investigators of a study of neonates which used a Zelen single-consent design for failing to obtain consent from parents of the control group neonates.23
Criticism of the single-consent procedure led to greater interest in the “double-consent Zelen design,” in which both the usual care and intervention arms are approached for consent. In double consent, in contrast to single consent, all participants are at least informed that they are participating in research. However, in addition to the obvious difference from a traditional RCT in obtaining consent after treatment assignment, Zelen double consent may include little or no information about the other arm of the trial, or indeed about the fact of randomization.13
Trials using Zelen double- and single-consent designs have remained relatively uncommon—as of 2006, two reviews suggest that approximately 83 unique studies employing Zelen designs had been conducted.24,25 This relative unpopularity has no definitive explanation, but the experiences of investigators who have used post-randomization consent designs reveal both ethical and logistical problems.
First, Zelen designs have attracted considerable ethical criticism.26 Even though patients assigned to the control group undergo no harm, and might actually be spared burdens related to a traditional consent process, they might still reasonably expect to know that a new intervention is being tested for their condition and that they have been randomly assigned to a group whose data are used for comparison. The perception that information is being withheld has been described as causing an “outcry” of concern about the ethics of the earliest Zelen proposals, and subsequent modifications have not fully allayed these concerns.27 As we note below, however, despite the 1990 reprimand by the OPRR, pragmatic RCTs are beginning to be conducted in the United States with post-randomization single-consent procedures with the apparent knowledge of the Office of Human Research Protections (the successor to the OPRR).28
Another problem with post-randomization consent is that it has not always proven to be as efficient as had been hoped. Analysis of a post-randomization study has to be done as intention-to-treat, including patients who declined the intervention, which reduces study power.27 Post-randomization designs must improve accrual and withdrawal rates sufficiently to make up for this loss of power; these improvements are difficult to predict and are not guaranteed.22,29,30
Ethics of informed consent for TwiCs and regulatory implications
There are variations in practice when it comes to the content of the initial consent procedures regarding future embedded RCTs within the cohort. We first describe a consent procedure31 which involves the greatest amount of disclosure regarding the elements of potential trials within a cohort. We then discuss other variations.
TwiCs with pre-randomization broad consent about TwiCs elements
In some jurisdictions, investigators implementing TwiCs have run into regulatory obstacles; this has led to the development of a consent model that includes explicit consent for some elements of future embedded trials.31 At the time of recruitment into the cohort, subjects provide specific consent for the cohort study and also provide broad consent—”broad” since the consent covers a range of unspecified future studies—that specifically includes information about randomizations for future TwiCs, for future contact if randomized to the intervention arm of TwiCs, and for use of their data in future TwiCs if randomized to the control arm (see Figure 1).
Figure 1.
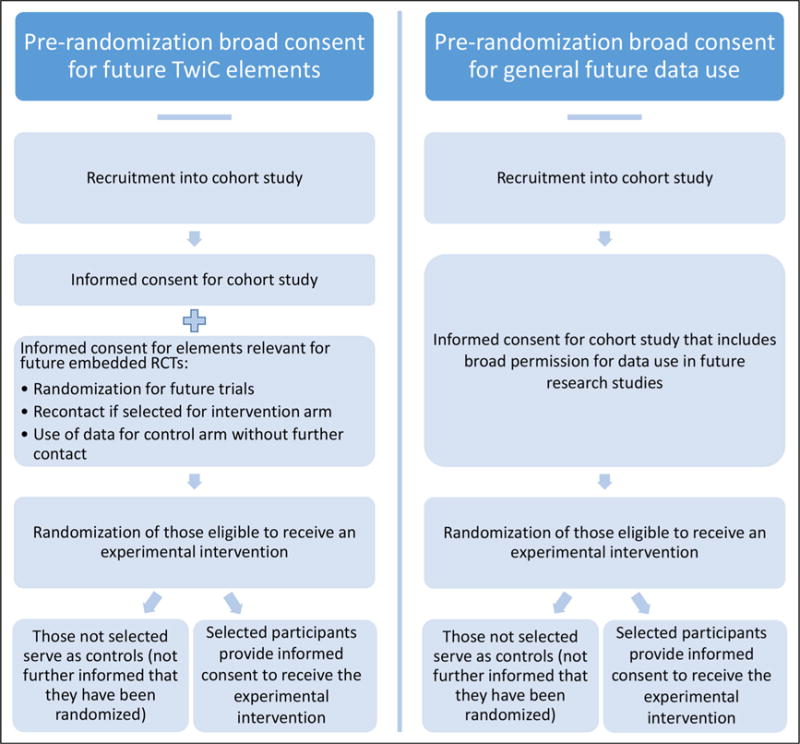
Comparison of a more detailed (left column) versus a general (right column) pre-randomization broad consent for Trials within Cohorts.
For those whose data will serve as the control arm data, their participation in an embedded trial is exhausted by two elements: (a) being randomly selected as a control and (b) use of their data in the embedded trial (usually collected from clinical medical records, or in some cases, from measurements that are part of the longitudinal cohort study10). Despite these elements, the entirety of their clinical experience will be decided by what their physicians consider to be the best care for them. When someone is randomized to the usual care arm, they have been randomized, not to a specific treatment, but to the ordinary interactions and decision-making processes with his or her doctor; thus, how his or her care is determined and provided is not disturbed at all. Furthermore, no additional research measures are needed; thus, no additional interactions or interventions of research are involved when these persons’ data are used in TwiCs. In sum, persons in the control arm receive care that is decided by usual clinical considerations and will have given consent to every element of their research participation.
The lack of specificity in broad consent (i.e. broad permission for future use without specific consent for each use) has led to some prominent controversies in other domains of research, such as the much publicized Havasupai case in which the controversy centered around researchers’ use of samples and data that went beyond the disease domains of initial focus of the research.32 The difference in the TwiCs context is that unlike in most biobank-based research, the cohorts are disease-based (or at risk of it) as are the trials within them; thus, given the specificity of the domain of research, there is little risk of violating any subject’s non-welfare interests such as their cultural, religious, and moral commitments.33 For instance, for a person in a diabetes cohort who provides broad consent for use of their electronic health record and other data for evaluation of future diabetes treatments, there is little danger of patients’ non-welfare interests (regarding the type of uses to which their data are put) being compromised. (However, it should be noted that if a cohort of interest were a very general one—for example, one encompassing all patients in an integrated health delivery system—conducting TwiCs in such a cohort would require further ethical analysis regarding the content of the initial broad consent.)
Those randomized to the intervention arms of TwiCs will have given consent to be approached for enrolment in such trials. After being told that they have been randomly selected to an embedded clinical trial, they will then provide informed consent for the trial intervention. They would not be enrolled in an embedded trial unless they explicitly give consent after they are provided all the usually required elements of informed consent for an RCT. Thus, everyone who enrolls in the intervention arm of the TwiCs will also have given informed consent to every aspect of their research participation in the TwiCs.
TwiCs with only general pre-randomization discussion of future research
Some TwiCs do not obtain explicit pre-randomization consent covering the possibility of future randomization and future contact for intervention studies. Consent is still obtained, but for unspecified future uses of their data (as part of the initial consent for enrolling in the cohort).4,10,16 The rationale is as follows. For the inter-vention arm group of a future embedded trial, when they are randomized into the intervention arm and then subsequently contacted to be asked if they wish to enroll in the trial, it is not that different from someone in a clinic being approached to participate in a traditional RCT. There is no “cold contact” involved; the subjects are aware that the clinic is a locus of clinical research, and they should not be surprised that they are being asked to consider participation in an RCT.
For the control arm, it might be argued that by enrolling in the cohort study (on, say, diabetes), their permission to the researchers to use their medical records and other data includes a variety of future research uses, and this is sufficient to permit their use for comparison purposes in a TwiC testing an intervention to treat diabetes.
There are two potential objections to not employing pre-randomization consent that explicitly includes relevant elements about future embedded trials. First, some may argue that randomization is a research procedure that always requires consent prior to the act of randomization. A contrary point of view would be that, if intervention recipients are being selected to be approached at random from a pool of patients receiving usual care, those not selected do not need to give consent for that random selection any more than individuals who are not selected in a random-digit-dial telephone survey need to give prior consent for randomization. Of course, research studies where the randomization leads to any potential alterations in the way the subjects are treated (e.g. using an experimental intervention) always require consent before such alterations are implemented. But in the control arm of the TwiCs study in question, there are no deviations from the usual way the subjects are treated and in the intervention arm informed consent is obtained before any deviations from the usual are implemented.
Second, it is plausible that some persons, enrolled in a cohort, who later find out that there are embedded randomized trials in that cohort may feel that the researchers could easily have made their plans for embedding trials in the cohort clearer from the beginning. Some of these participants may feel that the researchers were not as transparent as they could have been, even while recognizing that the lack of transparency has no impact on their welfare (benefits and harms/burdens).
Different people will have different moral intuitions about whether pre-randomization broad consent that specifically mentions elements about future embedded trials is ethically necessary. On one side of the argument, there is a potential for mistrust due to the lack of transparency such that it may be not only ethically right but prudent to obtain an explicit consent to future embedded trials and randomization, especially if the burden of obtaining it is low. On the other side is the view that consent is not only unnecessary but could cause confusion (since the idea of broad consent to future randomization with asymmetric consequences for the participants could be a challenging set of concepts to digest), then it may be better to avoid it. We suspect that a part of the answer will rest on the particular features of the TwiCs—the nature of the cohort, the interventions involved, and the setting in which the study is done and the reasonable expectations that researchers might anticipate in the participants.
Implications for US regulations
Although the use of TwiCs is gaining momentum, most of the activity has been in countries outside the United States. Given the potential advantages of the TwiCs design, it may prove useful for US researchers as well. However, the regulations do differ among jurisdictions, especially regarding the issue of when it is permissible to deviate from the traditional informed consent procedures.
How might IRBs apply the US research regulations to TwiCs? The task for the IRBs will be different depending on whether cohort studies employ pre-randomization broad consent for future embedded randomized trials in that cohort. We begin with the assumption that pre-randomization broad consent including explicit discussion of randomization is used.
First, unlike recent debates in the United States, in which the focus has been on whether traditional informed consent is necessary for pragmatic trials in learning health systems,34,35 TwiCs does not need to rely on waivers or alterations of informed consent. As noted above, the intervention arm participants, before consenting to the intervention in the TwiC, would have received all of the information that persons enrolling in traditional RCTs would receive. The only difference is that the information is given (and consent for the intervention obtained) after randomization while consent for the randomization would have been given separately at the time of enrolment into the cohort.
The control arm participants’ consent would not be waived or altered either. They would have provided informed consent for the cohort study, and also given broad consent for randomization and for the use of their data for TwiCs. Since those in the control group will have given consent to every element of their research participation, there is no need to invoke the criteria for waiver or alteration of consent in the Common Rule.
What about TwiCs that are proposed without a substantive pre-randomization broad consent? The regulatory situation could involve the IRBs requiring the investigator to show that the waiver or alteration criteria in the US regulations are met. As we saw above, the intuition concerning the need for pre-randomization broad consent that specifies the elements of future embedded trials varies, and will likely vary among IRBs. It is possible that some IRBs will see the lack of transparency regarding randomization and future TwiCs as implying at least an alteration of informed consent, and therefore will require that such a proposal meet the several regulatory criteria for waiver or alteration of informed consent in 45CFR46.116: (a) the research must be minimal risk, (b) the research would be impracticable to conduct without the waiver or alteration, (c) the participants’ rights or welfare would not be adversely affected by the waiver or alteration, and (d) whenever appropriate, participants are provided with additional pertinent information after participation.
How might these criteria apply to studies that forgo substantive pre-randomization broad consent? Although some TwiCs will be minimal risk, many will not be minimal risk; whether an IRB would or should analyze the risk-benefit issue separately for the intervention and the control arms is not clear. In terms of the practicability of research criterion, it would be difficult to argue that the trial is impracticable without an alteration or waiver since there are examples of TwiCs that are being successfully conducted with substantive pre-randomization broad consent. And we have already noted that some people may see the lack of transparency about randomization into TwiCs as something that goes against their legitimate expectations—this could be interpreted by some as at odds with the condition that waiver or alteration not adversely affect subjects’ rights and welfare.36 Finally, an IRB would need to determine whether debriefing after the embedded trial would be necessary for those assigned to the control arm. Thus, some IRBs could require the use of substantive pre-randomization broad consent for TwiCs.
It is, however, difficult to predict how this issue would finally be decided by the regulators. Of particular interest is a pragmatic clinical trial in the United States involving approximately 20,000 subjects comparing care management, skills training, and treatment as usual for the prevention of suicide attempts among out-patients who endorse suicidal thoughts on a routine clinical measure.28 According to the investigators, this study uses a modified Zelen design (control arm patients are unaware of the RCT; subjects in the intervention arms provide clinical consent to the interventions) that has been approved by IRBs of multiple institutions, and the investigators report having held “extensive discussions” with the Office of Human Research Protections. Thus, it appears that the study is deemed to pose no more than minimal incremental risk and also that it would have been impracticable to conduct without the waiver and alteration of consent, despite what amounts to a single-consent Zelen design. However, the authors do not provide further details about how their IRBs made these determinations.
Conclusion
For conditions in which longitudinal cohort studies can be valuable (which likely includes most chronic conditions), recruiting and conducting multiple randomized trials within such cohorts provide significant scientific and ethical advantages over both traditional and stand-alone Zelen designs. With the increasing emphasis on pragmatic trials,15,37 investigators from many countries are now using this design. One of the main obstacles to its use is the concern over the ethics of obtaining informed consent for the embedded trials after randomizing the subjects and only from the intervention arm. Pre-randomization consent to cohort participation as well as, in some cases, to more explicit broad consent to elements of future TwiCs (including for randomization, and use of data specifically for TwiCs) mitigates this ethical concern. However, regulatory policies vary among jurisdictions and interpretations of those policies vary among research ethics committees. Investigators who hope to benefit from the scientific and practical advantages of the TwiCs design will need to clearly articulate its ethical and scientific strengths and limitations.
Acknowledgments
The authors thank Frank Miller for helpful comments on an earlier draft. The opinions expressed in this article are the authors’ and do not represent the views or policies of the National Institutes of Health, Department of Health and Human Services, or the US government.
Funding
This research was supported in part by the Intramural Research Program at the National Institutes of Health (S.Y.H.K.). C.R. was funded and supported by the Wellcome Trust and the NIHR Collaboration for Leadership in Applied Health Research and Care Yorkshire and Humber (NIHR CLAHRC YH; www.clahrc-yh.nihr.ac.uk) and her views expressed in this paper are not necessarily those of the NHS, the NIHR, or the Department of Health.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Olsen L, Aisner D, McGinnis J. The learning health-care system (IOM roundtable on evidence-based medicine) Washington, DC: National Academies Press; 2007. [Google Scholar]
- 2.Califf RM, Platt R. Embedding cardiovascular research into practice. JAMA. 2013;310:2037–2038. doi: 10.1001/jama.2013.282771. [DOI] [PubMed] [Google Scholar]
- 3.Mitchell N, Hewitt C, Adamson J, et al. A randomised evaluation of CollAborative care and active surveillance for Screen-Positive EldeRs with sub-threshold depression (CASPER): study protocol for a randomized controlled trial. Trials. 2011;12:225. doi: 10.1186/1745-6215-12-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Relton C, Bissell P, Smith C, et al. South Yorkshire Cohort: a “cohort trials facility” study of health and weight—protocol for the recruitment phase. BMC Public Health. 2011;11:640. doi: 10.1186/1471-2458-11-640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clegg A, Relton C, Young J, et al. Improving recruitment of older people to clinical trials: use of the cohort multiple randomised controlled trial design. Age Ageing. 2015;44:547–550. doi: 10.1093/ageing/afv044. [DOI] [PubMed] [Google Scholar]
- 6.Cockayne S, Adamson J, Corbacho MB, et al. The REFORM study protocol: a cohort randomised controlled trial of a multifaceted podiatry intervention for the prevention of falls in older people. BMJ Open. 2014;4:e006977. doi: 10.1136/bmjopen-2014-006977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dickerson J, Bird PK, McEachan RR, et al. Born in Bradford’s Better Start: an experimental birth cohort study to evaluate the impact of early life interventions. BMC Public Health. 2016;15:711. doi: 10.1186/s12889-016-3318-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson JA, Al Sayah F, Wozniak L, et al. Controlled trial of a collaborative primary care team model for patients with diabetes and depression: rationale and design for a comprehensive evaluation. BMC Health Serv Res. 2012;12:258. doi: 10.1186/1472-6963-12-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jongbloed K, Friedman AJ, Pearce ME, et al. The Cedar Project WelTel mHealth intervention for HIV prevention in young Indigenous people who use illicit drugs: study protocol for a randomized controlled trial. Trials. 2016;17:128. doi: 10.1186/s13063-016-1250-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kwakkenbos L, Jewett LR, Baron M, et al. The Scleroderma Patient-centered Intervention Network (SPIN) Cohort: protocol for a cohort multiple randomised con-trolled trial (cmRCT) design to support trials of psycho-social and rehabilitation interventions in a rare disease context. BMJ Open. 2013;3:e003563. doi: 10.1136/bmjopen-2013-003563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mayo NE, Brouillette MJ, Fellows LK, et al. Under-standing and optimizing brain health in HIV now: protocol for a longitudinal cohort study with multiple randomized controlled trials. BMC Neurol. 2016;16:8. doi: 10.1186/s12883-016-0527-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burbach JP, Kurk SA, Van den Braak RRC, et al. Prospective Dutch colorectal cancer cohort: an infrastructure for long-term observational, prognostic, predictive and (randomized) intervention research. Acta Oncol. 2016;55:1273–1280. doi: 10.1080/0284186X.2016.1189094. [DOI] [PubMed] [Google Scholar]
- 13.Flory JH, Mushlin AI, Goodman ZI. Proposals to conduct randomized controlled trials without informed consent: a narrative review. J Gen Intern Med. 2016;31:1511–1518. doi: 10.1007/s11606-016-3780-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Relton C, Torgerson D, O’Cathain A, et al. Rethinking pragmatic randomised controlled trials: introducing the “cohort multiple randomised controlled trial” design. BMJ. 2010;340:c1066. doi: 10.1136/bmj.c1066. [DOI] [PubMed] [Google Scholar]
- 15.Ford I, Norrie J. Pragmatic trials. N Engl J Med. 2016;375:454–463. doi: 10.1056/NEJMra1510059. [DOI] [PubMed] [Google Scholar]
- 16.Uher R, Cumby J, MacKenzie LE, et al. A familial risk enriched cohort as a platform for testing early interventions to prevent severe mental illness. BMC Psychiatry. 2014;14:344. doi: 10.1186/s12888-014-0344-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van der Velden JM, Verkooijen HM, Seravalli E, et al. Comparing conVEntional RadioTherapy with stereotactIC body radiotherapy in patients with spinAL metastases: study protocol for an randomized controlled trial following the cohort multiple randomized controlled trial design. BMC Cancer. 2016;16:909. doi: 10.1186/s12885-016-2947-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Relton C, Burbach M, Collett C, et al. The ethics of “Trials within Cohorts” (TwiCs): 2nd international symposium. Trials. 18:244. [Google Scholar]
- 19.Vickers AJ, Young-Afat DA, Ehdaie B, et al. Just-in-time consent: the ethical case for an alternative to traditional informed consent in randomized trials comparing an experimental intervention with usual care. Clin Trials. 2018;15:3–8. doi: 10.1177/1740774517746610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van der Velden JM, Verkooijen HM, Young-Afat DA, et al. The cohort multiple randomized controlled trial design: a valid and efficient alternative to pragmatic trials? Int J Epidemiol. 2017;46:96–102. doi: 10.1093/ije/dyw050. [DOI] [PubMed] [Google Scholar]
- 21.Zelen M. A new design for randomized clinical trials. N Engl J Med. 1979;300:1242–1245. doi: 10.1056/NEJM197905313002203. [DOI] [PubMed] [Google Scholar]
- 22.Zelen M. Randomized consent designs for clinical trials: an update. Stat Med. 1990;9:645–656. doi: 10.1002/sim.4780090611. [DOI] [PubMed] [Google Scholar]
- 23.Marwick C. NIH “Research Risks Office” reprimands hospital institutional review board. JAMA. 1990;263:2420. [PubMed] [Google Scholar]
- 24.Schellings R, Kessels AG, Ter Riet G, et al. Randomized consent designs in randomized controlled trials: systematic literature search. Contemp Clin Trials. 2006;27:320–332. doi: 10.1016/j.cct.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 25.Adamson J, Cockayne S, Puffer S, et al. Review of randomised trials using the post-randomised consent (Zelen’s) design. Contemp Clin Trials. 2006;27:305–319. doi: 10.1016/j.cct.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 26.Hawkins JS. The ethics of Zelen consent. J Thromb Haemost. 2004;2:882–883. doi: 10.1111/j.1538-7836.2004.00782.x. [DOI] [PubMed] [Google Scholar]
- 27.Ellenberg SS. Informed consent: protection or obstacle? Some emerging issues. Control Clin Trials. 1997;18:628–636. doi: 10.1016/s0197-2456(96)00130-4. [DOI] [PubMed] [Google Scholar]
- 28.Simon GE, Beck A, Rossom R, et al. Population-based outreach versus care as usual to prevent suicide attempt: study protocol for a randomized controlled trial. Trials. 2016;17:452. doi: 10.1186/s13063-016-1566-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ellenberg SS. Randomization designs in comparative clinical trials. N Engl J Med. 1984;310:1404–1408. doi: 10.1056/NEJM198405243102141. [DOI] [PubMed] [Google Scholar]
- 30.Velthuis MJ, May AM, Monninkhof EM, et al. Alternatives for randomization in lifestyle intervention studies in cancer patients were not better than conventional randomization. J Clin Epidemiol. 2012;65:288–292. doi: 10.1016/j.jclinepi.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 31.Young-Afat DA, Verkooijen HM, Van Gils CH, et al. Staged-informed consent in the cohort multiple randomized controlled trial design. Epidemiology. 2016;27:389–392. doi: 10.1097/EDE.0000000000000435. [DOI] [PubMed] [Google Scholar]
- 32.Mello MM, Wolf LE. The Havasupai Indian tribe case—lessons for research involving stored biologic samples. N Engl J Med. 2010;363:204–207. doi: 10.1056/NEJMp1005203. [DOI] [PubMed] [Google Scholar]
- 33.Tomlinson T. Respecting donors to biobank research. Hastings Cent Rep. 2013;43:41–47. doi: 10.1002/hast.115. [DOI] [PubMed] [Google Scholar]
- 34.Platt R, Kass NE, McGraw D. Ethics, regulation, and comparative effectiveness research: time for a change. JAMA. 2014;311:1497–1498. doi: 10.1001/jama.2014.2144. [DOI] [PubMed] [Google Scholar]
- 35.Kim SYH, Miller FG. Varieties of standard-of-care treatment randomized trials: ethical implications. JAMA. 2015;313:895–896. doi: 10.1001/jama.2014.18528. [DOI] [PubMed] [Google Scholar]
- 36.Kim S, Miller F. Waivers and alterations to consent in pragmatic clinical trials: respecting the principle of respect for persons. IRB. 2016;38:1–5. [PubMed] [Google Scholar]
- 37.Loudon K, Treweek S, Sullivan F, et al. The PRECIS-2 tool: designing trials that are fit for purpose. BMJ. 2015;350:h2147. doi: 10.1136/bmj.h2147. [DOI] [PubMed] [Google Scholar]