

Abstract
Replication Protein A (RPA) and RAD51 are DNA binding proteins that help maintain genome stability during DNA replication. These proteins regulate nucleases, helicases, DNA translocases, and signaling proteins to control replication, repair, recombination and the DNA damage response. Their different DNA binding mechanisms, enzymatic activities, and binding partners provide unique functionalities that cooperate to ensure that the right activities are deployed at the right time to overcome replication challenges. Here we discuss the latest discoveries of the mechanisms by which these proteins work to preserve genome stability with a focus on their actions in fork reversal and fork protection.
Introduction
Accurately and completely copying the genome each cell division cycle is essential to prevent disease. This is a daunting challenge considering the trillions of cell divisions in a human lifetime during which billions of bases need to be replicated. Replication stress caused by DNA lesions and conflicts with transcription create additional challenges that must by overcome each cell division cycle1.
The DNA in mammalian cells is replicated by the replisome, which synthesizes DNA in discontinuous stretches on the lagging strand and long continuous sections on the leading strand. This mode of DNA synthesis results in small regions of single strand DNA (ssDNA) on the lagging strand template. When DNA replication is challenged by stress, replication forks “stall”. In many cases, this stalling generates larger stretches of ssDNA on the leading strand because of polymerase and helicase uncoupling2. In addition, ssDNA can be generated by exonucleases that enlarge ssDNA gaps3 or resect reversed or broken forks (see more below).
Management of ssDNA by DNA binding proteins is essential during replication. These proteins protect the DNA from nucleases, recruit replication stress response proteins like checkpoint kinases and regulate enzymes that control replication fork stability and restart. In this review we will focus on the functions of the major eukaryotic ssDNA binding protein RPA and the single- and double-strand DNA binding protein RAD51 in the regulation of replication fork reversal and fork protection.
First responder: RPA
As a highly abundant (~4 million molecules/cell4), high affinity ssDNA binding protein, RPA is expected to be the first responder to ssDNA whether it is formed during normal DNA replication or in response to replication stress. Below we discuss how its biochemical characteristics facilitate its activities in a variety of DNA metabolic processes with a focus on how it regulates replication fork reversal.
Biochemical characteristics of RPA
RPA is a heterotrimer of three subunits RPA70, RPA32 and RPA14 (Figure 1A). The three RPA subunits have a total of six OB (oligonucleotide/oligosaccharide binding) fold domains5. Four of these (70A, 70B, 70C, 32D) act as ssDNA binding domains (Figure 1A). 32D, 70C, and RPA14 help assemble the RPA trimer while 70N acts as a protein interaction domain. In addition to these OB-folds, RPA32 has a winged-helix C terminal domain (RPA32C) that is involved in mediating protein interactions.
Fig. 1. RPA and RAD51 have different biochemical characteristics.
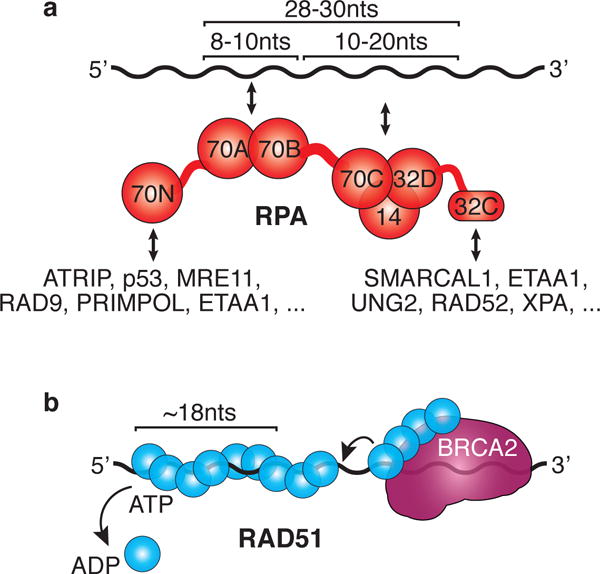
(A) RPA uses modular domain architecture to facilitate dynamic DNA and protein interactions. The size of the ssDNA that is bound by the RPA DNA binding domains is indicated as well as the major protein-protein interaction domains with example binding partners. (B) RAD51 forms a protein-ssDNA filament with the help of BRCA2 and is regulated by its ATPase activity. BRCA2 uses its own ssDNA binding activity to help deliver RAD51 and displace RPA (arrow). ATP hydrolysis promotes filament disassembly.
RPA binds ssDNA with a Kd as low as 10−10 M for optimal DNA ligands of approximately 30 nucleotides6. However, this tight affinity masks the dynamic nature of the binding that is made possible by its modular domain architecture. Individual domains can microscopically associate and dissociate from the DNA7. Two of the four DNA binding domains 70A and 70B act as a single unit to bind 8-10nts of ssDNA8. The other two DNA binding domains, 70C and 32D, are part of the trimer core and engage the ssDNA with lower affinities9. At least 20nts of DNA are required for all of the binding domains to be occupied, but the full DNA footprint is ~28-30nts6. The linker between the 70B and 70C domains also makes DNA contacts and likely participates in overcoming ssDNA secondary structure as RPA transitions between binding modes10. RPA can also diffuse along ssDNA, leading to melting of hairpins, other secondary structures and even short regions of duplex DNA11-13. Thus, RPA-ssDNA interactions should be viewed as highly dynamic with individual domains dissociating and re-associating as RPA adopts different conformations, destabilizes duplex regions and reveals small ssDNA stretches that can act as binding sites for other replication and repair proteins.
Importantly, the domain architecture also means that RPA binding to ssDNA is polar, with 70A-B-C and RPA32D domains binding DNA 5′-3′. The polarity of RPA binding facilitates the proper assembly of protein complexes in some repair contexts14, and as we will discuss later, dictates activities of fork reversal enzymes15.
RPA interacts with a large number of replication and repair proteins. These interactions are often mutually exclusive since many occupy the same 70N and 32C binding surfaces on RPA (Figure 1A). For example, the 70N domain binds to the ATRIP subunit of the ATR replication stress kinase to recruit it to regions of ssDNA16,17. The same 70N surface binds to RAD9, MRE11, p53, PRIMPOL, and ETAA118-21. The 32C domain interacts with several proteins including ETAA1, SMARCAL/HARP, UNG2, XPA, and RAD5221-24. The 70N and 32C domains are connected to the DNA binding domains via flexible, unstructured tethers. Thus, these interactions happen independently of DNA binding. The affinities of the interactions are low, usually in the micromolar range, indicating that they are likely to be transient. The picture that emerges is that both soluble and DNA-bound RPA molecules are constantly exchanging binding partners. Larger assemblies of proteins may form on the DNA in some circumstances to stabilize specific complexes.
The dynamic nature of both DNA and protein interactions allows RPA to fulfill multiple functions at replication forks. First, by binding the lagging strand template as the helicase extrudes it, RPA removes secondary structures25. Second, it promotes priming activity on the lagging strand26. Third, when replication forks stall, it acts as a platform to assemble checkpoint signaling proteins27. Finally, it recruits and regulates the function of numerous fork repair proteins. These activities are critical to prevent fork collapse28.
RPA as a regulator of fork remodeling
Replication fork remodeling is the process of inter-converting the replication fork from a three-way junction to four-way junction and back (Figure 2A). Conversion from a three-way to four-way junction is called fork reversal or fork regression, while the opposite reaction is fork restoration. During fork reversal, rewinding of the parental DNA duplex and the protrusion of the nascent DNA strands forms a reversed replication fork or “chicken-foot”.
Fig. 2. Replication fork reversal is regulated by RPA and RAD51.
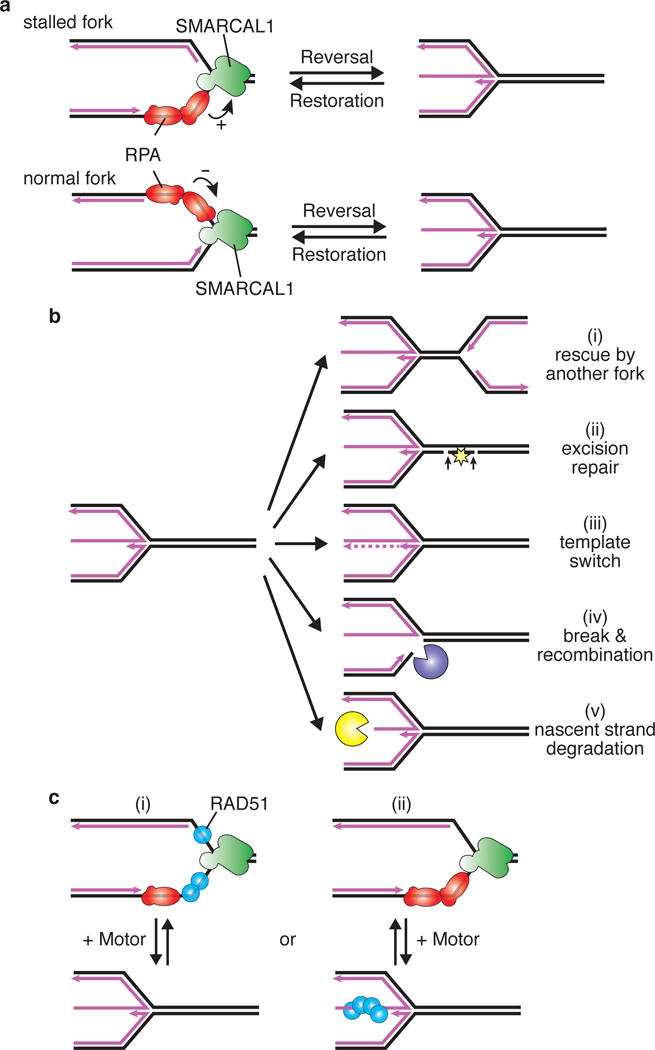
(A) RPA stimulates SMARCAL1 to catalyze fork reversal when bound to the leading strand template “stalled fork” but inhibits SMARCAL1 when bound to the lagging strand “normal fork”. The polarity difference of RPA bound to these substrates is illustrated since that contributes to SMARCAL1 regulation. Pink lines indicate nascent DNA while template DNA is illustrated in black. (B) Reversed forks are intermediates in fork stabilization and restart mechanisms, but are also susceptible to nuclease processing. Five outcomes of fork reversal are illustrated and described further in the text. (C) Two models for how RAD51 may promote fork reversal. First, RAD51 may bind the template ssDNA of the stalled fork to cooperate with motor proteins like SMARCAL1 and ZRANB3 to drive reversal. Second, RAD51 could capture the reversed fork, driving an equilibrium towards fork reversal.
There are at least four ways fork reversal can promote genetic stability (Figure 2B)29,30. First, fork reversal could stabilize the stalled fork until a converging fork from a nearby origin passively replicates the region. Second, the formation of the reversed fork places any DNA lesion on the template strand that caused polymerase stalling back into the context of duplex DNA. This would then allow repair of the damage by excision repair mechanisms. Third, annealing of the nascent strands could provide an undamaged template to synthesize past the lesion in a template switching mechanism. Finally, the reversed fork can be cleaved by endonucleases such as MUS81 or other SLX4-dependent nucleases to promote recombination-mediated repair31.
In contrast to an active replication fork, a subset of stalled forks would be expected to have RPA on the leading template strand due to the stalling of Polε. During the process of fork reversal, RPA must be removed from the parental ssDNA as the template strands re-form a DNA duplex32. The SNF2 family DNA translocase SMARCAL1/HARP can catalyze both reversal and restoration reactions on RPA-bound substrates33. SMARCAL1 interacts directly with the 32C domain of RPA, and this interaction is required for its recruitment to stalled replication forks22,23. Moreover, the interaction regulates SMARCAL1 activity. Specifically, RPA stimulates SMARCAL1 mediated fork reversal activity when it is bound to the leading template strand and inhibits its function when bound to the lagging strand32. The polar binding of RPA to the ssDNA facilitates this regulatory difference (Figure 2A)15. Thus, SMARCAL1 has exactly the kind of activity required to reverse replication forks that stall because of a leading strand template problem.
SMARCAL1 works in repetitive bursts in which small amounts of reversal are followed by pausing events32. Pausing provides opportunities for regulation, direction switching, or hand-off to another enzyme. For example, after SMARCAL1 engages the fork DNA, it is phosphorylated by ATR22,34-36. This phosphorylation inhibits SMARCAL1 activity and is important to prevent “excessive” fork reversal which can lead to fork breakage36. Second, pausing allows the fork to test whether the obstacle to replication has been resolved. The T4 UvsW protein, highly similar in structure to SMARCAL137, also exhibits repetitive fork reversal activity and an ability to switch branch migration direction to catalyze fork restoration38. Combined, these activities promote lesion bypass by template switching without replisome dissociation39. Thus, fork reversal may often be a transient intermediate that rapidly resolves back to an elongating fork unless the replication stress is persistent.
SMARCAL1 is only one of several proteins that can reverse replication forks. Other SNF2 family members including ZRANB3 and HLTF also perform this function in vitro and in cells32,40-42. Each has unique functional domains and regulation as reviewed elsewhere43. For example, in contrast to SMARCAL1, RPA bound to a leading strand gap that mimics a stalled fork acts as a block to ZRANB3 fork reversal32. HLTF has not been studied with the same substrates, but it does exhibit different DNA substrate preferences from SMARCAL1 and ZRANB341. Distinct substrate recognition and protein-interaction domains on these proteins generate these substrate differences32,33,37,41,44.
Second responder: RAD51
RAD51 binds both ssDNA and dsDNA, and is best known for its actions in catalyzing strand invasion during double strand break (DSB) repair by homologous recombination (HR). RAD51 also has essential functions at replication forks, including in regulating fork reversal, that are genetically separable from DSB repair, and these will be our focus.
RAD51 biochemical characteristics
RAD51 is the eukaryotic ortholog of the E. coli recombinase, RecA45. Like RecA, RAD51 forms nucleoprotein filaments on ssDNA through a cooperative DNA binding mechanism (Figure 1B)46,47. However, RAD51 has modest affinity (Kd~10−6M) for both ssDNA and dsDNA48,49. Each RAD51 monomer binds three nucleotides of ssDNA and six monomers generate one turn of a helical RAD51-ssDNA filament50. RAD51 is also a DNA-dependent ATPase. It is loaded onto DNA in its ATP-bound form, and after ATP hydrolysis the ADP-RAD51 filament is less stable51,52. Thus, by hydrolyzing ATP in response to DNA binding, RAD51 is self-inactivating. This inherent instability of the RAD51-DNA filament is only the first of many regulatory mechanisms that are important to ensure RAD51 function is directed properly.
RAD51 requires positively acting mediator proteins to access RPA-bound ssDNA53. In mammalian cells, a primary mediator is the tumor suppressor, BRCA253. Like RPA, BRCA2 uses OB folds to bind ssDNA in addition to a less well characterized second ssDNA binding region54. BRCA2 ssDNA binding helps to displace RPA and load RAD51. BRCA2 BRC repeats promote RAD51 filament nucleation by delivering ~4 RAD51 monomers to the DNA as a unit, inhibiting RAD51 ATPase activity, and reducing the association of RAD51 with dsDNA55-57. Additional BRC repeats and a C-terminal region help stabilize the RAD51-ssDNA filament. Other RAD51 mediators, including RAD51 paralogs, may cooperate with BRCA2 or act in different contexts to promote RAD51 filament formation and stabilization53.
RAD51 as a fork reversal enzyme
In E. coli, the RecG motor protein acts like SMARCAL1 to reverse replication forks58. This function is stimulated by the E. coli SSB with exactly the same leading vs. lagging strand preference as SMARCAL132,59. However, the RecA recombinase has also been implicated in fork reversal and can catalyze this reaction in vitro, a function that is independent of the RecFOR proteins (BRCA2 orthologs in E. coli)60. Unlike RecG, RecA catalyzed fork reversal is inhibited by E. coli SSB61.
In human cells, RAD51 is also required for fork reversal62. Interestingly, like in prokaryotes, this fork reversal function of RAD51 is independent of the BRCA2 mediator63,64. The diffusion properties of BRCA2 and RAD51 suggest that most cellular RAD51 is bound to BRCA2 all the time65, so it is surprising that BRCA2 is not needed for RAD51-dependent fork reversal. Unlike RecA, RAD51 does not have fork-remodeling activity on its own, although it may stimulate fork reversal by other proteins66. These observations raise a number of questions. Does RAD51 bind to the stalled replication fork and drive fork reversal in cells? If so, how does it access the ssDNA without the BRCA2 mediator protein? Are RPA and RAD51 binding the same stalled fork at the same time? How does RAD51 cooperate with motor proteins like SMARCAL1 and ZRANB3?
While some models have RAD51 binding to the parental ssDNA to catalyze fork reversal, thinking of fork reversal as a dynamic process with the reversed fork in equilibrium with the restored fork suggests a second possibility (Figure 2C). When the fork reverses because of a leading strand lesion, the expectation is that there will be ssDNA in the reversed arm since the lengths of the nascent leading and lagging strands are different. RAD51 binding to that ssDNA end could capture it and drive the equilibrium towards fork reversal, thus explaining the genetic requirement for RAD51 to observe reversed forks. This idea fits with the ability of RAD51 to inhibit fork restoration in some contexts66. Perhaps, the initial extruded ssDNA tail is too small (<8nts) for the 70AB domains of RPA to bind – thereby precluding the need for displacement by BRCA2. Alternatively, there could be another mediator protein, such as MMS22L-TONSL complex that is important for the fork reversal functions of RAD5167-69, or it is even possible that RAD51 dsDNA binding is involved in fork reversal. Biochemical reconstitution of these reactions and additional analysis of where and when RAD51 binds in comparison to RPA will be needed to test these ideas.
Excessive fork reversal can be detrimental to genome stability
Fork reversal preserves genome stability. However, as is typical of many DNA repair and tolerance mechanisms, this intermediate in DNA processing can be deleterious if not properly controlled. For example, reversed forks have been implicated as the cause of DSBs associated with transcription-replication collisions59, UV-induced DNA damage70, and in bacterial strains deficient in the helicase DnaB29. Additionally, reversed forks may cause the genome instability associated with the rDNA region in S. cerevisiae71,72 and the hyper-recombination in the E. coli termination region73. In mammalian cells, deregulation of the fork remodeler, SMARCAL1, either by overexpression22 or by ATR inhibition36, causes increased genome instability and breaks.
These deleterious consequences of fork reversal can be traced to its 4-way junction structure with an exposed DNA end. The junction itself can be targeted by structure-selective endonucleases (Figure 2B). Cleavage would generate a true DSB that would then require recombination for repair. While regulated recombination may be beneficial to resume replication, it can also lead to chromosomal rearrangements and instability through inappropriate recombination and microhomology mediated recombination events74. The reversed nascent-nascent duplex could also be bound by DSB repair proteins like KU70/80 or by telomerase if it contains telomeric sequences75. These proteins may interfere with fork restoration or cause telomere dysfunction, respectively. Finally, the end of the reversed arm is an access point for nucleases like MRE11 as part of the MRE11-RAD50-NBS1 (MRN) complex. MRE11 could remove end-binding proteins to facilitate fork restoration; however, excessive nuclease activity at persistently stalled forks can be problematic.
RAD51 as a fork protection enzyme
As first described for RecA in E. coli, RAD51 binding to the reversed arm of the chicken foot protects it from excessive degradation mediated by exonucleases. This process is called fork protection and is dependent on BRCA2-mediated stabilization of RAD51-ssDNA filaments76,77. Thus, BRCA2-deficient cells exhibit fork instability or nascent strand degradation. A BRCA2 C-terminal region that binds and stabilizes RAD51 filaments is required for fork protection even though it is dispensable for repair of site-specific DSBs76. Fork degradation in BRCA2-deficient cells can be restored by overexpression of a mutant RAD51 protein that cannot hydrolyze ATP and forms hyper-stable filaments. RAD51 binding can also protect DNA from MRE11-dependent degradation in vitro; a function not shared by RPA63.
Many additional HR proteins, including BRCA1, the RAD51 paralogs and the Fanconi Anemia (FA) proteins as well as proteins implicated in chromatin regulation like the histone methyltransferase EZH2 are also needed to prevent fork degradation (see Box 1 and Table 1). Adding to the complexity is that multiple nucleases are implicated in the degradation. While BRCA1/2 and the FA pathway prevent MRE11-dependent degradation76-78, other factors such as BOD1L prevent DNA2-mediated degradation, also by stabilizing RAD51 on ssDNA79. Thus, RAD51 loss can cause MRE11 or DNA2 dependent fork degradation. Other nucleases like EXO1 and MUS81 may also be involved in some cases80,81. The mechanisms governing the nuclease choice are unknown. Since both the leading and lagging nascent strands are degraded, one might expect two nucleases of different polarities (5′-3′ and 3′-5′) to be involved. Finally, there are RAD51-independent pathways that prevent fork degradation, raising the possibility that fork protection actually involves multiple different processes82.
Box 1. Replication fork protection.
While many proteins have been implicated in fork protection, multiple conceptual questions about this genome maintenance pathway remain to be answered.
First, is fork protection a single pathway or an assembly of multiple distinct mechanisms? Until recently, the unifying theme of fork protection was the need to stabilize a RAD51-ssDNA filament to prevent nuclease action. However, there are reports of RAD51-independent mechanisms of fork protection82. Combined with the involvement of different nucleases in different contexts, this observation suggests that the fork protection assay may be monitoring more than one type of fork processing.
Second, just about every double-strand break repair protein examined seems to regulate fork protection. Is the end of the reversed arm of the chicken foot subject to the same processing events as every other type of double strand break or are there specific mechanisms that discriminate these types of reversed fork ends? Also, what does the end look like? How much ssDNA is there and does it change during processing?
Third, how is fork protection physiologically important? The fork protection assay requires stalling all forks for extended periods of time, and even in these circumstances removal of the replication challenge leads to rapid fork restart. The non-physiological experimental perturbation nevertheless correlates with physiological significance since a patient-derived RAD51 mutant (T131P) is defective in fork protection but not HR86, BRCA1 heterozygosity causes fork degradation and may promote tumorigenesis97, and genetic backgrounds that restore fork protection to BRCA2-deficient cancer cells without restoring HR cause chemotherapy and PARP inhibitor resistance98. Further studies are needed to understand the importance of fork protection to determining cancer etiology and therapeutic response.
Table 1.
List of select proteins involved in fork reversal and fork protection.
| Protein | References |
|---|---|
| Fork Remodelers | |
| HLTF | 41,84 |
| RAD54 | 66 |
| SMARCAL1 | 33,63,84 |
| ZRANB3 | 32,40,42,44,84 |
| Homologous recombination and Fanconi Anemia proteins | |
| BRCA1 | 78 |
| BRCA2 | 76 |
| FANCA, B, D2 | 78,99 |
| PALB2 | 100 |
| RAD51 | 62,76,77 |
| RAD51C, XRCC2, XRCC3 | 101 |
| RAD52 | 64 |
| Nucleases and helicases | |
| BLM | 79,99,102 |
| DNA2 | 79,103 |
| EXO1 | 81 |
| FBH1 | 79 |
| MRE11 | 76,77 |
| MUS81 | 80 |
| RECQ1 | 103,104 |
| RECQL5 | 99 |
| WRN | 81,103 |
| Other regulators | |
| ABRO1 | 82 |
| ATR | 36,105 |
| ATRX-DAXX | 106 |
| BOD1L | 79 |
| CHD4 | 98 |
| EZH2 | 80 |
| MLL2/3 | 98 |
| MMS22L-TONSL | 69 |
| PARI | 107 |
| PARP1 | 104,108 |
| PTIP | 98 |
| RADX | 87 |
| REV1 | 109 |
| WRNIP1 | 110 |
| 53BP1 | 111,112 |
In all cases where it has been examined, a reversed fork is the entry point for nucleases to degrade the newly synthesized DNA. Thus, silencing any one of several fork reversal enzymes including SMARCAL1, ZRANB3, HLTF and even RAD51, restores fork protection to BRCA2-deficient cells63,64,83,84. Intriguingly, while nascent strand degradation removes thousands of bases, only small ssDNA gaps are visible by electron microscopy and degraded forks rapidly restart once the replication block is removed63,64,76.
To explain these observations, we suggest the following model (Figure 3): Upon encountering a replication challenge that exposes leading template strand ssDNA, RPA directs SMARCAL1 to initiate fork reversal. Often, this reversal will be transient and fork restoration will then lead to fork restart or a converging fork will complete DNA synthesis. Alternatively, RAD51 might capture the nascent lagging strand ssDNA that is exposed. If the ssDNA overhang is not large or if polymerases fill it in, the duplexed DNA end could be captured by end-binding proteins like KU85. Just as it does at DSBs, MRE11 may act to remove these proteins to allow fork restoration. Fork processing by polymerases and nucleases combined with post-translational modifications to replisome proteins like PCNA ubiquitylation, may generate good substrates for the other fork reversal enzymes like ZRANB3 and HLTF. If the replication blockage is persistent, then a more extensive RAD51-ssDNA filament may stabilize the reversed fork. However, in the absence of BRCA2, progressive rounds of reversal and nuclease action could slowly degrade large amounts of nascent ssDNA. Each time this happens, there is a chance that incorrect processing or premature entry into mitosis could generate the gaps, breaks, and chromosomal aberrations associated with nascent strand degradation. Nonetheless, most forks would be capable of rapidly resuming DNA synthesis once the replication stress challenge is removed.
Fig. 3. Nascent strand degradation may be due to multiple rounds of fork reversal and nuclease action.
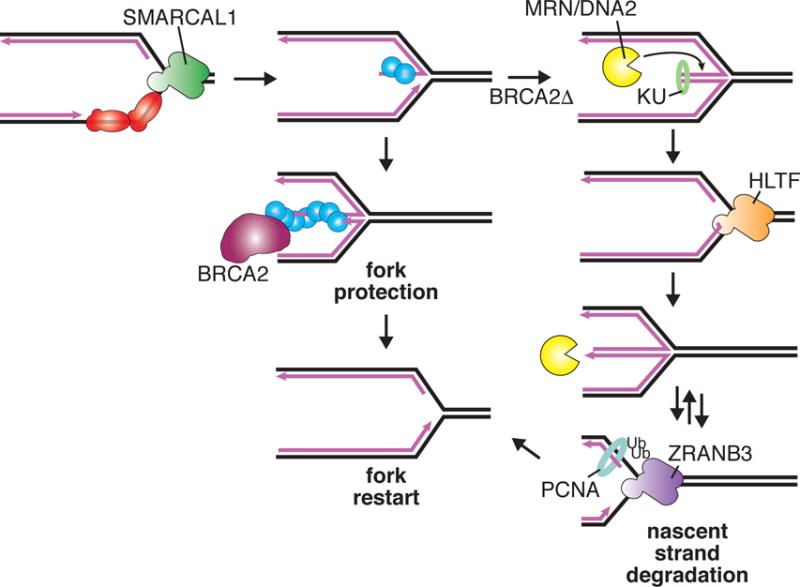
When RAD51-DNA filaments are not stabilized, nucleases including MRN and DNA2 can access the end of the reversed arm. Limited nascent strand degradation could be beneficial to remove end-binding proteins and allow fork restart. However, in the context of a persistent fork block, multiple rounds of end processing and reversal by motor proteins with different substrate specificities yields extensive degradation and repeated opportunities for mistakes that generate chromosomal abnormalities. Depicted is a speculative model of the motor proteins acting sequentially. Ub = ubiquitin.
Differential requirements for RAD51 in fork reversal, protection and HR
Since RAD51 is needed for fork reversal and fork degradation proceeds from the reversed fork, RAD51-deficient cells should not exhibit fork degradation because the substrate, the reversed fork, should not form. Indeed, silencing RAD51 by siRNA was reported to prevent fork degradation64,83. However, cells expressing a partial loss of function mutant RAD51 (T131P) and cells treated with a RAD51 inhibitor do exhibit fork degradation63,84,86. One possible explanation for this apparent discrepancy is that fork protection may be more sensitive to RAD51 inhibition than fork reversal because less RAD51 may be needed to perform reversal as compared to protection (Figure 4A). Consistent with this hypothesis, we found that at high concentrations, two potent RAD51 siRNAs prevented fork degradation. However, at intermediate concentrations of the same siRNAs, we observed nascent strand degradation (Bhat and Cortez, unpublished). This differential requirement of RAD51 for reversal vs. protection is also consistent with the need for BRCA2 for fork protection but not fork reversal and the observation that overexpression of a RAD51 antagonist, RADX, also causes fork degradation87.
Fig. 4. Different functions of RAD51 may require different amounts of protein and ssDNA.
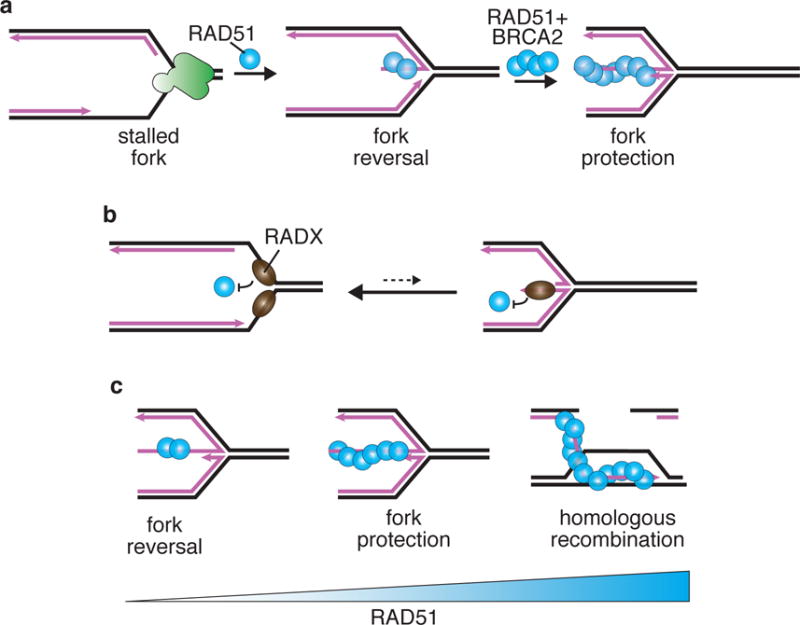
(A) RAD51 is needed for both fork reversal and fork protection. Fork reversal does not require BRCA2 and may require less cellular RAD51 protein than fork protection. (B) During normal replication, transient exposures of ssDNA at an elongating replication fork may be shielded from RAD51 by RADX to prevent unnecessary fork reversal. (B) Increasing amounts of RAD51 protein/activity may be needed for fork reversal, fork protection, and HR.
While RAD51 is required for replication and repair, too much RAD51 can be detrimental to cells. Indeed, RAD51 overexpression causes genome instability88,89 and RAD51 is frequently overexpressed in cancers90-92. Similarly, RecA overexpression is also detrimental to E. coli. RecA activity is regulated by RecX, a protein that caps RecA-ssDNA filaments93. RADX may be a functional analog of RecX and may operate with additional RAD51 antagonists like RecQ helicases and PARI to prevent inappropriate RAD51 activity94.
RADX is a ssDNA binding protein that contains three RPA-like OB-fold domains and is required to prevent fork cleavage by the MUS81 endonuclease87. This function is tied to RAD51 regulation since RADX-deficient cells accumulate excessive fork-bound RAD51 that is hypothesized to cause inappropriate fork reversal even in the absence of exogenous replication stress (Figure 4B). RAD51 and fork reversal might be a threat to normal elongating forks because leading and lagging strand polymerases may not always be coordinated and ssDNA intermediates could form even at active forks, as shown recently for the E. coli replisome95. If this is true in vertebrates, RADX may be needed to counteract RAD51 in the absence of stress. Indeed, reducing RAD51 levels suppresses the fork breakage phenotype of RADX-deficient cells87.
RADX functionally acts as a buffer for RAD51, supplementing its self-inactivating ATPase activity and other antagonists to prevent inappropriate RAD51 action at replication forks. Silencing RADX increases the amount of RAD51 that can access forks in cells even when RAD51 mediators like BRCA2 are inactive. Therefore, RADX depletion rescues the fork degradation observed in BRCA2 deficient cells87. This correlates with an increase in viability and partial resistance to PARP inhibitors. Consistent with only a partial effect on RAD51, RADX deficiency is unable to overcome the requirement for BRCA2 in HR and overexpression only modestly reduces HR efficiencies although it can cause fork degradation87. These data further argue that the three functions of RAD51 in fork reversal, fork protection and HR repair require increasing amounts of RAD51 function (Figure 4C). An important determinant of the differential requirements of RAD51 might be the amount or persistence of ssDNA. End resection at a DSB can yield long stretches of ssDNA (several hundred to thousands of nucleotides)96 while the ssDNA at a stalled fork averages less than 100 bases62.
Summary and Outlook
RAD51 and RPA have different biochemical characteristics and functions. However, both are essential regulators of replication fork stability through management of ssDNA. The ability of both proteins to promote fork reversal facilitates responses to replication challenges but is also dangerous since reversed forks are access points for nucleases. As with most DNA repair mechanisms, the management of intermediates is essential and failures can generate worse outcomes than not attempting the repair at all. Many unanswered questions about mechanisms remain, but the explosion of interest in replication stress responses will certainly speed our understanding of these complex and critical genome maintenance processes.
References
- 1.Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16:2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–52. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garcia-Rodriguez N, Morawska M, Wong RP, Daigaku Y, Ulrich HD. Spatial separation between replisome- and template-induced replication stress signaling. EMBO J. 2018 doi: 10.15252/embj.201798369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takai KK, Kibe T, Donigian JR, Frescas D, de Lange T. Telomere protection by TPP1/POT1 requires tethering to TIN2. Mol Cell. 2011;44:647–59. doi: 10.1016/j.molcel.2011.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flynn RL, Zou L. Oligonucleotide/oligosaccharide-binding fold proteins: a growing family of genome guardians. Crit Rev Biochem Mol Biol. 2010;45:266–75. doi: 10.3109/10409238.2010.488216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim C, Paulus BF, Wold MS. Interactions of human replication protein A with oligonucleotides. Biochemistry. 1994;33:14197–206. doi: 10.1021/bi00251a031. [DOI] [PubMed] [Google Scholar]
- 7.Gibb B, Ye LF, Gergoudis SC, Kwon Y, Niu H, Sung P, Greene EC. Concentration-dependent exchange of replication protein A on single-stranded DNA revealed by single-molecule imaging. PLoS One. 2014;9:e87922. doi: 10.1371/journal.pone.0087922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arunkumar AI, Stauffer ME, Bochkareva E, Bochkarev A, Chazin WJ. Independent and coordinated functions of replication protein A tandem high affinity single-stranded DNA binding domains. J Biol Chem. 2003;278:41077–82. doi: 10.1074/jbc.M305871200. [DOI] [PubMed] [Google Scholar]
- 9.Bochkareva E, Korolev S, Lees-Miller SP, Bochkarev A. Structure of the RPA trimerization core and its role in the multistep DNA-binding mechanism of RPA. Embo J. 2002;21:1855–63. doi: 10.1093/emboj/21.7.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fan J, Pavletich NP. Structure and conformational change of a replication protein A heterotrimer bound to ssDNA. Genes Dev. 2012;26:2337–47. doi: 10.1101/gad.194787.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nguyen B, Sokoloski J, Galletto R, Elson EL, Wold MS, Lohman TM. Diffusion of human replication protein A along single-stranded DNA. J Mol Biol. 2014;426:3246–3261. doi: 10.1016/j.jmb.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kemmerich FE, Daldrop P, Pinto C, Levikova M, Cejka P, Seidel R. Force regulated dynamics of RPA on a DNA fork. Nucleic Acids Res. 2016;44:5837–48. doi: 10.1093/nar/gkw187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen R, Subramanyam S, Elcock AH, Spies M, Wold MS. Dynamic binding of replication protein a is required for DNA repair. Nucleic Acids Res. 2016;44:5758–72. doi: 10.1093/nar/gkw339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Laat WL, Appeldoorn E, Sugasawa K, Weterings E, Jaspers NG, Hoeijmakers JH. DNA-binding polarity of human replication protein A positions nucleases in nucleotide excision repair. Genes Dev. 1998;12:2598–609. doi: 10.1101/gad.12.16.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhat KP, Betous R, Cortez D. High-affinity DNA-binding domains of replication protein A (RPA) direct SMARCAL1-dependent replication fork remodeling. J Biol Chem. 2015;290:4110–7. doi: 10.1074/jbc.M114.627083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–8. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 17.Ball HL, Myers JS, Cortez D. ATRIP Binding to RPA-ssDNA Promotes ATR-ATRIP Localization but Is Dispensable for Chk1 Phosphorylation. Mol Biol Cell. 2005;16:2372–2381. doi: 10.1091/mbc.E04-11-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu X, Vaithiyalingam S, Glick GG, Mordes DA, Chazin WJ, Cortez D. The basic cleft of RPA70N binds multiple checkpoint proteins including RAD9 to regulate ATR signaling. Mol Cell Biol. 2008;28:7345–7353. doi: 10.1128/MCB.01079-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bochkareva E, Kaustov L, Ayed A, Yi GS, Lu Y, Pineda-Lucena A, Liao JC, Okorokov AL, Milner J, Arrowsmith CH, Bochkarev A. Single-stranded DNA mimicry in the p53 transactivation domain interaction with replication protein A. Proc Natl Acad Sci U S A. 2005;102:15412–7. doi: 10.1073/pnas.0504614102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guilliam TA, Brissett NC, Ehlinger A, Keen BA, Kolesar P, Taylor EM, Bailey LJ, Lindsay HD, Chazin WJ, Doherty AJ. Molecular basis for PrimPol recruitment to replication forks by RPA. Nat Commun. 2017;8:15222. doi: 10.1038/ncomms15222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bass TE, Luzwick JW, Kavanaugh G, Carroll C, Dungrawala H, Glick GG, Feldkamp MD, Putney R, Chazin WJ, Cortez D. ETAA1 acts at stalled replication forks to maintain genome integrity. Nat Cell Biol. 2016;18:1185–1195. doi: 10.1038/ncb3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bansbach CE, Betous R, Lovejoy CA, Glick GG, Cortez D. The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev. 2009;23:2405–14. doi: 10.1101/gad.1839909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ciccia A, Bredemeyer AL, Sowa ME, Terret ME, Jallepalli PV, Harper JW, Elledge SJ. The SIOD disorder protein SMARCAL1 is an RPA-interacting protein involved in replication fork restart. Genes Dev. 2009;23:2415–25. doi: 10.1101/gad.1832309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mer G, Bochkarev A, Gupta R, Bochkareva E, Frappier L, Ingles CJ, Edwards AM, Chazin WJ. Structural basis for the recognition of DNA repair proteins UNG2, XPA, and RAD52 by replication factor RPA. Cell. 2000;103:449–56. doi: 10.1016/s0092-8674(00)00136-7. [DOI] [PubMed] [Google Scholar]
- 25.Sugiyama T, New JH, Kowalczykowski SC. DNA annealing by RAD52 protein is stimulated by specific interaction with the complex of replication protein A and single-stranded DNA. Proc Natl Acad Sci U S A. 1998;95:6049–54. doi: 10.1073/pnas.95.11.6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Melendy T, Stillman B. An interaction between replication protein A and SV40 T antigen appears essential for primosome assembly during SV40 DNA replication. J Biol Chem. 1993;268:3389–95. [PubMed] [Google Scholar]
- 27.Saldivar JC, Cortez D, Cimprich KA. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol. 2017 doi: 10.1038/nrm.2017.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, Mailand N, Bartek J, Lukas J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell. 2013;155:1088–103. doi: 10.1016/j.cell.2013.10.043. [DOI] [PubMed] [Google Scholar]
- 29.Atkinson J, McGlynn P. Replication fork reversal and the maintenance of genome stability. Nucleic Acids Res. 2009;37:3475–92. doi: 10.1093/nar/gkp244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neelsen KJ, Lopes M. Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat Rev Mol Cell Biol. 2015;16:207–20. doi: 10.1038/nrm3935. [DOI] [PubMed] [Google Scholar]
- 31.Sarbajna S, West SC. Holliday junction processing enzymes as guardians of genome stability. Trends Biochem Sci. 2014;39:409–19. doi: 10.1016/j.tibs.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 32.Betous R, Couch FB, Mason AC, Eichman BF, Manosas M, Cortez D. Substrate-selective repair and restart of replication forks by DNA translocases. Cell Rep. 2013;3:1958–69. doi: 10.1016/j.celrep.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Betous R, Mason AC, Rambo RP, Bansbach CE, Badu-Nkansah A, Sirbu BM, Eichman BF, Cortez D. SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes & development. 2012;26:151–62. doi: 10.1101/gad.178459.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Postow L, Woo EM, Chait BT, Funabiki H. Identification of SMARCAL1 as a component of the DNA damage response. J Biol Chem. 2009;284:35951–61. doi: 10.1074/jbc.M109.048330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuan J, Ghosal G, Chen J. The annealing helicase HARP protects stalled replication forks. Genes Dev. 2009;23:2394–9. doi: 10.1101/gad.1836409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Betous R, Carroll CM, Jung SY, Qin J, Cimprich KA, Cortez D. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 2013;27:1610–23. doi: 10.1101/gad.214080.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mason AC, Rambo RP, Greer B, Pritchett M, Tainer JA, Cortez D, Eichman BF. A structure-specific nucleic acid-binding domain conserved among DNA repair proteins. Proc Natl Acad Sci U S A. 2014 doi: 10.1073/pnas.1324143111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manosas M, Perumal SK, Bianco P, Ritort F, Benkovic SJ, Croquette V. RecG and UvsW catalyse robust DNA rewinding critical for stalled DNA replication fork rescue. Nat Commun. 2013;4:2368. doi: 10.1038/ncomms3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manosas M, Perumal SK, Croquette V, Benkovic SJ. Direct observation of stalled fork restart via fork regression in the T4 replication system. Science. 2012;338:1217–20. doi: 10.1126/science.1225437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ciccia A, Nimonkar AV, Hu Y, Hajdu I, Achar YJ, Izhar L, Petit SA, Adamson B, Yoon JC, Kowalczykowski SC, Livingston DM, Haracska L, Elledge SJ. Polyubiquitinated PCNA Recruits the ZRANB3 Translocase to Maintain Genomic Integrity after Replication Stress. Mol Cell. 2012;47:396–409. doi: 10.1016/j.molcel.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kile AC, Chavez DA, Bacal J, Eldirany S, Korzhnev DM, Bezsonova I, Eichman BF, Cimprich KA. HLTF’s Ancient HIRAN Domain Binds 3′ DNA Ends to Drive Replication Fork Reversal. Mol Cell. 2015;58:1090–100. doi: 10.1016/j.molcel.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vujanovic M, Krietsch J, Raso MC, Terraneo N, Zellweger R, Schmid JA, Taglialatela A, Huang JW, Holland CL, Zwicky K, Herrador R, Jacobs H, Cortez D, Ciccia A, Penengo L, Lopes M. Replication Fork Slowing and Reversal upon DNA Damage Require PCNA Polyubiquitination and ZRANB3 DNA Translocase Activity. Mol Cell. 2017;67:882–890 e5. doi: 10.1016/j.molcel.2017.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poole LA, Cortez D. Functions of SMARCAL1, ZRANB3, and HLTF in maintaining genome stability. Crit Rev Biochem Mol Biol. 2017;52:696–714. doi: 10.1080/10409238.2017.1380597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Badu-Nkansah A, Mason AC, Eichman BF, Cortez D. Identification of a Substrate Recognition Domain in the Replication Stress Response Protein Zinc Finger Ran-binding Domain-containing Protein 3 (ZRANB3) J Biol Chem. 2016;291:8251–7. doi: 10.1074/jbc.M115.709733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shinohara A, Ogawa H, Ogawa T. Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein [published erratum appears in Cell 1992 Oct 2;71(1):following 180] Cell. 1992;69:457–70. doi: 10.1016/0092-8674(92)90447-k. [DOI] [PubMed] [Google Scholar]
- 46.Mine J, Disseau L, Takahashi M, Cappello G, Dutreix M, Viovy JL. Real-time measurements of the nucleation, growth and dissociation of single Rad51-DNA nucleoprotein filaments. Nucleic Acids Res. 2007;35:7171–87. doi: 10.1093/nar/gkm752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hilario J, Amitani I, Baskin RJ, Kowalczykowski SC. Direct imaging of human Rad51 nucleoprotein dynamics on individual DNA molecules. Proc Natl Acad Sci U S A. 2009;106:361–8. doi: 10.1073/pnas.0811965106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Benson FE, Stasiak A, West SC. Purification and characterization of the human Rad51 protein, an analogue of E. coli RecA. EMBO J. 1994;13:5764–71. doi: 10.1002/j.1460-2075.1994.tb06914.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sung P. Catalysis of ATP-dependent homologous DNA pairing and strand exchange by yeast RAD51 protein. Science. 1994;265:1241–3. doi: 10.1126/science.8066464. [DOI] [PubMed] [Google Scholar]
- 50.Stasiak A, Egelman EH. Structure and function of RecA-DNA complexes. Experientia. 1994;50:192–203. doi: 10.1007/BF01924002. [DOI] [PubMed] [Google Scholar]
- 51.Chi P, Van Komen S, Sehorn MG, Sigurdsson S, Sung P. Roles of ATP binding and ATP hydrolysis in human Rad51 recombinase function. DNA Repair (Amst) 2006;5:381–91. doi: 10.1016/j.dnarep.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 52.Bugreev DV, Mazin AV. Ca2+ activates human homologous recombination protein Rad51 by modulating its ATPase activity. Proc Natl Acad Sci U S A. 2004;101:9988–93. doi: 10.1073/pnas.0402105101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kowalczykowski SC. An Overview of the Molecular Mechanisms of Recombinational DNA Repair. Cold Spring Harb Perspect Biol. 2015;7 doi: 10.1101/cshperspect.a016410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.von Nicolai C, Ehlen A, Martin C, Zhang X, Carreira A. A second DNA binding site in human BRCA2 promotes homologous recombination. Nat Commun. 2016;7:12813. doi: 10.1038/ncomms12813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carreira A, Hilario J, Amitani I, Baskin RJ, Shivji MK, Venkitaraman AR, Kowalczykowski SC. The BRC repeats of BRCA2 modulate the DNA-binding selectivity of RAD51. Cell. 2009;136:1032–43. doi: 10.1016/j.cell.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu J, Doty T, Gibson B, Heyer WD. Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat Struct Mol Biol. 2010;17:1260–2. doi: 10.1038/nsmb.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jensen RB, Carreira A, Kowalczykowski SC. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature. 2010;467:678–83. doi: 10.1038/nature09399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McGlynn P, Lloyd RG. Rescue of stalled replication forks by RecG: simultaneous translocation on the leading and lagging strand templates supports an active DNA unwinding model of fork reversal and Holliday junction formation. Proc Natl Acad Sci U S A. 2001;98:8227–34. doi: 10.1073/pnas.111008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McGlynn P, Lloyd RG. Modulation of RNA polymerase by (p)ppGpp reveals a RecG-dependent mechanism for replication fork progression. Cell. 2000;101:35–45. doi: 10.1016/S0092-8674(00)80621-2. [DOI] [PubMed] [Google Scholar]
- 60.Robu ME, Inman RB, Cox MM. RecA protein promotes the regression of stalled replication forks in vitro. Proc Natl Acad Sci U S A. 2001;98:8211–8. doi: 10.1073/pnas.131022698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gupta S, Yeeles JT, Marians KJ. Regression of replication forks stalled by leading-strand template damage: I. Both RecG and RuvAB catalyze regression, but RuvC cleaves the holliday junctions formed by RecG preferentially. J Biol Chem. 2014;289:28376–87. doi: 10.1074/jbc.M114.587881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, Lopes M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol. 2015;208:563–79. doi: 10.1083/jcb.201406099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Techer H, Baldi G, Shen R, Ciccia A, Pellegrini L, Krejci L, Costanzo V. Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol Cell. 2017 doi: 10.1016/j.molcel.2017.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mijic S, Zellweger R, Chappidi N, Berti M, Jacobs K, Mutreja K, Ursich S, Ray Chaudhuri A, Nussenzweig A, Janscak P, Lopes M. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat Commun. 2017;8:859. doi: 10.1038/s41467-017-01164-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reuter M, Zelensky A, Smal I, Meijering E, van Cappellen WA, de Gruiter HM, van Belle GJ, van Royen ME, Houtsmuller AB, Essers J, Kanaar R, Wyman C. BRCA2 diffuses as oligomeric clusters with RAD51 and changes mobility after DNA damage in live cells. J Cell Biol. 2014;207:599–613. doi: 10.1083/jcb.201405014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bugreev DV, Rossi MJ, Mazin AV. Cooperation of RAD51 and RAD54 in regression of a model replication fork. Nucleic Acids Res. 2011;39:2153–64. doi: 10.1093/nar/gkq1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Duro E, Lundin C, Ask K, Sanchez-Pulido L, MacArtney TJ, Toth R, Ponting CP, Groth A, Helleday T, Rouse J. Identification of the MMS22L-TONSL complex that promotes homologous recombination. Mol Cell. 2010;40:632–44. doi: 10.1016/j.molcel.2010.10.023. [DOI] [PubMed] [Google Scholar]
- 68.O’Donnell L, Panier S, Wildenhain J, Tkach JM, Al-Hakim A, Landry MC, Escribano-Diaz C, Szilard RK, Young JT, Munro M, Canny MD, Kolas NK, Zhang W, Harding SM, Ylanko J, Mendez M, Mullin M, Sun T, Habermann B, Datti A, Bristow RG, Gingras AC, Tyers MD, Brown GW, Durocher D. The MMS22L-TONSL complex mediates recovery from replication stress and homologous recombination. Mol Cell. 2010;40:619–31. doi: 10.1016/j.molcel.2010.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Piwko W, Mlejnkova LJ, Mutreja K, Ranjha L, Stafa D, Smirnov A, Brodersen MM, Zellweger R, Sturzenegger A, Janscak P, Lopes M, Peter M, Cejka P. The MMS22L-TONSL heterodimer directly promotes RAD51-dependent recombination upon replication stress. EMBO J. 2016;35:2584–2601. doi: 10.15252/embj.201593132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Courcelle J, Donaldson JR, Chow KH, Courcelle CT. DNA damage-induced replication fork regression and processing in Escherichia coli. Science. 2003;299:1064–7. doi: 10.1126/science.1081328. [DOI] [PubMed] [Google Scholar]
- 71.Defossez PA, Prusty R, Kaeberlein M, Lin SJ, Ferrigno P, Silver PA, Keil RL, Guarente L. Elimination of replication block protein Fob1 extends the life span of yeast mother cells. Mol Cell. 1999;3:447–55. doi: 10.1016/s1097-2765(00)80472-4. [DOI] [PubMed] [Google Scholar]
- 72.Zou H, Rothstein R. Holliday junctions accumulate in replication mutants via a RecA homolog-independent mechanism. Cell. 1997;90:87–96. doi: 10.1016/s0092-8674(00)80316-5. [DOI] [PubMed] [Google Scholar]
- 73.Louarn J, Cornet F, Francois V, Patte J, Louarn JM. Hyperrecombination in the terminus region of the Escherichia coli chromosome: possible relation to nucleoid organization. J Bacteriol. 1994;176:7524–31. doi: 10.1128/jb.176.24.7524-7531.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Carr AM, Lambert S. Replication stress-induced genome instability: the dark side of replication maintenance by homologous recombination. J Mol Biol. 2013;425:4733–44. doi: 10.1016/j.jmb.2013.04.023. [DOI] [PubMed] [Google Scholar]
- 75.Margalef P, Kotsantis P, Borel V, Bellelli R, Panier S, Boulton SJ. Stabilization of Reversed Replication Forks by Telomerase Drives Telomere Catastrophe. Cell. 2018;172:439–453 e14. doi: 10.1016/j.cell.2017.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-Strand Break Repair-Independent Role for BRCA2 in Blocking Stalled Replication Fork Degradation by MRE11. Cell. 2011;145:529–42. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hashimoto Y, Ray Chaudhuri A, Lopes M, Costanzo V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol. 2010;17:1305–11. doi: 10.1038/nsmb.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell. 2012;22:106–16. doi: 10.1016/j.ccr.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Higgs MR, Reynolds JJ, Winczura A, Blackford AN, Borel V, Miller ES, Zlatanou A, Nieminuszczy J, Ryan EL, Davies NJ, Stankovic T, Boulton SJ, Niedzwiedz W, Stewart GS. BOD1L Is Required to Suppress Deleterious Resection of Stressed Replication Forks. Mol Cell. 2015;59:462–77. doi: 10.1016/j.molcel.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 80.Rondinelli B, Gogola E, Yucel H, Duarte AA, van de Ven M, van der Sluijs R, Konstantinopoulos PA, Jonkers J, Ceccaldi R, Rottenberg S, D’Andrea AD. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat Cell Biol. 2017;19:1371–1378. doi: 10.1038/ncb3626. [DOI] [PubMed] [Google Scholar]
- 81.Iannascoli C, Palermo V, Murfuni I, Franchitto A, Pichierri P. The WRN exonuclease domain protects nascent strands from pathological MRE11/EXO1-dependent degradation. Nucleic Acids Res. 2015 doi: 10.1093/nar/gkv836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xu S, Wu X, Wu L, Castillo A, Liu J, Atkinson E, Paul A, Su D, Schlacher K, Komatsu Y, You MJ, Wang B. Abro1 maintains genome stability and limits replication stress by protecting replication fork stability. Genes Dev. 2017;31:1469–1482. doi: 10.1101/gad.299172.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lemacon D, Jackson J, Quinet A, Brickner JR, Li S, Yazinski S, You Z, Ira G, Zou L, Mosammaparast N, Vindigni A. MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat Commun. 2017;8:860. doi: 10.1038/s41467-017-01180-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Taglialatela A, Alvarez S, Leuzzi G, Sannino V, Ranjha L, Huang JW, Madubata C, Anand R, Levy B, Rabadan R, Cejka P, Costanzo V, Ciccia A. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol Cell. 2017;68:414–430 e8. doi: 10.1016/j.molcel.2017.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Teixeira-Silva A, Ait Saada A, Hardy J, Iraqui I, Nocente MC, Freon K, Lambert SAE. The end-joining factor Ku acts in the end-resection of double strand break-free arrested replication forks. Nat Commun. 2017;8:1982. doi: 10.1038/s41467-017-02144-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang AT, Kim T, Wagner JE, Conti BA, Lach FP, Huang AL, Molina H, Sanborn EM, Zierhut H, Cornes BK, Abhyankar A, Sougnez C, Gabriel SB, Auerbach AD, Kowalczykowski SC, Smogorzewska A. A Dominant Mutation in Human RAD51 Reveals Its Function in DNA Interstrand Crosslink Repair Independent of Homologous Recombination. Mol Cell. 2015;59:478–90. doi: 10.1016/j.molcel.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dungrawala H, Bhat KP, Le Meur R, Chazin WJ, Ding X, Sharan SK, Wessel SR, Sathe AA, Zhao R, Cortez D. RADX Promotes Genome Stability and Modulates Chemosensitivity by Regulating RAD51 at Replication Forks. Mol Cell. 2017 doi: 10.1016/j.molcel.2017.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Richardson C, Stark JM, Ommundsen M, Jasin M. Rad51 overexpression promotes alternative double-strand break repair pathways and genome instability. Oncogene. 2004;23:546–53. doi: 10.1038/sj.onc.1207098. [DOI] [PubMed] [Google Scholar]
- 89.Klein HL. The consequences of Rad51 overexpression for normal and tumor cells. DNA Repair (Amst) 2008;7:686–93. doi: 10.1016/j.dnarep.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tennstedt P, Fresow R, Simon R, Marx A, Terracciano L, Petersen C, Sauter G, Dikomey E, Borgmann K. RAD51 overexpression is a negative prognostic marker for colorectal adenocarcinoma. Int J Cancer. 2013;132:2118–26. doi: 10.1002/ijc.27907. [DOI] [PubMed] [Google Scholar]
- 91.Vispe S, Cazaux C, Lesca C, Defais M. Overexpression of Rad51 protein stimulates homologous recombination and increases resistance of mammalian cells to ionizing radiation. Nucleic Acids Res. 1998;26:2859–64. doi: 10.1093/nar/26.12.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hansen LT, Lundin C, Spang-Thomsen M, Petersen LN, Helleday T. The role of RAD51 in etoposide (VP16) resistance in small cell lung cancer. Int J Cancer. 2003;105:472–9. doi: 10.1002/ijc.11106. [DOI] [PubMed] [Google Scholar]
- 93.Drees JC, Lusetti SL, Chitteni-Pattu S, Inman RB, Cox MM. A RecA filament capping mechanism for RecX protein. Mol Cell. 2004;15:789–98. doi: 10.1016/j.molcel.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 94.Marians KJ. Lesion Bypass and the Reactivation of Stalled Replication Forks. Annu Rev Biochem. 2018 doi: 10.1146/annurev-biochem-062917-011921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Graham JE, Marians KJ, Kowalczykowski SC. Independent and Stochastic Action of DNA Polymerases in the Replisome. Cell. 2017;169:1201–1213 e17. doi: 10.1016/j.cell.2017.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Symington LS. End resection at double-strand breaks: mechanism and regulation. Cold Spring Harb Perspect Biol. 2014;6 doi: 10.1101/cshperspect.a016436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pathania S, Bade S, Le Guillou M, Burke K, Reed R, Bowman-Colin C, Su Y, Ting DT, Polyak K, Richardson AL, Feunteun J, Garber JE, Livingston DM. BRCA1 haploinsufficiency for replication stress suppression in primary cells. Nat Commun. 2014;5:5496. doi: 10.1038/ncomms6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, John S, Day A, Crespo AV, Shen B, Starnes LM, de Ruiter JR, Daniel JA, Konstantinopoulos PA, Cortez D, Cantor SB, Fernandez-Capetillo O, Ge K, Jonkers J, Rottenberg S, Sharan SK, Nussenzweig A. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature. 2016;535:382–7. doi: 10.1038/nature18325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kim TM, Son MY, Dodds S, Hu L, Luo G, Hasty P. RECQL5 and BLM exhibit divergent functions in cells defective for the Fanconi anemia pathway. Nucleic Acids Res. 2015;43:893–903. doi: 10.1093/nar/gku1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hartford SA, Chittela R, Ding X, Vyas A, Martin B, Burkett S, Haines DC, Southon E, Tessarollo L, Sharan SK. Interaction with PALB2 Is Essential for Maintenance of Genomic Integrity by BRCA2. PLoS Genet. 2016;12:e1006236. doi: 10.1371/journal.pgen.1006236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Somyajit K, Saxena S, Babu S, Mishra A, Nagaraju G. Mammalian RAD51 paralogs protect nascent DNA at stalled forks and mediate replication restart. Nucleic Acids Res. 2015;43:9835–55. doi: 10.1093/nar/gkv880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Patel DS, Misenko SM, Her J, Bunting SF. BLM helicase regulates DNA repair by counteracting RAD51 loading at DNA double-strand break sites. J Cell Biol. 2017 doi: 10.1083/jcb.201703144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Thangavel S, Berti M, Levikova M, Pinto C, Gomathinayagam S, Vujanovic M, Zellweger R, Moore H, Lee EH, Hendrickson EA, Cejka P, Stewart S, Lopes M, Vindigni A. DNA2 drives processing and restart of reversed replication forks in human cells. J Cell Biol. 2015;208:545–62. doi: 10.1083/jcb.201406100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Berti M, Ray Chaudhuri A, Thangavel S, Gomathinayagam S, Kenig S, Vujanovic M, Odreman F, Glatter T, Graziano S, Mendoza-Maldonado R, Marino F, Lucic B, Biasin V, Gstaiger M, Aebersold R, Sidorova JM, Monnat RJ, Jr, Lopes M, Vindigni A. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat Struct Mol Biol. 2013;20:347–54. doi: 10.1038/nsmb.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yazinski SA, Comaills V, Buisson R, Genois MM, Nguyen HD, Ho CK, Todorova Kwan T, Morris R, Lauffer S, Nussenzweig A, Ramaswamy S, Benes CH, Haber DA, Maheswaran S, Birrer MJ, Zou L. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017;31:318–332. doi: 10.1101/gad.290957.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Huh MS, Ivanochko D, Hashem LE, Curtin M, Delorme M, Goodall E, Yan K, Picketts DJ. Stalled replication forks within heterochromatin require ATRX for protection. Cell Death Dis. 2016;7:e2220. doi: 10.1038/cddis.2016.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mochizuki AL, Katanaya A, Hayashi E, Hosokawa M, Moribe E, Motegi A, Ishiai M, Takata M, Kondoh G, Watanabe H, Nakatsuji N, Chuma S. PARI regulates stalled replication fork processing to maintain genome stability upon replication stress in mice. Mol Cell Biol. 2017 doi: 10.1128/MCB.00117-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ding X, Ray Chaudhuri A, Callen E, Pang Y, Biswas K, Klarmann KD, Martin BK, Burkett S, Cleveland L, Stauffer S, Sullivan T, Dewan A, Marks H, Tubbs AT, Wong N, Buehler E, Akagi K, Martin SE, Keller JR, Nussenzweig A, Sharan SK. Synthetic viability by BRCA2 and PARP1/ARTD1 deficiencies. Nat Commun. 2016;7:12425. doi: 10.1038/ncomms12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yang Y, Liu Z, Wang F, Temviriyanukul P, Ma X, Tu Y, Lv L, Lin YF, Huang M, Zhang T, Pei H, Chen BP, Jansen JG, de Wind N, Fischhaber PL, Friedberg EC, Tang TS, Guo C. FANCD2 and REV1 cooperate in the protection of nascent DNA strands in response to replication stress. Nucleic Acids Res. 2015;43:8325–39. doi: 10.1093/nar/gkv737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Leuzzi G, Marabitti V, Pichierri P, Franchitto A. WRNIP1 protects stalled forks from degradation and promotes fork restart after replication stress. EMBO J. 2016;35:1437–51. doi: 10.15252/embj.201593265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Her J, Ray C, Altshuler J, Zheng H, Bunting SF. 53BP1 Mediates ATR-Chk1 Signaling and Protects Replication Forks under Conditions of Replication Stress. Mol Cell Biol. 2018;38 doi: 10.1128/MCB.00472-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Villa M, Bonetti D, Carraro M, Longhese MP. Rad9/53BP1 protects stalled replication forks from degradation in Mec1/ATR-defective cells. EMBO Rep. 2018;19:351–367. doi: 10.15252/embr.201744910. [DOI] [PMC free article] [PubMed] [Google Scholar]