

Abstract
Orphan receptors for whom cognate ligands have not yet been identified form a large subclass within the nuclear receptor superfamily. To address one aspect of how they might regulate transcription, we analyzed the mode of interaction between the Drosophila orphan receptor FTZ-F1 (NR5A3) and a segmentation gene product Fushi tarazu (FTZ). Strong interaction between these two factors was detected by use of the mammalian one- and two-hybrid interaction assays without addition of ligand. This interaction required the AF-2 core and putative ligand-binding domain of FTZ-F1 and the LXXLL motif of FTZ. The requirement of these elements was further confirmed by examination of their target gene expression in Drosophila embryos and observation of a cuticle phenotype in transgenic fly lines that express mutated factors. In Drosophila cultured cells, FTZ is required for FTZ-F1 activation of a FTZ-F1 reporter gene. These results reveal a resemblance in the mode of interaction between FTZ-F1 and FTZ and that of nuclear receptor-coactivator and indicate that direct interaction is required for regulation of gene expression by FTZ-F1. Thus, we propose that FTZ may represent a category of LXXLL motif-dependent coactivators for nuclear receptors.
More than 150 members of the nuclear receptor superfamily have been identified to date in animals ranging from hydra to human. These transcription factors are generally characterized by the presence of two conserved structural features, a DNA-binding domain (DBD) composed of two zinc fingers and a putative ligand-binding domain (LBD) located at the C-terminal region. Various lipophilic ligands have been found to interact with the cognate LBDs in apo-type receptors, converting them into an active holo-type conformation that can dynamically regulate transcription (1–7).
It has been shown that the general structure of the LBD is composed mainly of 12 helices. Interaction with ligand induces allosteric changes in conformation, especially in the configuration of helix 12 at the C terminus of the LBD, leading to transcriptional activation (8, 9) or repression (10, 11). Helix 12 is often referred to as the AF-2 core (or AF-2 activation domain, τc or τ4) that stands in some receptors as a conserved domain essential for ligand-dependent transcriptional activation (4, 7). Transcriptional coactivators such as CBP/p300, TRAP220, and p160 family factors, SRC-1/NcoA-1, TIF2/GRIP1/NcoA-2, and p/CIP/ACTR/AIB1, have been shown to mediate activating signals through binding to nuclear receptors in a ligand-dependent manner (ref. 12 and references therein; see also ref. 13). For this receptor–coactivator interaction, conserved sequences containing a short signature motif of LXXLL (where L is leucine and X can be any amino acid) have been implicated (14–17). The conserved leucines in these so-called LXXLL motifs, or NR boxes, appear indispensable for interaction with nuclear receptors. In nuclear receptors, the importance of helices 3, 5, and 12 (AF-2 core) in the LBD has been demonstrated, and computational modeling studies have predicted that helices 3, 5, and an appropriately realigned helix 12 form an interacting surface for the LXXLL (18, 19).
The majority of nuclear receptors, however, are “orphans” (20), for which cognate ligands have not yet been identified and the molecular mechanisms of their transcriptional regulation remain unclear. From an evolutionary aspect, the extension of the structural conservation to domains including the LBD strongly suggests a functional significance, raising a naive question. Despite large-scale ligand screenings that have been undertaken by many groups, why are there still so many “orphans” remaining? One answer may be that the structural conservation in the LBD implicates an importance for interactions with various intracellular factors other than small lipophilic molecules.
FTZ-F1 is a Drosophila orphan nuclear receptor (21) that was originally identified as an activator of the pair-rule class segmentation gene fushi tarazu (ftz) (22). In blastoderm embryos, one maternally derived isoform, αFTZ-F1, is expressed uniformly (23, 24), whereas ftz is expressed as seven stripes in the even-numbered parasegments (25). ftz is required for formation of these parasegments, and thus ftz mutant embryos possess only half of the normal number of body segments (26). Guichet et al. (23) and Yu et al. (24) reported that surprisingly, FTZ is expressed normally in ftz-f1 mutants, although both mutants exhibit similar larval cuticle phenotypes and similar expression patterns of ftz target genes. To explain this, they showed direct interaction between FTZ and FTZ-F1 by using biochemical analyses. These results suggest that FTZ-F1 can regulate some of its target genes through direct interaction with FTZ.
In this study, we analyzed the interaction between FTZ-F1 and FTZ to better understand the regulatory mechanisms of orphan receptors. We found that the mode of their interaction is similar to mammalian nuclear receptor–coactivator interaction but is unique in that their interaction seems to be ligand-independent. Our results provide a clear example that protein–protein interaction plays a definitive role in transcriptional regulation by nuclear receptors.
Materials and Methods
Plasmids.
For expression of nonfusion proteins in mammalian cells, pCMX (27) expression constructs were used. pCMX-FTZ-F1 and pCMX-FTZ contain Drosophila melanogaster FTZ-F1 (NR5A3) and FTZ (28) cDNA sequences, respectively, in pCMX. pCMX-FTZ-F1ΔAF2C encodes a truncated FTZ-F1. pCMX-GAL4 (29) encodes the GAL4-DBD (amino acids 1–147). pCMX-GAL4-FTZ-F1-LBD and pCMX-GAL4-FTZ contain the corresponding cDNA fragments of FTZ-F1 and FTZ in pCMX-GAL4. pCMX-GAL4-mutFTZ was constructed by substitution of a 0.18-kb StyI fragment from the coding region of pCMX-mutFTZ into pCMX-GAL4-FTZ. pCMX-VP16-FTZ-F1-LBD, pCMX-VP16-FTZ-F1-LBDΔAF2C, and pCMX-VP16-FTZ contain cDNA fragments derived from the corresponding regions in pCMX-GAL4-FTZ-F1-LBD, pCMX-FTZ-F1ΔAF2C, and pCMX-GAL4-FTZ in pCMX-VP16 (30) that encodes the VP16 transactivation domain (VP16-AD; amino acids 1–78). pCMX-VP16-FTZ-F1-LBDΔN-BstXI, pCMX-VP16-FTZ-F1-LBDΔN-SalI, pCMX-VP16-FTZ-N, pCMX-VP16-FTZ-C, pCMX-VP16-hRXRα-LBD, and pCMX-VP16-hRXRα-LBDΔAF2C contain corresponding cDNAs of FTZ-F1, FTZ, human RXRα (hRXRα; NR2B1), and hRXR443 (a kind gift from D. Mangelsdorf, Univ. of Texas Southwestern Medical Center, Dallas), respectively, in pCMX-VP16. For expression in Drosophila S2 cells, pAc5.1 (Invitrogen) expression constructs were used. pAc5.1-FTZ-F1, pAc5.1-FTZ, and pAc5.1-mutFTZ were constructed by inserting EcoRI fragments from the corresponding pCMX plasmids, pAc5.1-FTZ-F1ΔAF2C was constructed by inserting an EcoRI–XbaI fragment from phs-FTZ-F1ΔAF2C, and pAc5.1-FTZ-N was constructed by inserting a 0.7-kb EcoRI–SalI fragment from pCMX-FTZ (corresponding to amino acid positions 1–172) into the respective sites of pAc5.1. The pCaSpeR-hs (phs) vector was used for protein expression under the heat shock promoter in transgenic fly lines. phs-αFTZ-F1 was constructed with a strategy similar to hsLFTZ-F1 (31). phs-FTZ-F1ΔAF2C was constructed by inserting the corresponding restriction fragment from pCMX-FTZ-F1ΔAF2C into phs-αFTZ-F1. phs-FTZ and phs-mutFTZ were constructed by inserting EcoRI fragments of pCMX-FTZ and pCMX-mutFTZ, respectively. Point mutations on pCMX-mutFTZ, pCMX-FTZ-F1ΔAF2C, and pCMX-VP16-FTZ-F1-LBD mutant constructs were introduced by PCR-mediated mutagenesis by using primers containing the mutations and verified by DNA sequencing. FTZ-F1REx3-TK-LUC was constructed by inserting three copies of FTZ-F1RE (5′-TGAGTTTTTCAAGGTCATGCTCAATTT) with HindIII overhangs into a TK-LUC reporter containing the herpes virus thymidine kinase promoter (−105/+51). pCMX-βGAL (27), UASG-TK-LUC (32), and pCMX-SAH/Y145F (33) have been described. The unified nomenclature system used here for nuclear receptors was established by the Nuclear Receptors Nomenclature Committee (34).
Cell Culture and Cotransfection Assays.
CV-1 cells were grown in DMEM (ICN) supplemented with 10% FBS (DMEM-FBS) at 37°C in 5% CO2. Cells were plated to 10–20% confluency 1 day before transfection, and transfections were performed in 24-well plates in triplicate by the calcium phosphate–DNA precipitation method. Each well received 75 ng of the indicated expression vectors, 325 ng of reporter plasmid, and 350 ng of reference pCMX-βGAL. pCMX-SAH/Y145F containing the humanized green fluorescent protein gene was added to equalize amounts of plasmid DNA. After washing of DNA precipitates, cells were incubated for 36–48 h with DMEM-FBS alone or with 1 μM 4-[1-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydro-2-napthyl)ethenyl]-benzoic acid (LG69) and then harvested and assayed for luciferase and β-galactosidase activities. All luciferase activities were normalized with β-galactosidase activities. The presented data in each panel represent one of at least two independent transfection experiments with similar results. Values and bars represent the averages and standard deviations, respectively, of obtained values in triplicate for each data point, and the value obtained without effector was set as 1 in each experiment. Schneider's Drosophila line 2 (S2) cells were grown at 24°C in DES expression media (Invitrogen) supplemented with heat-inactivated (56°C for 30 min) 10% FBS. Cells were plated at a density of 2–4 × 106 cells per ml 1 day before transfection in 24-well plate. Transfections were performed as described above except that pAc5.1/V5-His/lacZ (Invitrogen) was used for reference, and cells were kept for 3 days without washing the DNA precipitate and then harvested. pAc5.1 was used to equalize amounts of transfected plasmid DNA concentrations.
Drosophila Strains.
ftz-f1ex7 (35), which carries a deletion including one entire exon by P element excision, was used as the ftz-f1 mutant. The deleted exon encodes amino acids 401–578 of αFTZ-F1 including the Zn finger DBD, and this mutant expresses an αFTZ-F1 that is 30 kDa smaller than wild type. To produce maternal mutants of ftz-f1, germ-line clones were produced by using ovo-FRT system as described by Chou and Perrimon (36). ftz9H34 and ftz11 were used as ftz mutants. For identification of homozygous ftz mutant embryos, balancer chromosomes that contain hb-lacZ or Ubx-lacZ transgenes were used. Transgenic fly lines carrying the heat shock promoter fusion gene were established as described (31).
Histochemical Analyses.
Antibody detection was performed with mouse monoclonal anti-Engrailed or anti-β-galactosidase by using the TSA direct system (NEN). ftz mutant embryos were identified as negative for lacZ expression. In situ hybridization to wg mRNA (37) and cuticle preparations (38) was carried out as previously described.
Results
Determination of FTZ-F1–FTZ Interaction Domain in FTZ-F1.
To examine the interaction between FTZ and FTZ-F1, the mammalian one- and two-hybrid interaction assay systems offer the advantage of quantitative comparison under physiological conditions. We first attempted to detect interaction using CV-1 cells (derived from monkey kidney fibroblasts) in terms of potentiation of FTZ-F1-dependent transcription upon coexpression of FTZ fused to the VP16 transactivation domain (VP16-AD) (Fig. 1A). Interaction was measured either as VP16-dependent transcriptional activation of a reporter gene containing FTZ-F1-binding sites (Fig. 1C) or as activation of a reporter gene containing GAL4-binding sites with FTZ fused to GAL4-DBD and FTZ-F1 LBD fused to VP16-AD (Fig. 1D). The above results indicate the apparent dispensability of the FTZ-F1 DBD and suggest that the C-terminal region of FTZ-F1 (amino acids 574-1029) encompassing the putative LBD is sufficient to support interaction with FTZ. To further define the sequences required for this interaction, we constructed fusion proteins containing only the conserved LBD region (FTZ-F1 LBDΔN-BstXI) and made further deletions from the N terminus (FTZ-F1 LBDΔN-SalI) or from the C terminus (FTZ-F1 LBDΔAF2C) (Fig. 1A). The proteins encoding the intact LBD retained the FTZ-interacting ability, but both proteins containing deletions within the LBD region lost their ability to interact with FTZ (Fig. 1D). Similar results were obtained using FTZ-F1 fused to GAL4-DBD and FTZ to VP16-AD (data not shown), and the importance of the FTZ-F1 C-terminal region was confirmed with an expression construct encoding a full-length protein carrying the same truncation (FTZ-F1ΔAF2C) (Fig. 1C). These results indicate that a relatively intact LBD is essential for target recognition by FTZ. A point to keep in mind is that FTZ-F1ΔAF2C lacks part of the highly conserved putative AF-2 core that reportedly stands as an essential region for interaction with coactivators and for the resulting ligand-dependent activation (11, 18, 19, 39).
Figure 1.

The LXXLL motif in FTZ and the AF-2 core in FTZ-F1 are required for interaction. (A) Schematic structures of LBD/AF-2 truncation mutant FTZ-F1 and hRXRα. Alignment of amino acid sequences around the conserved AF-2 core is shown. Arrows indicate C-terminal positions of truncated proteins. Numbers represent amino acid positions from the N terminus. (B) Structures of point mutant FTZ-F1-LBD and FTZ proteins. Mutations were introduced into pCMX-VP16-FTZ-F1-LBD or FTZ constructs (pCMX-GAL4-mutFTZ, pAc5.1-mutFTZ, and phs-mutFTZ). Positions of the mutations are shown by arrows with alignment of amino acid sequences for comparison. Residues marked by black boxes have been reported to be critical for interaction with coactivators (18, 19). Asterisks indicate amino acids conserved among all three FTZ proteins. Numbers represent amino acid positions from the N terminus. (C) Detection of interaction between FTZ-F1 and FTZ by mammalian one-hybrid assay. CV-1 cells were cotransfected with indicated pCMX expression plasmids along with the FTZ-F1REx3-TK-LUC reporter. The same levels of FTZ and VP-FTZ protein expressions in CV-1 cells were confirmed by Western blotting (data not shown). (D) The FTZ-F1 LBD is required for interaction with FTZ. CV-1 cells were transfected with indicated pCMX expression plasmids along with FTZ-F1REx3-TK-LUC. (E) The LXXLL motif in FTZ is required for interaction with FTZ-F1 LBD. CV-1 cells were transfected with indicated expression plasmids along with UASG-TK-LUC. (F) RXRα interacts with FTZ in a ligand- and AF-2-dependent manner. CV-1 cells were cotransfected with indicated plasmids and treated with or without 1 μM LG69, an RXR-specific agonist. (G) Mutations within the putative coactivator-binding surface abolish interaction with FTZ. CV-1 cells were transfected with indicated expression plasmids along with UASG-TK-LUC.
FTZ Interacts with FTZ-F1 in a Manner Similar to the Interaction Between Mammalian Nuclear Receptors and Their Coactivators.
A similar strategy was used for defining the essential domain in FTZ that supports interaction with FTZ-F1. Preliminary results revealed that the N-terminal half (amino acids 2–172) could interact with the FTZ-F1 LBD at a level comparable to the full-length construct, whereas no activity was observed with the C-terminal (amino acids 171–413); (data not shown). These results support the observation by Guichet et al. (23) who found that amino acids 101–150 in FTZ are necessary for interaction with FTZ-F1 in vitro. They also showed that the corresponding region of a distant FTZ homologue derived from Tribolium castaneum interacts with FTZ-F1 and thus pointed out the importance of a conserved amino acid motif, LRALLT, in this region (Fig. 1B), which had also been noted by sequence comparison between the two species by Brown et al. (40).This conserved sequence conforms to the typical LXXLL motif generally found in the nuclear receptor interaction domain of nuclear receptor coactivators (14–16). To examine the functional importance of this putative motif in FTZ, we made a construct (mutFTZ) containing substitutions in the tandem leucines (Fig. 1B). As shown in Fig. 1E, mutFTZ completely lost the ability to interact with the FTZ-F1 LBD. Interestingly, we found that the mammalian nuclear receptor RXRα (NR2B1) can also interact with FTZ in a ligand- and AF-2 core-dependent manner that requires the tandem leucines (Fig. 1F). Together, these observations strongly suggest that the conserved LXXLL motif in FTZ may allow interaction with FTZ-F1 in a manner resembling ligand-bound nuclear receptor interactions with coactivators. This idea was supported by results showing that introduction of amino acid substitutions into FTZ-F1 LBD (Fig. 1B) drastically reduced its ability to interact with FTZ (Fig. 1G); corresponding positions in helices 3 and 5 of the ERα and TRβ1 LBDs (marked by black boxes in Fig. 1B) have been shown to be important for interaction with p160 family coactivators (18, 19).
Role of FTZ-F1 AF-2 and FTZ LXXLL for in Vivo Interaction.
To evaluate the importance of the observed interaction through the LXXLL motif in FTZ and the AF-2 core in FTZ-F1 in vivo, we established transgenic fly lines that can express FTZ, mutFTZ, FTZ-F1, and/or FTZ-F1ΔAF2C under control of the hsp70 heat shock promoter and tested the effect of these factors on early fly embryogenesis. As FTZ activates en and represses wg in even-numbered parasegments, even-numbered en stripes are missing (Fig. 2A-1, compare with Fig. 2G-1) and wg stripes expand (Fig. 2A-2, compare with Fig. 2G-2) in ftz mutant embryos (41, 42). The above phenotypes are rescued by ubiquitous expression of FTZ under control of the heat shock promoter (Fig. 2B) but not by mutFTZ (Fig. 2C) carrying a mutation(s) in the LXXLL motif, indicating the requirement of the LXXLL motif for this function. Likewise, in embryos derived from ftz-f1ex7 mutant germ-line clones, even-numbered en stripes were not observed, and expanded wg stripes were present (Fig. 2D) as previously reported in other ftz-f1 mutants (23, 24). To see whether the AF-2 core region in FTZ-F1 is necessary for mutant rescue, either FTZ-F1 or FTZ-F1ΔAF2C was expressed under control of the heat shock promoter. We also expressed FTZ in conjunction with FTZ-F1 or FTZ-F1ΔAF2C, as the level of FTZ expression is slightly reduced in the ftz-f1ex7 mutant (H.K., T.S., S. Hirose, and H.U., unpublished observations). Independent of FTZ coexpression, the even-numbered en stripes and repression of wg could be recovered by expression of wild-type FTZ-F1 (Fig. 2 E and J) but not by that of FTZ-F1ΔAF2C (Fig. 2 F and L), indicating the importance of the AF-2 core region for this function. As expected from the rescue of en and wg expression, the cuticle phenotype in ftz-f1 mutants was also rescued by expression of FTZ-F1 under heat shock control (Fig. 3B) but not by that of FTZ-F1ΔAF2C (Fig. 3C). These results strongly suggest that direct interaction through the LXXLL motif in FTZ and the AF-2 core region in FTZ-F1 in vivo is important for en activation and wg repression in the ftz-dependent parasegments and for segment formation.
Figure 2.

Effect of FTZ-F1 and FTZ interaction for en and wg expression. Expression patterns of En proteins (1) and wg mRNA (2) in ftz mutant embryos (A–C), embryos from ftz-f1 mutant germ-line clones (D–F and J–L) and wild-type embryos (G–I) carrying hs-FTZ (B, H, J, and L), hs-mutFTZ (C, I, and K), hs-FTZ-F1 (E, J, and K), and/or hs-FTZ-F1ΔAF2C (F and L). Embryos carrying heat shock promoter fusion constructs were incubated at 37°C for 15 min and then for 30–60 min at 25°C before staining. The same levels of wild-type and mutant protein expression in vivo were confirmed by Western blotting and immunohistochemical staining. White dots indicate En stripes rescued by expression of FTZ. White bars indicate expanded regions resulting from ectopic expression of FTZ at a typical stripe. Black bars indicate expanded regions in either ftz-f1 or ftz mutants at a typical stripe.
Figure 3.

Interaction between FTZ-F1 and FTZ is required to produce ftz-dependent cuticle phenotypes. Larval cuticle structures of embryos derived from ftz-f1 mutant germ-line clones (A–C and G–I, and of wild-type embryos (D–F) carrying hs-FTZ (E, G, and I), hs-mutFTZ (F and H), hs-FTZ-F1 (B, G, and H), and/or hs-FTZ-F1ΔAF2C (C and I). Embryos carrying heat shock promoter fusion constructs were incubated at 37°C for 15 min and then for 1 day at 25°C.
Ubiquitous expression of FTZ in early embryos under control of the heat shock promoter broadens even-numbered en stripes (underlined region in Fig. 2H-1, compare with Fig. 2G-1), represses alternate wg stripes (Fig. 2H-2, compare with 2G-2) (43), and results in a so-called anti-ftz cuticle phenotype (Fig. 3E), in which roughly reciprocal segments are missing compared with the ftz larval cuticle phenotype (44). However, ectopic en induction or wg repression was not observed by expressing mutFTZ (Fig. 2I), indicating that the LXXLL motif in FTZ is necessary for the ectopic expression of en and the repression of wg. FTZ-dependent en induction and wg repression were also observed by expression of both FTZ-F1 and FTZ under control of the heat shock promoter in the ftz-f1 mutant embryo as shown in Fig. 2J (compare with Fig. 2K) but not when FTZ-F1ΔAF2C was used instead of FTZ-F1 (Fig. 2L). An anti-ftz cuticle phenotype was produced by forced expression of wild-type FTZ (Fig. 3E) but not by expression of mutFTZ (Fig. 3F). In ftz-f1 mutant embryos, anti-ftz phenotypes were obtained when FTZ-F1 and FTZ were coexpressed under heat shock control (Fig. 3G) but not upon replacement of FTZ-F1 with FTZ-F1ΔAF2C (Fig. 3I). These observations indicate that interaction through the LXXLL motif in FTZ and AF-2 core in FTZ-F1 is necessary for producing an anti-ftz phenotype and further support the results of the one- and two-hybrid assays.
It should be noted that the ftz-f1ex7 mutant used in this experiment expresses a FTZ-F1 protein in which one entire exon encoding the zinc finger DBD region is deleted. Because this mutant FTZ-F1 still contains the LBD, the possibility exists that it is able to bind FTZ and antagonize FTZ function. We do not exclude the possibility that some of the phenotypes observed in the ftz-f1ex7 mutant are caused by this antagonistic effect. Even in this case, however, the results still indicate the importance of the AF-2 core in FTZ-F1 and the LXXLL motif in FTZ and support their interaction in vivo.
FTZ Serves as a Coactivator for FTZ-F1 in Drosophila-Cultured Cell Line.
To see whether FTZ works as a coactivator or a corepressor for FTZ-F1, the transcriptional function of FTZ-F1 and FTZ was examined by using Schneider's Drosophila line 2 cells (S2 cells). As shown in Fig. 4, strong activation of a FTZ-F1REx3-TK-LUC reporter gene containing FTZ-F1-binding sites was observed in the presence of both FTZ-F1 and FTZ but not with either factor alone. Furthermore, this activation was dependent on both the AF-2 core of FTZ-F1 and the LXXLL motif in FTZ but did not appear to require the C-terminal homeodomain in FTZ. These results strongly suggest that FTZ functions as a coactivator for FTZ-F1 independent of its DNA-binding activity and that the interaction is mediated through the FTZ-F1 AF-2 core and FTZ LXXLL motif.
Figure 4.
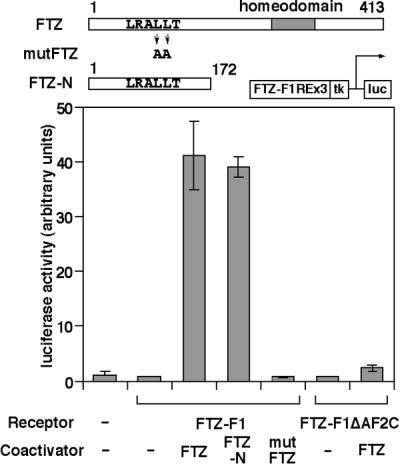
FTZ works as a coactivator for FTZ-F1 through direct interaction. Characterization of FTZ-F1 and FTZ-dependent transcriptional activation detected in S2 cells. Cells were transfected with indicated pAc5.1 expression plasmids along with FTZ-F1REx3-TK-LUC. Structures of FTZ proteins are also shown schematically.
Discussion
In this study, we demonstrated that the interaction between FTZ-F1 and FTZ, which has previously been reported by Guichet et al. and Yu et al. (23, 24), depends on the LXXLL motif in FTZ and the AF-2 core in FTZ-F1 by using the mammalian one- and two-hybrid assays. Requirement for these motifs was confirmed by analyses of target gene expression in fly embryos in vivo and reporter gene activation in Drosophila cultured cells. Truncation of the region corresponding to helix 3 of the hRXRα LBD or amino acid substitutions in helices 3 and 5 abolished the interaction (Fig. 1 D and G), as is the case with liganded receptor–coactivator interactions (11, 15, 16, 18, 19, 39). Some groups have also shown that a short flanking region adjacent to the LXXLL motif is important for preferential interaction with a subset of nuclear receptors (19, 45, 46); the importance of a similar region in FTZ was also observed (data not shown). Together, these support the view that FTZ-F1 may interact with FTZ in a manner similar to the interaction that occurs between ligand-bound nuclear receptors and their coactivators.
In the case of FTZ-F1 and FTZ, however, interaction was detected without addition of any putative ligand, in contrast to mammalian coactivators that generally interact only with ligand-bound holo-type nuclear receptors. We observed this to be the case in all cell lines tested including JEG-3 (human choriocarcinoma), EPC (fish epithelial), and S2 (fly embryonic), as well as for CV-1 using serum-free media (data not shown). This is consistent with studies showing their interaction by in vitro affinity chromatography, far-Western blotting, and gel mobility shift assay (23, 24) and suggests that a ligand may not be necessary for the interaction, although its existence cannot be excluded. We propose the following three possibilities. (i) FTZ-F1 maintains a holo-type conformation at all times. If this is the case, FTZ-F1 may not be competent to activate transcription with ubiquitously expressed general coactivators. (ii) FTZ-F1 is structurally unstable so that it adopts both apo- and holo-type conformations in a stochastic manner; FTZ may bind to and stabilize the holo-type FTZ-F1. This is similar to the equilibrium model proposed by Schulman et al. (47) for ligand-dependent transactivation by the thyroid hormone receptor, in which a role of the ligand may be to stabilize the holo-type structure and/or destabilize the apo-type. A similar possibility for orphan receptors was also discussed by Escriva et al. (48). (iii) Apo-type FTZ-F1 is switched to holo-type by a ubiquitous FTZ-F1 ligand or other intracellular signals that are present in all cell lines used, but FTZ-type factors are further required for transcriptional activation. It is possible that FTZ itself contributes to changing conformation of FTZ-F1. On the basis of the second and third hypotheses, the observation that RXRα interacted with the specific cofactor FTZ in a ligand-dependent manner can be explained by the determinative role of ligand for the conformation of RXRα. Further analysis is necessary to understand the mechanism of their interaction.
Some groups reported that FTZ functions in a homeodomain-independent manner in Drosophila cultured cells and in embryos (49–52). Our results in S2 cells were consistent with these results and indicated that FTZ works as a coactivator when it interacts with FTZ-F1 through the LXXLL motif. Interestingly, FTZ functions as a coactivator only when fused to VP16-AD in CV-1 cells (Fig. 1C) and in other vertebrate cells so far examined (data not shown). This difference suggests that species- and/or cell-type specific factor(s) are necessary for this activation function.
Our results using embryos strongly suggest that FTZ-F1 activates en in vivo through the AF-2–LXXLL-dependent direct interaction. In early fly embryos, FTZ-F1 seems to function as an activator for en only in regions where FTZ is also present despite the uniform expression of FTZ-F1 (23, 24). Such situations mimic that of the requirement for a ligand by a nuclear receptor in controlling its function and specificity in gene expression. The characteristic cooperation of FTZ-F1 and FTZ provides a novel example of transcriptional regulation by a nuclear receptor, which may be an alternative pathway to the conventional one using lipophilic ligands. From an evolutionary aspect, Escriva et al. (48) proposed that the ancestral nuclear receptor had no ligand and the ability to bind a ligand was acquired by a subset of descendent receptors later in evolution. They also presumed FTZ-F1 to be one of the most ancient receptors based on its distribution among species. We believe that transcriptional activation by FTZ-F1 through binding to FTZ might represent a primitive style of regulation by nuclear receptors before the acquisition of ligand-binding ability. We also assume the existence of yet unidentified corresponding factors for other orphan receptors as well as for ligand-responsive receptors, which may form a new group of nuclear receptor coactivators and play critical roles for development and metabolism.
Acknowledgments
We thank A. Guichet (European Molecular Biology Laboratory, Germany), S. Goto, S. Hayashi, Y. Hiromi, M. Okabe (National Institute of Genetics, Japan), M. Mangelsdorf, N. Pate (University of Chicago, IL), and C. Schwartz (University of Toronto, Canada) for providing materials, Drosophila strains, and/or technical advice; R. Evans (Salk Institute for Biological Studies, La Jolla, CA), S. Hirose (National Institute of Genetics), and T. Perlmann (Karolinska Institute, Stockholm) for helpful discussions; R. Kageyama and S. Narumiya (Kyoto University) for generous support; and Y. Hayashi, H. Imai, and the members of Umesono laboratory for practical help and suggestions. We especially thank K. Morohashi (National Institute for Basic Biology, Okazaki, Japan) and the members of his laboratory for generosity and continuous encouragement. This paper was supported by Research for the Future from the Japan Society for the Promotion of Science (Japan) and Grants-in-Aid for Scientific Research from the Ministry of Education, Science, Sports, and Culture (Japan). This paper is dedicated to the memory of K.U.
Abbreviations
- LBD
ligand-binding domain
- DBD
DNA-binding domain
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Beato M, Herrlich P, Schutz G. Cell. 1995;83:851–857. doi: 10.1016/0092-8674(95)90201-5. [DOI] [PubMed] [Google Scholar]
- 2.Kastner P, Mark M, Chambon P. Cell. 1995;83:859–869. doi: 10.1016/0092-8674(95)90202-3. [DOI] [PubMed] [Google Scholar]
- 3.Mangelsdorf D J, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, et al. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mangelsdorf D J, Evans R M. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- 5.Thummel C S. Cell. 1995;83:871–877. doi: 10.1016/0092-8674(95)90203-1. [DOI] [PubMed] [Google Scholar]
- 6.Blumberg B, Evans R M. Genes Dev. 1998;12:3149–3155. doi: 10.1101/gad.12.20.3149. [DOI] [PubMed] [Google Scholar]
- 7.Giguere V. Endocr Rev. 1999;20:689–725. doi: 10.1210/edrv.20.5.0378. [DOI] [PubMed] [Google Scholar]
- 8.Bourguet W, Ruff M, Chambon P, Gronemeyer H, Moras D. Nature (London) 1995;375:377–382. doi: 10.1038/375377a0. [DOI] [PubMed] [Google Scholar]
- 9.Renaud J P, Rochel N, Ruff M, Vivat V, Chambon P, Gronemeyer H, Moras D. Nature (London) 1995;378:681–689. doi: 10.1038/378681a0. [DOI] [PubMed] [Google Scholar]
- 10.Brzozowski A M, Pike A C, Dauter Z, Hubbard R E, Bonn T, Engstrom O, Ohman L, Greene G L, Gustafsson J A, Carlquist M. Nature (London) 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 11.Shiau A K, Barstad D, Loria P M, Cheng L, Kushner P J, Agard D A, Greene G L. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 12.Xu L, Glass C K, Rosenfeld M G. Curr Opin Genet Dev. 1999;9:140–147. doi: 10.1016/S0959-437X(99)80021-5. [DOI] [PubMed] [Google Scholar]
- 13.Yuan C X, Ito M, Fondell J D, Fu Z Y, Roeder R G. Proc Natl Acad Sci USA. 1998;95:7939–7944. doi: 10.1073/pnas.95.14.7939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Le Douarin B, Nielsen A L, Garnier J M, Ichinose H, Jeanmougin F, Losson R, Chambon P. EMBO J. 1996;15:6701–6715. [PMC free article] [PubMed] [Google Scholar]
- 15.Heery D M, Kalkhoven E, Hoare S, Parker M G. Nature (London) 1997;387:733–736. doi: 10.1038/42750. [DOI] [PubMed] [Google Scholar]
- 16.Torchia J, Rose D W, Inostroza J, Kamei Y, Westin S, Glass C K, Rosenfeld M G. Nature (London) 1997;387:677–684. doi: 10.1038/42652. [DOI] [PubMed] [Google Scholar]
- 17.Ren Y, Behre E, Ren Z, Zhang J, Wang Q, Fondell J D. Mol Cell Biol. 2000;20:5433–5446. doi: 10.1128/mcb.20.15.5433-5446.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng W, Ribeiro R C, Wagner R L, Nguyen H, Apriletti J W, Fletterick R J, Baxter J D, Kushner P J, West B L. Science. 1998;280:1747–1749. doi: 10.1126/science.280.5370.1747. [DOI] [PubMed] [Google Scholar]
- 19.Mak H Y, Hoare S, Henttu P M, Parker M G. Mol Cell Biol. 1999;19:3895–3903. doi: 10.1128/mcb.19.5.3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Enmark E, Gustafsson J A. Mol Endocrinol. 1996;10:1293–1307. doi: 10.1210/mend.10.11.8923456. [DOI] [PubMed] [Google Scholar]
- 21.Lavorgna G, Ueda H, Clos J, Wu C. Science. 1991;252:848–851. doi: 10.1126/science.1709303. [DOI] [PubMed] [Google Scholar]
- 22.Ueda H, Sonoda S, Brown J L, Scott M P, Wu C. Genes Dev. 1990;4:624–635. doi: 10.1101/gad.4.4.624. [DOI] [PubMed] [Google Scholar]
- 23.Guichet A, Copeland J W, Erdelyi M, Hlousek D, Zavorszky P, Ho J, Brown S, Percival-Smith A, Krause H M, Ephrussi A. Nature (London) 1997;385:548–552. doi: 10.1038/385548a0. [DOI] [PubMed] [Google Scholar]
- 24.Yu Y, Li W, Su K, Yussa M, Han W, Perrimon N, Pick L. Nature (London) 1997;385:552–555. doi: 10.1038/385552a0. [DOI] [PubMed] [Google Scholar]
- 25.Carroll S B, Scott M P. Cell. 1985;43:47–57. doi: 10.1016/0092-8674(85)90011-x. [DOI] [PubMed] [Google Scholar]
- 26.Wakimoto B T, Turner F R, Kaufman T C. Dev Biol. 1984;102:147–172. doi: 10.1016/0012-1606(84)90182-9. [DOI] [PubMed] [Google Scholar]
- 27.Umesono K, Murakami K K, Thompson C C, Evans R M. Cell. 1991;65:1255–1266. doi: 10.1016/0092-8674(91)90020-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laughon A, Scott M P. Nature (London) 1984;310:25–31. doi: 10.1038/310025a0. [DOI] [PubMed] [Google Scholar]
- 29.Perlmann T, Jansson L. Genes Dev. 1995;9:769–782. doi: 10.1101/gad.9.7.769. [DOI] [PubMed] [Google Scholar]
- 30.Perlmann T, Rangarajan P N, Umesono K, Evans R M. Genes Dev. 1993;7:1411–1422. doi: 10.1101/gad.7.7b.1411. [DOI] [PubMed] [Google Scholar]
- 31.Murata T, Kageyama Y, Hirose S, Ueda H. Mol Cell Biol. 1996;16:6509–6515. doi: 10.1128/mcb.16.11.6509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Forman B M, Umesono K, Chen J, Evans R M. Cell. 1995;81:541–550. doi: 10.1016/0092-8674(95)90075-6. [DOI] [PubMed] [Google Scholar]
- 33.Ogawa H, Umesono K. Acta Histochem Cytochem. 1998;31:303–308. [Google Scholar]
- 34.Nuclear Receptors Nomenclature Committee. Cell. 1999;97:161–163. doi: 10.1016/s0092-8674(00)80726-6. [DOI] [PubMed] [Google Scholar]
- 35.Yamada M, Murata T, Hirose S, Lavorgna G, Suzuki E, Ueda H. Development (Cambridge, UK) 2000;127:5083–5092. doi: 10.1242/dev.127.23.5083. [DOI] [PubMed] [Google Scholar]
- 36.Chou T B, Perrimon N. Genetics. 1996;144:1673–1679. doi: 10.1093/genetics/144.4.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tautz D, Pfeifle C. Chromosoma. 1989;98:81–85. doi: 10.1007/BF00291041. [DOI] [PubMed] [Google Scholar]
- 38.Wieschaus E, Nusslein-Volhard C. In: Drosophila: A Practical Approach. Roberts D B, editor. Oxford: IRL; 1986. pp. 199–227. [Google Scholar]
- 39.Nolte R T, Wisely G B, Westin S, Cobb J E, Lambert M H, Kurokawa R, Rosenfeld M G, Willson T M, Glass C K, Milburn M V. Nature (London) 1998;395:137–143. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- 40.Brown S J, Hilgenfeld R B, Denell R E. Proc Natl Acad Sci USA. 1994;91:12922–12926. doi: 10.1073/pnas.91.26.12922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DiNardo S, O'Farrell P H. Genes Dev. 1987;1:1212–1225. doi: 10.1101/gad.1.10.1212. [DOI] [PubMed] [Google Scholar]
- 42.Ingham P W, Baker N E, Martinez-Arias A. Nature (London) 1988;331:73–75. doi: 10.1038/331073a0. [DOI] [PubMed] [Google Scholar]
- 43.Ish-Horowicz D, Pinchin S M, Ingham P W, Gyurkovics H G. Cell. 1989;57:223–232. doi: 10.1016/0092-8674(89)90960-4. [DOI] [PubMed] [Google Scholar]
- 44.Struhl G. Nature (London) 1985;318:677–680. doi: 10.1038/318677a0. [DOI] [PubMed] [Google Scholar]
- 45.Darimont B D, Wagner R L, Apriletti J W, Stallcup M R, Kushner P J, Baxter J D, Fletterick R J, Yamamoto K R. Genes Dev. 1998;12:3343–3356. doi: 10.1101/gad.12.21.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McInerney E M, Rose D W, Flynn S E, Westin S, Mullen T M, Krones A, Inostroza J, Torchia J, Nolte R T, Assa-Munt N, et al. Genes Dev. 1998;12:3357–3368. doi: 10.1101/gad.12.21.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schulman I G, Juguilon H, Evans R M. Mol Cell Biol. 1996;16:3807–3813. doi: 10.1128/mcb.16.7.3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Escriva H, Safi R, Hanni C, Langlois M C, Saumitou-Laprade P, Stehelin D, Capron A, Pierce R, Laudet V. Proc Natl Acad Sci USA. 1997;94:6803–6808. doi: 10.1073/pnas.94.13.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hyduk D, Percival-Smith A. Genetics. 1996;142:481–492. doi: 10.1093/genetics/142.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Copeland J W, Nasiadka A, Dietrich B H, Krause H M. Nature (London) 1996;379:162–165. doi: 10.1038/379162a0. [DOI] [PubMed] [Google Scholar]
- 51.Fitzpatrick V D, Percival-Smith A, Ingles C J, Krause H M. Nature (London) 1992;356:610–612. doi: 10.1038/356610a0. [DOI] [PubMed] [Google Scholar]
- 52.Ananthan J, Baler R, Morrissey D, Zuo J, Lan Y, Weir M, Voellmy R. Mol Cell Biol. 1993;13:1599–1609. doi: 10.1128/mcb.13.3.1599. [DOI] [PMC free article] [PubMed] [Google Scholar]