

Abstract
Keutel syndrome is caused by mutations in the matrix gamma-carboxyglutamic acid (MGP) gene (OMIM 154870) and is inherited in an autosomal recessive fashion. It is characterized by brachydactyly, pulmonary artery stenosis, a distinctive facial phenotype, and cartilage calcification. To date, only 36 cases have been reported worldwide. We describe clinical and molecular findings of the first Brazilian patient with Keutel syndrome. Keutel syndrome was suspected based on clinical and morphological evaluation, so we sequenced the MGP gene using the TruSight One Sequencing Panel (Illumina). The obtained MGP gene sequence was then validated by Sanger sequencing. We identified a novel pathogenic homozygous variant of the MGP gene (c.2T>C; p.Met1Thr) confirming Keutel syndrome. Proper diagnosis of this syndrome is important for clinical management and is an indication for genetic counseling. Keutel syndrome should be suspected in patients with cartilage calcifications and brachydactyly when associated with a distinctive facial phenotype and pulmonary artery stenosis.
Keywords: Brachydactyly, Cartilage calcifications, Keutel syndrome, MGP mutation, Pulmonary artery stenosis
Keutel syndrome (OMIM 245150) was first described in 2 siblings presenting with brachytelephalangy, diffuse calcification of the cartilages, mixed hearing loss, multiple peripheral pulmonary artery stenoses, and midface retrusion with depressed nasal bridge [Keutel, 1972]. Subsequent cases demonstrated that Keutel syndrome is a rare autosomal recessive disorder caused by mutations in the MGP gene encoding matrix Gla protein [Munroe et al., 1999], a calcification inhibitor expressed by chondrocytes and smooth muscle cells [Hale et al., 1998]. The matrix Gla protein requires post-translational modifications (serine phosphorylation and gamma-glutamate carboxylation) for inhibition of calcification [Schurgers et al., 2007]. To date, only 36 confirmed cases have been documented worldwide [Hur et al., 2005; Sun et al., 2013] with no known cases in Brazil.
As a rare autosomal recessive disease, this condition is reported predominantly in children of consanguineous parents. We report the case of a Brazilian patient referred to our Medical Genetics Department at the Federal University of São Paulo with peripheral pulmonary artery stenosis and morphological signs of Keutel syndrome. Our purpose is to highlight the importance of clinical suspicion for timely Keutel syndrome diagnosis, appropriate clinical follow-up, and genetic counseling.
Case Report
The patient was a 13-year-old Brazilian female referred from the cardiology clinic at the Federal University of São Paulo for genetic evaluation due to peripheral pulmonary artery stenosis. The pregnancy was uneventful, and her parents are first-degree cousins. She has a younger sister. Neurodevelopmental milestones were unremarkable.
She had complained of dyspnea and was referred for cardiac treatment due to a murmur. Transthoracic echocardiography suggested right pulmonary artery stenosis without pulmonary hypertension. A thoracic angiotomography was conducted, which revealed hypoplasia of the right pulmonary artery (Fig. 1d).
Fig. 1.
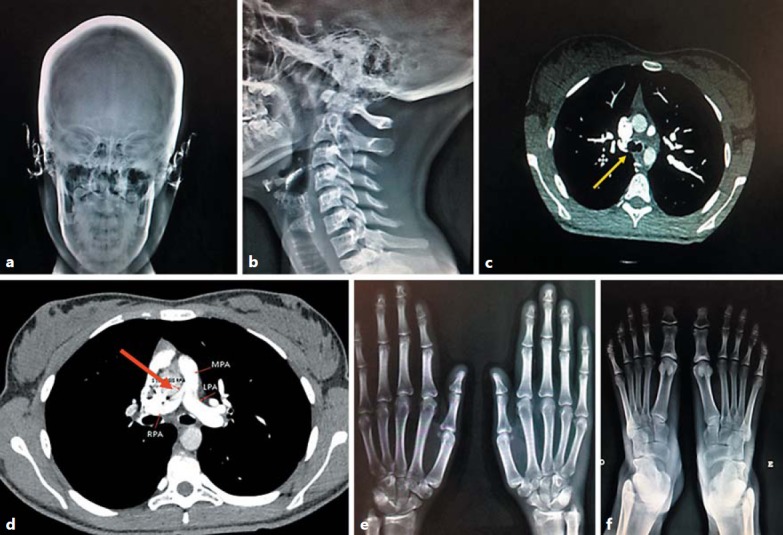
Radiographs showing features of suspected Keutel syndrome. a Cranial thickness and calcifications in the ears. b Laryngeal calcifications. c Tracheal calcifications revealed by angiotomography (yellow arrow). d Right pulmonary artery hypoplasia (red arrow). e Shortening of the second, third, and fourth distal phalanges of the hands. f Shortening of the second, third, and fourth distal phalanges of the feet. LPA, left pulmonary artery; MPA, main pulmonary artery; RPA, right pulmonary artery.
Morphological examination showed normal weight (51.5 kg; 50–75th percentile), height (1.62 m; 50–75th percentile), and OFC (54.8 cm; 50–75th percentile). She had a broad and depressed nasal bridge, low-hanging columella, short philtrum, underdeveloped alae nasi, midface retrusion, stiff and rigid ears and nasal cartilage, posteriorly rotated ears, and brachytelephalangism (middle finger length: 6.2 cm; <3rd percentile) (Fig. 1e).
Skeletal X-ray showed cranium thickness, ear and laryngeal calcifications, and shortening of the 2nd, 3rd, and 4th distal phalanges of hands and feet (Fig. 1a, e, f). Review of angiotomography images revealed tracheal calcifications (Fig. 1b, c). Thyroid hormone levels (thyroid-stimulating hormone, thyroxine) and coagulation cascade evaluation showed no abnormalities. Pulmonary function tests were normal [forced expiratory volume (FEV) ≥80%, forced vital capacity (FVC) ≥80%, and FEV1/FVC ≥80%]. Keutel syndrome was suspected based on these clinical features.
Materials and Methods
Genomic DNA was isolated from a peripheral blood sample using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) following the manufacturer's instructions and quantified using the Qubit dsDNA HS Assay kit for the Qubit 2.0 Fluorometer (Life Technologies, Carlsbad, CA, USA). The Qubit readings were used as a guide for dilution of the DNA samples.
The MGP gene was sequenced using the TruSight™ One Sequencing Panel (Illumina, Inc., San Diego, CA, USA) for paired-end sequencing with a v3 600 cycles kit on a MiSeq System platform (Illumina) according to the manufacturer's guidelines. This kit includes approximately 5,000 genes. We used 10 μL of sample DNA (50 ng total) according to the manufacturer's instructions. Although Keutel syndrome was suspected, this 5,000-gene panel was used as part of the validation process for this platform in our laboratory.
The mutation identified as pathogenic was confirmed by Sanger sequencing. Briefly, PCR products were enzymatically purified to remove residual primers and dNTPs using Illustra GFX PCR DNA and a Gel Band purification kit (GE Healthcare, UK) following the manufacturer's instructions. All sequencing reactions were carried out using the BigDye Terminator v.3.1 Cycle Sequencing Kit and were performed on a 3500 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) according to the manufacturer's instructions. The sequencing data were analyzed using Chromas software v.2.5.1 (Technelysium Pty Ltd, Brisbane, Australia).
Data Analysis
FASTQ files generated from the MiSeq System platform were subjected to several preprocessing steps. Files were first aligned to the Human Genome Reference Consortium build 37 (hg19) using the Burroughs-Wheeler Alignment (BWA) algorithm as implemented in the BWA software package, yielding BAM files of the aligned reads. After genome alignment, we performed the Variant Call. In this step, single nucleotide polymorphisms and short indels were identified using the Genome Analysis Toolkit and compared with known variants. Variants were annotated with ANNOVAR and read alignments were visualized using Integrative Genomics Viewer. ANNOVAR produces a VCF output file, which contains information from databases regarding the annotated variants. Each single variant reported in the VCF output file was visualized and evaluated for coverage and Q score. Based on the guidelines of the American College of Medical Genetics and Genomics (ACMG) [Richards et al., 2015], all regions with a sequencing depth <20 were considered unsuitable for analysis. Furthermore, we established a minimum threshold Phred-like quality score of 30 (base call accuracy of 99.9%).
Results
Sequence analysis identified a novel homozygous missense variant in the MGP gene, c.2T>C, which predicts p.Met1Thr in exon 1. This variant is not present in population control databases (Exome Aggregation Consortium, 1000 genomes, Exome Variant Server, and Brazilian Archive of Mutation). According to the ACMG guidelines [Richards et al., 2015], this variant is classified as null (initiation codon variant) and thus pathogenic. Furthermore, algorithms in silico (PolyPhen2, Mutation Taster, and SIFT) predicted that this variant causes damage to the protein. The variant found by panel sequencing was confirmed by Sanger sequencing (Fig. 2).
Fig. 2.
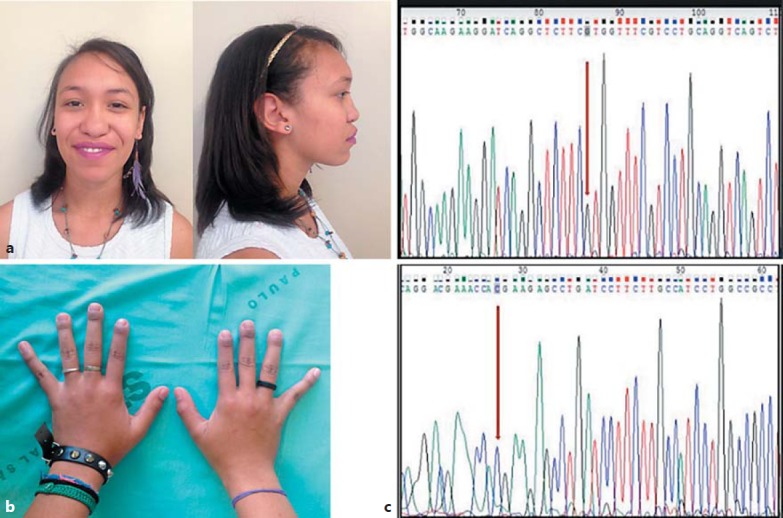
Dysmorphic features of the patient and molecular confirmation of Keutel syndrome. a Lateral and frontal view of the patient. b Brachytelephalangism (from the 2nd to 4th fingers). c Electropherogram showing forward and reverse Sanger sequencing of the MGP gene, confirming the c.2T>C homozygous variant (red arrows).
Discussion
To the best of our knowledge, we identified the first case of Keutel syndrome in a Brazilian patient with confirmed molecular diagnosis. The pathogenic variant found in the MGP gene has not been previously reported.
To date, only 36 confirmed cases of Keutel syndrome have been described [Hur et al., 2005; Sun et al., 2013]. Abnormal cartilage calcification and brachytelephalangism are cardinal findings, although other clinical features are common, including a typical facial phenotype (broad and depressed nasal bridge and midface retrusion), intellectual disability, short stature, hearing loss, peripheral pulmonary artery stenosis, and respiratory diseases [Cormode et al., 1986; Ziereisen et al., 1993; Teebi et al., 1998; Hur et al., 2005; Sun et al., 2013]. In a report of 3 new cases and literature review, Hur et al. [2005] identified additional clinical features such as optic atrophy and areas of encephalomalacia observed by brain MRI. While it is not clear if these additional features are related to the Keutel syndrome causative mutation, it is possible that encephalomalacia is a consequence of cerebrovascular compromise caused by calcification. In another case report and review of 26 cases, Sun et al. [2013] found that the most common clinical features were cartilage calcification (100% of the cases), brachytelephalangism (100%), facial dysmorphisms such as midface retrusion and depressed nasal bridge (94%), and peripheral pulmonic stenosis (84%). Our patient also presented with the main features of Keutel syndrome, namely abnormal cartilage calcification, facial dysmorphisms, brachytelephalangism, and pulmonary artery stenosis.
The siblings originally described by Keutel et al. [1972] were subsequently reevaluated by Meier et al. [2001]. The affected male had died at age 38 from chronic obstructive pulmonary disease, and postmortem examination revealed extensive calcification of the tracheobronchial tree with significant obstruction up to the level of the lobular bronchi and chronic cor pulmonale. Khosroshahi et al. [2014] also monitored 4 Keutel syndrome patients for 26 years, although examinations were irregular (not strictly periodic). Follow-up included physical examination (facial appearance, skin lesions) and complementary exams, including pulmonary function, thyroid function, anatomy, hearing function, and assessment of upper and lower airway cartilage ossification. These examinations showed that all patients suffered from recurrent upper and lower respiratory tract diseases, chronic sinusitis, chronic obstructive pulmonary disease, asthma, and emphysema. Moreover, the pulmonary functional tests revealed an obstructive lung disease pattern. The patients also exhibited progressive unilateral or bilateral hearing loss (mild, moderate, or profound), thyroid nodules (including one case of papillary microcarcinoma), and multiple erythematous lesions. In our patient, thyroid hormone level and hearing were normal. Our patient also showed no evidence of abnormal lung function. Therefore, dyspnea and heart murmur are likely related to pulmonary artery stenosis.
The homozygous variant found in MGP (c.2T>C, which predicts p.Met1Thr in exon 1) is classified as pathogenic based on the ACMG guidelines criteria [Richards et al., 2015]:
Null variant (nonsense, frameshift, canonical ±1 or 2 splice sites, initiation codon, single or multiexon deletion) in a gene where loss of function (LOF) is a known mechanism of disease
Absent from controls (or at extremely low frequency if recessive) in Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium
Multiple lines of computational evidence support a deleterious effect on the gene or gene product (conservation, evolutionary, splicing impact, etc.).
The variant found in our patient is a novel mutation. However, literature review revealed only 10 patients with molecular diagnosis [Munroe et al., 1999; Hur et al., 2005; Cranenburg et al., 2011; Weaver et al., 2014; Tüysüz et al., 2015; Bayramoğlu et al., 2016]. These patients shared 7 distinct pathogenic variants: 3 were donator or acceptor splice site type (IVS1-2A>G/c.62-2A>G; IVS2+1G>A; IVS1+1G>A/c.61+1G>A), 3 were deletion variants [1 missense (c.113T>A), 1 nonsense (c.79G>T; p.Glu27X), and 1 frameshift (c.69delG)], and 1 was a partial deletion of exon 4. The variant IVS1+1G>A/c.61+1G>A was found in 2 unrelated patients, and the variant IVS1-2A>G/c.62-2A>G was found in 3 probands (2 of them from the same sibship).
Considering that 5 of 7 variants were null mutations (splice site, nonsense, frameshift deletion, and exon deletion), the syndrome is likely caused by LOF of the MGP gene. The mutation found in our patient is at the initiation codon, consistent with a LOF mechanism as causative of Keutel syndrome. However, given the paucity of molecular information, it is not possible to establish a well-founded genotype-phenotype association.
Conclusion
We report the first Brazilian patient with a molecular diagnosis of Keutel syndrome. The patient was referred to our genetic clinic due to peripheral pulmonary artery stenosis, a frequent feature of Keutel syndrome. The variant identified (c.2T>C; p. Met1Thr) is novel, but in accordance with previous cases, it appears to be LOF.
Identifying the syndromic etiology of this cardiac anomaly is crucial to provide the best clinical follow-up. Furthermore, proper genetic counseling is required for the parents considering the 25% risk of recurrence for future sibship. Therefore, it is important to evaluate the patient's younger sister.
This clinical report highlights the importance of genetic testing for patients with pulmonary artery stenosis and accompanying dysmorphisms. Keutel syndrome should be considered in the differential diagnosis when brachydactyly and abnormal cartilage calcifications are associated with this cardiac anomaly.
Statement of Ethics
Informed consent for manuscript publication and photographs was obtained from the patient and her mother. Procedures performed were in accordance with ethical standards (Helsinki Declaration).
Disclosure Statement
The authors have no conflicts of interest to disclose.
Acknowledgments
We thank the patient and her mother for participating in this study; we also thank Salomão Zoppi and Genomika Diagnostics for allowing us to perform molecular diagnosis without costs.
References
- Bayramoğlu A, Saritemur M, Tasdemir S, Omeroglu M, Erdem HB, Sahin I. A rare cause of dyspnea in emergency medicine: Keutel syndrome. Am J Emerg Med. 34((2016)):935. doi: 10.1016/j.ajem.2015.09.020. [DOI] [PubMed] [Google Scholar]
- Cormode EJ, Dawson M, Lowry RB. Keutel syndrome: clinical report and literature review. Am J Med Genet. 24((1986)):289–294. doi: 10.1002/ajmg.1320240209. [DOI] [PubMed] [Google Scholar]
- Cranenburg EC, VAN Spaendonck-Zwarts KY, Bonafe L, Mittaz Crettol L, Rödiger LA, et al. Circulating matrix γ-carboxyglutamate protein (MGP) species are refractory to vitamin K treatment in a new case of Keutel syndrome. J Thromb Haemost. 9((2011)):1225–1235. doi: 10.1111/j.1538-7836.2011.04263.x. [DOI] [PubMed] [Google Scholar]
- Hale JE, Fraser JD, Price PA. The identification of matrix Gla protein in cartilage. J Biol Chem. 263((1998)):5820–5824. [PubMed] [Google Scholar]
- Hur DJ, Raymond GV, Kahler SG, Riegert-Johnson DL, Cohen BA, Boyadjiev SA. A novel MGP mutation in a consanguineous family: review of the clinical and molecular characteristics of Keutel syndrome. Am J Med Genet A. 135((2005)):36–40. doi: 10.1002/ajmg.a.30680. [DOI] [PubMed] [Google Scholar]
- Keutel J, A new autosomal recessive syndrome: peripheral pulmonary stenosis, brachytelephalangism, neural hearing loss and abnormal cartilage calcification/ossification . Part XV. Cardiovascular System. In: Bergsma D, editor. Birth Defects. No. 5. vol VIII. Baltomore: Williams & Wilkins; 1972. [Google Scholar]
- Khosroshahi HE, Sahin S, Akyuz Y, Ede H. Long term follow-up of four patients with Keutel syndrome. Am J Med Genet A. 164A((2014)):2849–2856. doi: 10.1002/ajmg.a.36699. [DOI] [PubMed] [Google Scholar]
- Meier M, Weng LP, Alexandrakis E, Ruschoff J, Goeckenjan G. Tracheobronchial stenosis in Keutel syndrome. Eur Respir J. 17((2001)):566–569. doi: 10.1183/09031936.01.17305660. [DOI] [PubMed] [Google Scholar]
- Munroe PB, Olgunturk RO, Fryns JP, Van Maldergem L, Ziereisen F, et al. Mutations in the gene encoding the human matrix Gla protein cause Keutel syndrome. Nat Genet. 21((1999)):142–144. doi: 10.1038/5102. [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 17((2015)):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurgers LJ, Spronk HM, Skepper JN, Hackeng TM, Shanahan CM, et al. Post-translational modifications regulate matrix Gla protein function: importance for inhibition of vascular smooth muscle cell calcification. J Thromb Haemost. 5((2007)):2503–2511. doi: 10.1111/j.1538-7836.2007.02758.x. [DOI] [PubMed] [Google Scholar]
- Sun LF, Ju YF, Fu GJ, Wang , JR, Feng YZ, Chen X. Keutel syndrome with tracheal stenosis as the major symptom: case report and literature review (in Chinese) Zhonghua Er Ke Za Zhi. 51((2013)):527–530. [PubMed] [Google Scholar]
- Teebi AS, Lambert DM, Kaye GM, Al-Fifi S, Tewfik TL, Azouz EM. Keutel syndrome: further characterization and review. Am J Med Genet. 78((1998)):182–187. [PubMed] [Google Scholar]
- Tüysüz B, Cinar B, Laçiner S, Onay H, Mittaz-Crettol L. Clinical variability in two sisters with Keutel Syndrome due to a homozygous mutation in MGP gene. Genet Couns. 26((2015)):187–194. [PubMed] [Google Scholar]
- Weaver KN, El Hallek M, Hopkin RJ, Sund KL, Henrickson M, et al. Keutel syndrome: report of two novel MGP mutations and discussion of clinical overlap with arylsulfatase E deficiency and relapsing polychondritis. Am J Med Genet A. 164A((2014)):1062–1068. doi: 10.1002/ajmg.a.36390. [DOI] [PubMed] [Google Scholar]
- Ziereisen F, De Munter C, Perlmutter N. The Keutel syndrome. Report of a case and review of the literature. Pediatr Radiol. 23((1993)):314–315. doi: 10.1007/BF02010925. [DOI] [PubMed] [Google Scholar]