

Abstract
Pathogenic variants in WAC are uncommon causes of developmental delay and neurobehavioral phenotypes. The clinical features associated with WAC haploinsufficiency include recognizable dysmorphic facial features that were recently delineated as DeSanto-Shinawi syndrome (DESSH; OMIM 616708). Additional clinical features include hypotonia, hearing and vision abnormalities, gastrointestinal problems, and behavioral difficulties. Here, we report a case of a 4-year-old Colombian male patient with typical dysmorphic facial features, developmental delay, hyperactivity, and recurrent respiratory infections. His immune workup revealed hypogammaglobulinemia, and clinical exome sequencing revealed a novel intronic variant in WAC (c.1437+1G>A). To the best of our knowledge, this is the first case of DESSH in South America, underlining the accumulating evidence of the significant role of WAC haploinsufficiency in neurobehavioral phenotypes. Although this report suggested the potential involvement of WAC in immune regulation, additional reports are required to confirm our observations.
Keywords: Dysmorphic facial features, Intellectual disability, WAC
Intellectual disability (ID) is a neurodevelopmental disorder which is strongly related to a genetic component [Ropers, 2010]. Technological advances, such as whole-exome sequencing (WES), have facilitated the identification of novel ID genes in patients with unknown etiologies. Although pathogenic genetic variants in hundreds of genes have been associated with ID, most variants have a very low prevalence, and the clinical features are frequently indistinguishable [de Ligt et al., 2012; Gilissen et al., 2014].
Recently, DeSanto et al. [2015] described a rare and recognizable ID syndrome called DeSanto-Shinawi syndrome (DESSH; OMIM 616708) in association with loss-of-function variants of WAC. Later, additional patients were identified [Lugtenberg et al., 2016] with similar clinical phenotypes, including ID, hypotonia, behavioral problems, gastrointestinal abnormalities, and distinctive facial features (square face, deep-set eyes, long palpebral fissures, and broad mouth) [DeSanto et al., 2015; Lugtenberg et al., 2016]. Complementary experimental research in Drosophila has shown the role of WAC in cognitive processes [Dietzl et al., 2007; Lugtenberg et al., 2016]. Other studies have also demonstrated the functions of WAC in transcription elongation, microtubule formation, and histone H2B ubiquitination regulation [Shahdadpuri et al., 2008; Zhang and Yu, 2011].
Here, we report the case of a patient with DESSH due to a novel pathogenic genetic variant of WAC identified using WES. The patient presented with dysmorphic facial features, gastrointestinal abnormalities, recurrent respiratory infections, and hypotonia. In addition, he exhibited hypogammaglobulinemia, which has not been described in DESSH. This report compares the findings in our patient with those in previously reported cases and discusses a potential link between WAC haploinsufficiency and immune dysfunction.
Case Report
The proband was a 4-year-old male born at 38 weeks of gestation to nonconsanguineous Colombian parents. The patient's mother was 29 years old and healthy; his father was 34 years of age and had hyperthyroidism, but was otherwise healthy. Both pregnancy and delivery were uncomplicated with Apgar scores of 8 and 9 at 1 and 5 min, respectively. His birth weight was 2.7 kg (50th centile) and length was 47 cm (40th centile). The patient exhibited 6 documented infections and 4 additional mild respiratory infections which were reported by his family and resolved spontaneously at home. These infections started at 6 months of age and included 1 episode of bronchiolitis, which required management with corticosteroids, 3 episodes of pneumonia, which responded to intravenous antibiotics, and abscesses in his gluteus and leg.
The developmental delay was first noted at 6 months of age when he started exhibiting motor delay: sitting at 10 months, crawling at 2 years, and walking at 3 years. Later, his speech development was also noted as not being age appropriate, and at present, he can speak only 5–6 words. Since birth, the patient displayed feeding difficulties, swallowing problems, and gastroesophageal reflux symptoms. In addition, he displayed behavioral problems, including night terrors and hyperactivity. However, he did not exhibit problems in his social interaction and could play with peers, maintain eye contact, and obey instructions. The patient is receiving physical, occupational, and language therapies.
On physical examination at 4 years of age, his weight was 16 kg (6th centile), height was 99 cm (<1st centile), and OFC was 49 cm (2nd centile). He presented with dysmorphic features, including a prominent broad forehead, hypertelorism, epicanthic folds, posteriorly rotated ears, depressed and broad nasal bridge, bulbous nasal tip, thin upper lip, macroglossia, hirsutism on the back, truncal hypotonia, bilateral single palmar creases, and brachydactyly (Fig. 1, 2).
Fig. 1.
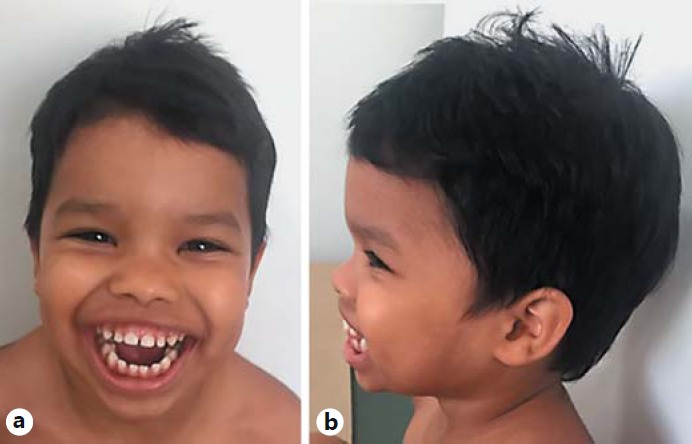
Facial features of the 4-year-old proband. Frontal view (a) and lateral view (b) showing facial dysmorphism, coarse facies, broad forehead, synophrys, deep-set eyes, hypertelorism, broad and depressed nasal bridge, epicanthic folds, posteriorly rotated ears, and a wide mouth.
Fig. 2.
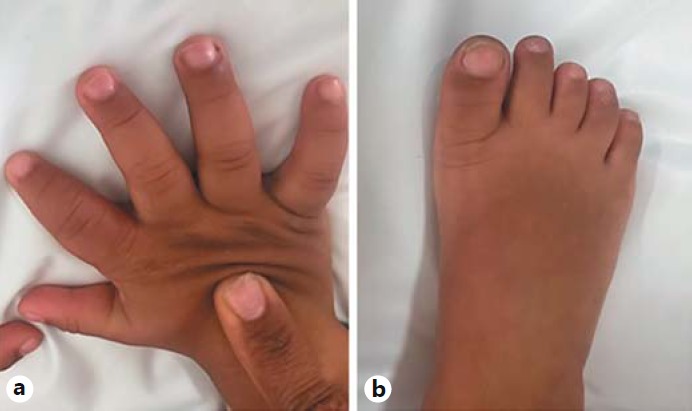
Extremities of the patient. a Right hand showing short and thick fingers and fifth finger clinodactyly. b Right foot showing short third to fifth toes.
His initial diagnostic examinations included normal comparative genomic hybridization, brain MRI, and echocardiography. Due to recurrent infections, he underwent an immune evaluation at 4 years of age, which included complete blood cell count evaluation. His lymphocyte subpopulation counts were as follows: CD4, 1,058 (range: 700–2,020) cells/mm3; CD3, 1,823 (1,400–3,700) cells/mm3, and CD8, 624 (490–1,300) cells/mm3. In addition, other blood component counts were as follows: natural killer cells, 233 (150–250) cells/mm3; immunoglobulin (Ig) M, 46 (54–392) mg/dL; IgG, 631 (805–2,421) mg/dL, and IgA, 44 (58–311) mg/dL. Repeated studies conducted at 5 years of age revealed low Ig levels. Although chronic granulomatous disease was suspected, the dihydrorhodamine (DHR) test result was negative. Furthermore, lactic acid and ammonia levels were normal. While paranasal CT at the age of 2.5 years revealed sinusitis and chronic mastoiditis, the boy's chest CT revealed small foci of opacity in the lung bases suggestive for atelectasis.
Methods
We performed WES on DNA obtained from the peripheral blood of the patient and his parents, and variants found were compared and filtered.
The coding and flanking intronic regions were enriched using Agilent solution (Agilent Technologies) and sequenced using the HiSeq 2500/4000 system (Illumina). Conventional Sanger sequencing was used to sequence at least 1 causative or rare variant for a second, independent confirmation. Only variants in the coding and flanking intronic regions (±8 bp) with a minor allele frequency (MAF) of <1.5% were evaluated. According to the Human Gene Mutation Database (HGMD), known disease-causing variants were evaluated in up to 30 bp of flanking regions and up to MAF of 5%. MAFs were based on the following databases: 1000 Genomes, dbSNP and Exome Variant Server and Exome Aggregation Consortium (ExAc). We covered at least 30 high-quality sequencing reads per base using high-throughput sequencing.
Results
WES revealed a heterozygous novel splicing mutation in WAC (NM_016628.4:c.1437+1G>A), which was confirmed by Sanger sequencing. Both parents tested negative for this genetic variant, confirming its de novo nature. This variant was not reported in ExAc. The genomic position was highly evolutionarily conserved and validated in the Human Splicing Finder (http://www.umd.be/HSF3/). The mutation is located in an essential splicing site and therefore is expected to have a deleterious effect on splicing donor site, adding an intronic region at the end of exon 10. Furthermore, it was considered pathogenic with a Functional Analysis through Hidden Markov Models v2.3 (FATHMM) score of 1 (Fig. 3).
Fig. 3.
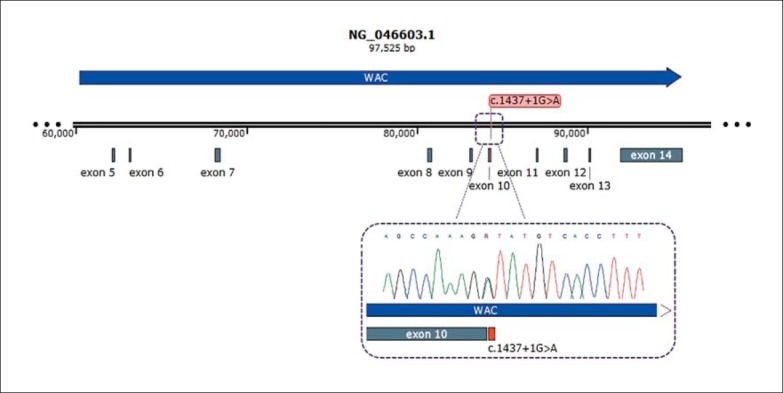
Diagram of WAC and the location of exons 5-14 showing the splicing variant designated as NM_016628.4:c.1437+1G>A in red found in the proband. The window below shows the Sanger sequencing results.
Discussion
DESSH is a rare genetic condition caused by loss-of-function mutations in WAC and characterized by motor and speech delays, behavioral abnormalities, and recognizable dysmorphic facial features, including a broad forehead, low nasal bridge, bulbous nasal tip, posteriorly rotated ears, deep-set eyes, brachycephaly as well as hearing and visual abnormalities [Shahdadpuri et al., 2008; Okamoto et al., 2012; Mroczkowski et al., 2014]. In addition, mutations in WAC were described in large cohorts of patients with autism and ID [Okamoto et al., 2012]. However, the mechanism by which WAC genetic variants lead to these phenotypes remains unclear.
Here, we report a case of DESSH caused by a novel heterozygous splicing mutation (NM_016628.4:c.1437+ 1G>A) in WAC. The phenotype of this patient was similar to those described in previously reported cases, including facial features such as synophrys, bilateral epicanthic folds, broad forehead, posteriorly rotated ears, low nasal bridge, and a thin upper lip. In addition, the patient exhibited ID, behavioral impairment, and feeding difficulties that were also noted in previous cases. However, behavioral abnormalities were less pronounced in our patient than in previously reported patients, and he did not display other major clinical findings [Okamoto et al., 2012; Mroczkowski et al., 2014; DeSanto et al., 2015]. In addition, our patient displayed hypogammaglobulinemia associated with recurrent infections, which to the best of our knowledge has not been previously described.
The severity of phenotype and clinical features could vary depending on the position of the mutation. Mutations that cluster in the C-terminal of WAC result in a milder phenotype compared with N-terminal mutations [Ropers, 2010]. This may elucidate the relatively milder phenotype in our patient; however, the effect of the mutation type on the phenotype remains unclear [Suñé-Pou et al., 2017] (Table 1).
Table 1.
Frequency of clinical features in patients with mutations in WAC
| DeSanto et al., 2015 | Lugtenberg et al., 2016 | Current report | |
|---|---|---|---|
| Normal prenatal period | 6/6 | NR | + |
| Hearing impairment | 6/6 | NR | + |
| Language delay | 6/6 | NR | + |
| Learning difficulties | 6/6 | 8/10 | + |
| Motor delay | 6/6 | 9/10 | + |
| Weight in normal percentile | 6/6 | 4/10 | + |
| Height in normal percentile | 6/6 | 6/10 | + |
| Respiratory infections | 0/6 | 7/10 | + |
| Behavioral abnormalities | 6/6 | 9/10 | + |
| Anxiety | 3/6 | 3/10 | + |
| ADHD | 3/6 | 4/10 | + |
| Autistic features | 1/6 | 4/10 | - |
| Sleep disturbances | 1/6 | 6/10 | - |
| Feeding difficulties | 3/6 | 4/10 | + |
| Short fingers | NR | 3/10 | + |
| Hypotonia | 6/6 | 6/10 | + |
| Dysmorphic facial features | 6/6 | 10/10 | + |
| Prominent forehead | 6/6 | 10/10 | + |
| Posteriorly rotated ears | 3/6 | 10/10 | + |
| Low-set ears | 3/6 | NR | + |
| Preauricular pit | 1/6 | NR | - |
| Low nasal bridge | 6/6 | NR | + |
| Bulbous nasal tip | 5/6 | NR | + |
| Malar hypoplasia | 1/6 | NR | + |
| Synophrys | 3/6 | 10/10 | + |
| Visual impairment | 2/6 | 3/10 | - |
| Deep-set eyes | 2/6 | 10/10 | + |
| Hypertelorism | 2/6 | 10/10 | + |
| Macroglossia | NR | 10/10 | + |
| Thin upper lip | 2/6 | NR | + |
| Flat philtrum | 1/6 | NR | + |
| Hirsutism | 2/6 | NR | + |
| Hypogammaglobulinemia | NR | NR | + |
ADHD, attention deficit hyperactivity disorder; NR, not reported; +, present; –, absent.
WAC encodes a protein that plays an essential role in multiple cell processes, including transcription, microtubule development, autophagy, and regulation of the Golgi apparatus function [Joachim et al., 2012; Lugtenberg et al., 2016]. Furthermore, it regulates histone transcription and ubiquitination by mediating the interaction between E3 ligase and RNF20-RNF40, thereby inhibiting chromatin organization and gene transcription [Totsukawa et al., 2011]. Moreover, WAC's role in transcription is essential for the activation of cell cycle checkpoints in response to DNA damage [Zhang and Yu, 2011]. Autophagy is essential for immune system regulation for maintaining cellular homeostasis and activating innate and adaptive immunities [Zhou and Zhang, 2012]. Furthermore, WAC facilitates pathogen recognition, antigen presentation and delivery to major histocompatibility complex type II [Joachim et al., 2012; Zhou and Zhang, 2012]. Thus, immunological findings and recurrent infections in our patient could be related to WAC haploinsufficiency because of the significant role of autophagy in different cellular processes, including immunity [Joachim et al., 2012; Zhou and Zhang, 2012; Harris et al., 2017]. Hence, further investigations are warranted to determine the frequency of infections and immune dysregulation in patients with DESSH and to elucidate the potential link between impaired autophagy and immune dysregulation as well as susceptibility to infections.
In conclusion, we report a case of a novel mutation in WAC with recognizable clinical and craniofacial dysmorphic features. The patient also exhibited recurrent infections and hypogammaglobulinemia, which may suggest an association between WAC haploinsufficiency and immunological impairments. However, further reports and experimental studies are required to confirm this observation and demonstrate this association.
Statement of Ethics
We obtained written informed consent from the patient's parents for publication of this case report and accompanying images. This study was approved by the ethics committee at our institution (Comite de Ética en Investigación Biomédica in Fundación Clínica Valle del Lili, Cali, Colombia).
Disclosure Statement
The authors declare no conflicts of interest. In addition, they have no financial conflicts relevant to this article to disclose, as no external funding was used to formulate this report.
Acknowledgments
We thank the Universidad ICESI and the Clinical Foundation Valle del Lili for their constant support.
References
- de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 367((2012)):1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- DeSanto C, D'Aco K, Araujo GC, Shannon N, DDD Study, et al. WAC loss-of-function mutations cause a recognisable syndrome characterised by dysmorphic features, developmental delay and hypotonia and recapitulate 10p11.23 microdeletion syndrome. J Med Genet. 52((2015)):754–761. doi: 10.1136/jmedgenet-2015-103069. [DOI] [PubMed] [Google Scholar]
- Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 448((2007)):151–156. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
- Gilissen C, Hehir-Kwa JY, Thung DT, van de Vorst M, van Bon BW, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature. 511((2014)):344–347. doi: 10.1038/nature13394. [DOI] [PubMed] [Google Scholar]
- Harris J, Lang T, Thomas JPW, Sukkar MB, Nabar NR, Kehrl JH. Autophagy and inflammasomes. Mol Immunol. 86((2017)):10–15. doi: 10.1016/j.molimm.2017.02.013. [DOI] [PubMed] [Google Scholar]
- Joachim J, Wirth M, McKnight NC, Tooze SA. Coiling up with SCOC and WAC: two new regulators of starvation-induced autophagy. Autophagy. 8((2012)):1397–1400. doi: 10.4161/auto.21043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugtenberg D, Reijnders MR, Fenckova M, Bijlsma EK, Bernier R, et al. De novo loss-of-function mutations in WAC cause a recognizable intellectual disability syndrome and learning deficits in Drosophila. Eur J Hum Genet. 24((2016)):1145–1153. doi: 10.1038/ejhg.2015.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mroczkowski HJ, Arnold G, Schneck FX, Rajkovic A, Yatsenko SA. Interstitial 10p11.23-p12.1 microdeletions associated with developmental delay, craniofacial abnormalities, and cryptorchidism. Am J Med Genet A. 164A((2014)):2623–2626. doi: 10.1002/ajmg.a.36627. [DOI] [PubMed] [Google Scholar]
- Okamoto N, Hayashi S, Masui A, Kosaki R, Oguri I, et al. Deletion at chromosome 10p11.23-p12.1 defines characteristic phenotypes with marked midface retrusion. J Hum Genet. 57((2012)):191–196. doi: 10.1038/jhg.2011.154. [DOI] [PubMed] [Google Scholar]
- Ropers HH. Genetics of early onset cognitive impairment. Annu Rev Genomics Hum Genet. 11((2010)):161–187. doi: 10.1146/annurev-genom-082509-141640. [DOI] [PubMed] [Google Scholar]
- Shahdadpuri R, de Vries B, Pfundt R, de Leeuw N, Reardon W. Pseudoarthrosis of the clavicle and copper beaten skull associated with chromosome 10p11.21p12.1 microdeletion. Am J Med Genet A. 146A((2008)):233–237. doi: 10.1002/ajmg.a.32088. [DOI] [PubMed] [Google Scholar]
- Suñé-Pou M, Prieto-Sánchez S, Boyero-Corral S, Moreno-Castro C, El Yousfi Y, et al. Targeting splicing in the treatment of human disease. Genes (Basel) 8((2017)):E87. doi: 10.3390/genes8030087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totsukawa G, Kaneko Y, Uchiyama K, Toh H, Tamura K, Kondo H. VCIP135 deubiquitinase and its binding protein, WAC, in p97ATPase-mediated membrane fusion. EMBO J. 30((2011)):3581–3593. doi: 10.1038/emboj.2011.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Yu X. WAC, a functional partner of RNF20/40, regulates histone H2B ubiquitination and gene transcription. Mol Cell. 41((2011)):384–397. doi: 10.1016/j.molcel.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XJ, Zhang H. Autophagy in immunity: implications in etiology of autoimmune/autoinflammatory diseases. Autophagy. 8((2012)):1286–1299. doi: 10.4161/auto.21212. [DOI] [PMC free article] [PubMed] [Google Scholar]