

Graphical abstract
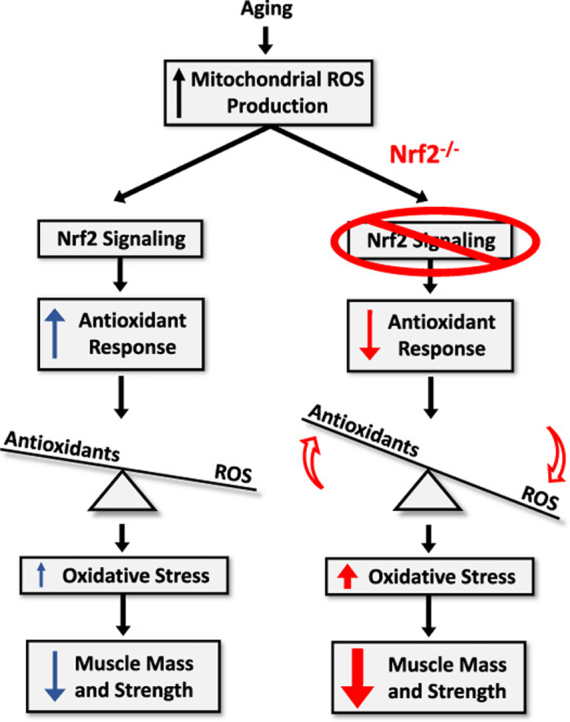
1. Introduction
Age-related loss of skeletal muscle mass and contractile dysfunction, or sarcopenia, reduces independence and quality of life in the elderly and leads to increased risk of comorbidities with chronic diseases, including diabetes [1] and cardiovascular disease [2]. Our prior work established that increased mitochondrial reactive oxygen species (ROS) production and oxidative damage [3], [4] occur in muscle atrophy and are correlated with the extent of atrophy [5]. Deletion of the antioxidant enzyme CuZn superoxide dismutase (Sod1) leads to mitochondrial dysfunction, increased oxidative damage, neuromuscular junction disruption, and muscle atrophy and contractile dysfunction [3], [6], [7], [8]. However, deficiency of other antioxidant enzymes, such as the mitochondrial superoxide dismutase MnSOD (Sod2) and glutathione peroxidase 1 (Gpx1), does not worsen muscle atrophy despite increased oxidative stress [9], [10], [11], and overexpression of Sod2 does not delay muscle atrophy despite a reduction of oxidative damage [12]. In contrast to our results in the Sod1 null mouse model, these results argue against a causal role for muscle oxidative stress in sarcopenia pathogenesis. To further explore this question, in the current study we ask whether loss of the transcriptional regulator Nrf2 can modulate muscle atrophy and contractile dysfunction elicited by aging.
The transcription factor nuclear factor (erythroid-derived 2)-like 2 (Nrf2) is a primary regulator of antioxidant enzyme and redox regulating protein expression that is induced in response to oxidative stress [13]. Nrf2 signaling is activated in skeletal muscle from aged active but not sedentary humans, and the lack of induction in sedentary individuals is associated with increased oxidative damage [14]. Mice deficient in Nrf2 (Nrf2-/- mice) are a useful tool to study the role of Nrf2 antioxidant response induction in muscle aging. Previous studies indicate that young (2–3 month old) Nrf2-/- mice have a reduced antioxidant response and increased levels of oxidative damage following hindlimb skeletal muscle denervation compared to wildtype mice [15]. Skeletal muscle from young (5 month old) Nrf2-/- mice shows no difference in maximum tetanic force, time to peak tension, or half relaxation time in situ but does show increased fatigue compared to wildtype controls associated with increased ROS production [16]. Previous studies have shown that skeletal muscle from aged (23–24 months) Nrf2-/- mice has reduced levels of basal and exercise-induced antioxidant enzymes, a reduced muscle stem cell population, and increased ROS production, oxidative stress, ubiquitinated proteins, and apoptotic signaling [17], [18]. Together these data suggest that Nrf2 plays an important role in muscle homeostasis during aging.
We hypothesized that Nrf2 deficiency in skeletal muscle of aged mice would contribute to a sarcopenia phenotype through mitochondrial dysfunction, dysregulation of cellular redox balance, increased levels of oxidative damage, and contractile dysfunction. To test our hypothesis, we measured these parameters in skeletal muscle from young (4 month) wildtype and old (24 month) wildtype and Nrf2-/- mice. Our data show reduced muscle mass and contractile force generation in old Nrf2-/- mice compared to age-matched wildtype mice associated with reduced mitochondrial oxygen consumption, increased mitochondrial ROS production, increased protein nitrosylation, cellular redox dysregulation, and reduced acetylcholine receptor expression. This work provides evidence that skeletal muscle Nrf2 plays a protective role against sarcopenia pathogenesis.
2. Materials and methods
2.1. Animals
The study was approved by the Institutional Animal Care and Use Committees at University of Oklahoma Health Sciences Center and OMRF. Young (~ 4 month old) C57Bl6/J and aged (~ 24 month old) B6.129X1-Nfe2l2tm1Ywk/J male mice were purchased from Jackson Laboratory (Bar Harbor, ME). Age-matched controls (~ 24 month old) C57Bl6/J were obtained from the National Institute on Aging.
2.2. Grip strength and grid hanging time
To evaluate the effect of Nrf2 deficiency on in vivo muscle function, we determined forelimb grip strength using a computerized electronic pull strain gauge (Grip Strength Meter-Columbus Instruments, Columbus, OH, USA), measured by the same observer three consecutive times. The four-limb grid hang test is a well-established test which seeks to evaluate muscle performance and motor deficits in rodent models. A four limb hang test was performed to measure the animals’ ability to exhibit sustained limb tension to overcome the gravitational pull. A standard metal grid was placed and fixed approximately 25 cm above soft bedding to prevent the mice from hurting themselves upon falling down. The mice were placed on the grid, and a short time interval was allowed for each mouse to grasp and familiarize himself with the grid to allow for consistent paw grasp with all four paws. After this the grid was inverted, and the latency-to-fall for each animal was recorded. The average of three measurements per mouse was used to determine grip strength and hang test values.
2.3. In vitro contractile properties of EDL
Studies of limb muscle isometric function were conducted as previously described [19]. Briefly, mice were sacrificed using gaseous carbon dioxide, and EDL muscle was excised and immediately placed in buffer solution for dissection. One end of the muscle was tied to a Dual-Mode Muscle Lever System (300C-LR, Aurora Scientific Inc, Aurora, Canada) and the other end onto a hook. Muscles were placed at optimal length and allowed 20 min of thermoequilibration to the desired temperature (32 °C), during which D-tubocurarine (25 μM) was added to the solution. Thereafter, measurements of force–frequency were initiated. In all electrical stimulations, a supramaximal current (600–800 mA) of 0.25 ms pulse duration was delivered through a stimulator (701 C, Aurora Scientific Inc.), while train duration for isometric contractions was 300 ms. All data were recorded and analyzed using commercial software (DMC and DMA, Aurora Scientific).
2.4. Fiber permeabilization
Preparation for skeletal muscle fiber permeabilization was performed as previously described [19], [20]. Briefly, a small piece (~ 3–5 mg) of red gastrocnemius muscle was carefully dissected, and we separated fibers in ice-cold solution (buffer X), containing (in mM) 7.23 K2EGTA, 2.77 CaK2EGTA, 20 imidazole, 0.5 DTT, 20 taurine, 5.7 ATP, 14.3 PCr, 6.56 MgCl2–6H2O, 50 K-MES (pH 7.1). The muscle bundle was permeabilized in saponin solution (30 µg/ml) for 30 mins, followed by 3 × 5 min washes in ice-cold wash buffer Z containing (in mM) 105 K-MES, 30 KCl, 10 K2HPO4, 5 MgCl2–6H2O, 0.5 mg/ml Bovine Serum Albumin (BSA), 0.1 EGTA (pH 7.1) [21].
2.5. Simultaneous measurement of respiration and hydrogen peroxide production
Oxygen consumption rate (OCR) and the rate of mitochondrial hydrogen peroxide production were simultaneously determined using the Oxygraph-2k (O2k, OROBOROS Instruments, Innsbruck, Austria) by a previously described method [22] with minor modifications. OCR was determined using an oxygen probe, while rates of hydrogen peroxide generation were determined using the O2k-Fluo LED2-Module Fluorescence-Sensor Green. Measurements were performed on permeabilized fibers in buffer Z media at 37 °C containing 10 µM Amplex® UltraRed (Molecular Probes, Eugene, OR), 1 U/ml horseradish peroxidase (HRP), and blebbistatin (25 µM). 10 µM Amplex® UltraRed has been shown not to impair mitochondrial respiration in permeabilized cells [23]. HRP catalyzes the reaction between hydrogen peroxide and Amplex UltraRed to produce the fluorescent resorufin (excitation: 565 nm; emission: 600 nM). The fluorescent signal was converted to nanomolar H2O2 via a standard curve established on each day of experiments. Background resorufin production was subtracted from each measurement. Rates of respiration and H2O2 production were determined using sequential additions of substrates and inhibitors as follows: glutamate (10 mM), malate (2 mM), pyruvate (5 mM), ADP (5 mM), succinate (10 mM), rotenone (1 µM), Antimycin A (1 µM), and TMPD (0.5 mM) immediately followed by ascorbate (5 mM). We added pyruvate in the sequence to test its impact on the mouse skeletal muscle fibers due to its previously reported impact on complex I-linked respiration [24], [25]. All respiration measurements were normalized to Antimycin A to account for non-mitochondrial oxygen consumption. Data for both OCR and rates of hydrogen peroxide generation were normalized by milligrams of muscle bundle wet weights.
2.6. Determination of GSH:GSSG ratio
Levels of GSH and GSSG were determined using reverse-phase HPLC and electrochemical detection [26]. GSH and GSSG were extracted from gastrocnemius tissue homogenate by treatment with 5% metaphosphoric acid (w/v). Proteins were precipitated upon incubation on ice (20 min) and then pelleted by centrifugation (10 min at 16,000g). The supernatant was filtered (0.45-μm syringe filters) before analysis of GSH and GSSG by HPLC and electrochemical detection (ESA HPLC system, ESA Coularray electrochemical detector 5600 A set at 750 mV). GSH and GSSG were eluted through a C18 column (Phenomenex Luna C18(2), 100 A, 3 µm, 150 × 4.6 mm) at 0.5 ml/min using an isocratic mobile phase consisting of 25 mM NaH2PO4, 0.5 mM 1-octane sulfonic acid, 4% acetonitrile (w/v), pH 2.7. GSH and GSSG concentrations were calculated employing GSH and GSSG standard curves constructed from peak areas.
2.7. Markers of lipid peroxidation
Levels of F2-isoprostanes in quadriceps were determined by a previously described method [27] with minor modifications to skeletal muscles. Briefly, 200 mg of tissue was homogenized in 10 ml of ice-cold Folch solution (CHCl3:MeOH, 2:1) containing butylated hydroxytoluene (BHT). The mixture was incubated at room temperature for 30 min. 2 ml of 0.9% NaCl (w/v) was added and mixed well. The homogenate was centrifuged at 3000g for 5 min at 4 °C. The aqueous layer was discarded while the organic layer was secured and evaporated to dryness under N2 at 37 °C. F2-isoprostanes were extracted and quantified by gas chromatography-mass spectrometry using the internal standard [2H4]8-Iso-PGF2α, which was added to the samples at the beginning of extraction to correct yield of the extraction process. Esterified F2-isoprostanes were measured using gas chromatography–mass spectrometry. The level of F2-isoprostanes in muscle tissues was expressed as nanograms of 8-Iso-PGF2α, per gram of muscle mass.
2.8. Redox status
Indicators of cellular redox status in gastrocnemius muscles were measured as previously described [27], [28]. All analytes were extracted from tissue by treatment with 150 mM potassium hydroxide. Proteins were precipitated upon incubation on ice (20 min), then pelleted by centrifugation (10 min at 16,000g) and the supernatant was filtered using 0.45-μm PVDF syringe filters. High-performance liquid chromatography and a UV/VIS diode array spectrometer were used to resolve and detect the analytes. The mobile phase consisted of 100 mM KH2PO4 and 1.0 mM tetrabutylammonium sulfate (TBAS) at pH 6.0 (buffer A) and CH3CN (buffer B) with a flow rate of 1.0 ml/min over an Eclipse Plus C18 column with 5 μM diameter beads, 4.6 × 150 mM in length (Agilent Technologies). Concentrations of NADH and NADPH were detected by fluorescence at excitation 340 nm and emission 430 nm. All analytes were quantified on the basis of the integrated area of standards.
2.9. Quantification of protein abundance using amino acid sequence
We used targeted quantitative mass spectrometry to measure protein abundance as previously described [29], [30] with minor modifications. Gastrocnemius samples were homogenized in a lysis buffer containing 10 mM Tris-Cl (pH 8.0), 1 mM EDTA, 1% Triton X-100 (v/v), 0.1% sodium deoxycholate (w/v), 0.1% SDS (w/v), 140 mM NaCL, and 1 mM PMSF, with protease inhibitor cocktail (Calbiochem Set III, EDTA-free; EMD Millipore; Billerica, MA, USA). Protein concentration was determined and a volume equivalent to 100 μg of total protein processed for analysis. BSA was added to samples to serve as an internal standard. The samples were incubated at 80 °C for 15 min in 1% SDS (w/v). Proteins were precipitated with 80% acetone (v/v) at − 20 °C overnight. The protein pellet was dissolved at 1 µg/µl in Laemmli sample buffer and 20 µg protein separated using SDS-PAGE. The gel was fixed and stained with GelCode Blue, and the 1.5-cm lane was cut out of the gel. Gel pieces were washed, reduced with DTT, alkylated with iodoacetamide, and digested with trypsin at room temperature overnight. Digested peptides were extracted with 50% methanol (v/v) and 10% formic acid (v/v). This extract was evaporated and dissolved in 1% acetic acid (v/v) for the LC-MS analysis.
Protein abundance was determined using selected reaction monitoring (SRM) with a triple quadrupole mass spectrometer (ThermoScientific TSQ Vantage, San Jose, CA, USA) with a splitless capillary column HPLC system (Eksigent, Dublin, CA, USA). Samples were injected into a C18 column (Phenomenex, Jupiter C18) and eluted with a gradient of acetonitrile in 0.1% formic acid (v/v). Absolute protein concentrations were determined using multiple validated peptide markers to determine abundance of each protein. The program Skyline [31] was used to monitor and process data from each peptide. The housekeeping proteins malate dehydrogenase 1 (Mdh1), glucose-6-phosphate isomerase (Gpi), heat-shock protein 1 (Hspd1), and BSA were used to normalize peptides representing glycolysis, Kreb's cycle, fatty acid oxidation, oxidative phosphorylation, and antioxidant enzyme proteins using the best flyer approach. Results for all detected peptides are listed in Supplemental Table 1.
2.10. Immunoblot and qRT-PCR
Information on primer sequences and primary antibodies are shown in Supplemental Table 2. For Nrf2, we used a primary antibody provided by Dr. Scott Plafker as described previously [32]. We followed standard procedures for qRT-PCR and western blot in homogenized gastrocnemius muscle. Procedures for the mitochondrial gene panel (Supplemental Table 3) [33], [34] and inflammation gene panel (Supplemental Table 4) were previously described [35].
2.11. Statistical analyses
Data analysis was performed using GraphPad Prism version 7.0b, GraphPad Software, La Jolla California USA. Outliers were removed from SRM and qRT-PCR data using Grubbs’ outlier test (Alpha = 0.05). Normal distribution of data was analyzed by D′Agostino and Pearson normality test. Non-normally distributed data were compared by Kruskal Wallis with Dunn's multiple comparisons test, while normally distributed data were analyzed using unpaired 1-way ANOVA followed by Tukey's post hoc test. Significance was established a priori at p < 0.05 in all cases. Values are presented as mean ± standard error of the mean (SEM).
3. Results
3.1. Nrf2 deficiency exacerbates age-related loss of muscle mass and contractile dysfunction
Nrf2 protein and mRNA expression were undetected in old Nrf2-/- mice (Fig. 1A, Supplemental Fig. 1). To determine the impact of Nrf2 deficiency on skeletal muscle mass and strength with age, we measured body weight, muscle mass for several hindlimb muscles, and muscle strength. As expected, body mass is increased in old wildtype and Nrf2-/- mice (24 months) compared to young (4 months) (Fig. 1B). However, the mass of individual hindlimb muscles normalized to body mass declines with age (Fig. 1E). Muscle mass declines to a greater extent in old Nrf2-/- mice compared to old wildtype mice in the gastrocnemius, tibialis anterior, and quadriceps femoris muscles, but not the soleus or extensor digitorum longus (EDL) (Fig. 1E). The age-related decline in muscle mass is associated with reduced forelimb grip strength in both old wildtype and old Nrf2-/- mice compared to young wildtype mice, and a trend toward a decline in grip strength in old Nrf2-/- mice compared to old wildtype (Fig. 1C). Grid hanging time is also significantly reduced by age in wildtype mice, which is further decreased by Nrf2 deficiency (Fig. 1D).
Fig. 1.

Impacts of Nrf2 deficiency on age-associated loss of skeletal muscle mass and in vivo muscle strength. Body weight in young, old, and old-Nrf2-/- mice at study endpoint. Young and old mice are aged ~ 4 and ~ 24 months for wildtype and Nrf2-/- mice. n = 9–15. B) Grip strength is normalized to mouse body weight (mN/kg). n = 7–17. C) Grid hanging time in seconds. n = 6–11. D) Isolated skeletal muscle mass normalized to body weight. n = 9–15. Young wildtype (white); old wildtype (grey); old-Nrf2-/- (black). Abbreviations: Gastroc, gastrocnemius; TA, tibialis anterior; EDL, extensor digitorum longus, Quad, quadriceps. Data are mean ± SEM. *p < 0.05.
To determine the impact of Nrf2 deficiency on skeletal muscle function in aged mice, we examined in vitro contractile properties of isolated EDL muscle, which tests intrinsic force generating capacity of muscle without neuronal impacts. Absolute force (mN) in the EDL muscle tends to decline with age and is significantly reduced in old Nrf2-/- mice by ~ 50% and ~ 40% compared to young and age-matched wildtype respectively (Figs. 2A, 2B). Maximum isometric tetanic specific force (N/cm2), absolute force normalized to estimated EDL cross sectional area, declines with age but is further reduced in old Nrf2-/- mice by ~ 50% and ~ 40% compared to young and old wildtype (Fig. 2C,D). Peak twitch tension is unchanged with age in wildtype mice consistent with existing literature [36] but decreases by ~ 35% in old Nrf2-/- mice (Fig. 2E). Time to peak twitch also remains similar by aging in wildtype mice, but old Nrf2-/- mice exhibit a ~ 40% increase (Fig. 2F). Half relaxation time is also increased by ~ 40% in old Nrf2-/- mice, but remains similar during aging in wildtype mice (Fig. 2G).
Fig. 2.

Effects of age and Nrf2 deficiency on EDL contractile properties in vitro. A) Force-frequency relationship of isolated EDL muscle from young, old, old-Nrf2-/- mice. Absolute force is expressed in mN. B) Maximum isometric force output at 300 Hz. C) Specific force-frequency relationship. Specific force, force normalized to estimated EDL bundle cross-sectional area. D) Maximum isometric specific force at 300 Hz. E) Peak tension normalized by estimated muscle cross-sectional area. F) Time to peak twitch in milliseconds. G) Half relaxation time (½ RT). n = 5–6. Young wildtype (white); old wildtype (grey); old-Nrf2-/- (black). Data are mean ± SEM. *p < 0.05.
3.2. Nrf2 deficiency is associated with reduced mitochondrial respiration and increased ROS production and oxidative stress in skeletal muscle
We have previously shown that muscle mitochondrial dysfunction and ROS production is increased in isolated mitochondria following denervation-induced muscle atrophy [5]. Here we studied muscle mitochondrial function using permeabilized gastrocnemius muscle fibers in the Oroboros Oxygraph-2k. We measured mitochondrial oxygen consumption rates (OCR) linked to electron transport chain (ETC) complex driven respiration and reactive oxygen species (ROS) production simultaneously. Fig. 3C demonstrates OCR and ROS production traces following sequential additions of substrates and inhibitors. While mitochondrial respiration remains similar with age in wildtype mice, we observe significantly reduced complex I- and complex I & II-linked respiration in muscle fibers from old Nrf2-/- mice (Fig. 3A). However, proton leak, CII-linked, and CIV-linked respiration are not significantly reduced in old Nrf2-/- compared to old wildtype fibers (Fig. 3A). Addition of pyruvate also does not affect CI-linked respiration. Thus, Nrf2 deficiency primarily affects CI-linked respiration. We found that ROS production during basal respiration is significantly increased in muscle from Nrf2-/- mice compared to wildtype control mice, and there is also a trend for an increase in ROS production for proton leak, CI-linked, and CI and CII-linked respiration in Nrf2-/- compared to wildtype control mice (Fig. 3B). Old wildtype mice have an age-related increase in mRNA expression of the complex I subunit mt-Nd1 in skeletal muscle that is not found in muscle from Nrf2-/- mice, and Ndufs3 expression is significantly reduced in skeletal muscle from Nrf2-/- mice compared to old wildtype mice (Fig. 3D). The decline in complex I expression is consistent with our mitochondrial function data showing reduced complex I-linked OCR (Fig. 3A). Changes in mRNA expression also occur in complex II and IV subunits (Supplemental Table 3). However, no significant change is observed in ETC protein subunit abundance measured by targeted mass spectrometry (Supplemental Table 1). The limited number of observations (2 out of 45) of complex I subunits does not support the functional data. While we observe no evidence of difference in F2-isoprostane levels in skeletal muscle from old Nrf2-/- mice (Fig. 3E), we find a significant age-related increase in protein nitrotyrosine level, a measure of protein oxidative damage, which is further increased by Nrf2 deficiency (Fig. 3F, G). In summary, long term Nrf2 deficiency results in age-related ETC dysfunction, increased ROS production, and oxidative stress in mouse skeletal muscle.
Fig. 3.

Mitochondrial function, abundances of mitochondrial enzymes, and oxidative modifications of skeletal muscle. A) Oxygen consumption rate (OCR) in permeabilized red gastrocnemius muscle fibers. To assess activities of individual mitochondrial complexes, substrates and inhibitors were sequentially added. B) Simultaneous measurements of hydrogen peroxide generation rate using Amplex UltraRed probe. Abbreviations: G, glutamate; M, malate; P, pyruvate; Rot, rotenone; AmA, antimycin A. n = 5–8. C) Representative data tracings from wildtype fiber bundle for mitochondrial OCR (red) and ROS (green) in response to sequential addition of substrates and inhibitors. D) mRNA levels of mitochondrial proteins normalized to 18S rRNA (Rn18s). Data are presented as log2 fold change relative to young. n = 7–8. E) F2-isoprostane level in ng/g quadriceps muscle tissues. n = 7–8. F) Abundances of proteins with tyrosine nitration normalized to total proteins. N = 5–6. G) Immunoblots showing nitrosylated proteins (top) and Ponceau-stained total proteins (bottom). Young wildtype (white); old wildtype (grey); old-Nrf2-/- (black). Data are mean ± SEM. *p < 0.05 young vs. old or old-Nrf2-/-. # p < 0.05 old vs. old-Nrf2-/-.
3.3. Nrf2 deficiency reduces muscle antioxidants, chaperones, and inflammatory cytokines
Aging activates induction of antioxidant enzymes and inflammatory cytokines [37], [38]. To determine the effect of Nrf2 deficiency on antioxidants and chaperones in aging skeletal muscles, we measured expression of a selected group of proteins representing antioxidants and chaperones using targeted mass spectrometry and selective reaction monitoring (SRM) in gastrocnemius muscle. We find that skeletal muscle from old Nrf2-/- mice does not show an induction of several antioxidants and heat shock proteins that are induced in old wildtype mice. For example, age-related induction of glutathione pathway enzymes is abrogated by Nrf2 deficiency except for glutathione peroxidase 1 (Gpx1) (Fig. 4A). Glutathione disulfide reductase (Gsr), glutathione S-transferase A3 (Gsta3), and glutathione S-transferase pi 1 (Gstp1) protein levels are increased with age in wildtype mice but decreased in old Nrf2-/- mice compared to age-matched wildtype controls, while glutathione S-transferase mu 1 (Gstm1) is also decreased in old Nrf2-/- muscle compared to young (Fig. 4A). Glutathione peroxidase 4 (Gpx4) is reduced in Nrf2-/- mice compared to both young and old wildtype controls (Fig. 4A). Peroxiredoxin 1 (Prdx1) abundance is unchanged with age but was reduced in old Nrf2-/- mice compared to young and old wildtype controls (Fig. 4A) while peroxiredoxin 2, 3, 5, and 6 are unchanged by aging or Nrf2 deficiency (Supplemental Table 1). Thioredoxin (Txn1) is increased with age in wildtype mice, but this increase is prevented by Nrf2 deficiency (Fig. 4A). Additionally, thioredoxin reductase 1 (Txnrd1) is decreased in old Nrf2-/- mice compared to both young and old wildtype controls (Fig. 4A). No change is observed in CuZnSOD or MnSOD (Supplemental Table 1). Nrf2 deficiency prevents or reduces the age-related induction of heat shock proteins, including heat shock protein 90 β family member 1 (Hsp90b1), heat shock protein family A member 1A (Hspa1a), and heat shock protein family A member 5 (Hspa5) (Fig. 4A). Age-related induction of muscle antioxidant systems and heat shock proteins is prevented or reduced by Nrf2 deficiency in old mice.
Fig. 4.

Antioxidant, heat shock protein, and glycolytic enzyme abundances. A) Protein abundances of antioxidant enzymes and heat shock proteins in gastrocnemius muscle by targeted mass spectrometry and SRM. B) Abundances of the proteins involved in glycolysis and fatty acid oxidation in gastrocnemius muscle. Proteins labeled using their respective gene symbols. Old wildtype (grey); old-Nrf2-/- (black). n = 6. Data are presented as mean ± SEM log2 fold change relative to young. *p < 0.05 young vs. old or old-Nrf2-/-. # p < 0.05 old vs. old-Nrf2-/-.
We measured expression of inflammatory cytokines and chemokines by qRT-PCR. Most are unchanged by aging (7 out of 8) except chemokine ligand 9 (Cxcl9). Age-induced upregulation of Cxcl9 is abrogated by Nrf2 deficiency (Supplemental Table 4). Although Cxcl9 has been shown to be a biomarker for heart failure patients [39], its importance in skeletal muscle pathology remains unclear. Our findings suggest that antioxidant and chaperones play a protective role against increased ROS production and oxidative stress in aging muscle, and their reduction could contribute to the increased age-related muscle atrophy observed in Nrf2-/- mice.
3.4. Nrf2 deficiency causes minor alterations in glycolysis and fatty acid metabolism enzymes
We also measured changes in expression of proteins involved in glycolysis, fatty acid oxidation, the tricarboxylic acid (TCA) cycle, and the electron transport chain (ETC) using SRM. Only a few significant changes occur in metabolic pathways in old wildtype or Nrf2-/- skeletal muscle when compared to young, and there are no observed changes in the assayed TCA cycle or ETC proteins (Supplemental Table 1). The muscle glucose transporter type 4 (Slc2a4) increases with age regardless of genotype (Fig. 4B). Phosphoglycerate Kinase 1 (Pgk1) is decreased in old wildtype but not old-Nrf2-/- muscle. However, two glycolysis enzymes, glucose 6-phosphate isomerase (Gpi1) and phosphofructokinase, muscle (Pfkm), are decreased in old Nrf2-/- compared to age-matched wildtype suggesting a potential reduction in glycolysis (Fig. 4B). Nrf2 deficiency alters abundance of proteins involved in fatty acid anabolism, catabolism, and transport. The peroxisomal β-oxidation enzyme Acetyl-CoA acyltransferase 1 (Acaa1a/b), peroxisomal trans-2-enoyl-CoA reductase (Pecr), and fatty acid binding protein 4 (Fabp4) are increased in old Nrf2-/- muscle compared to young wildtype (Fig. 4B). 3-hydroxymethyl-3-methylglutaryl-CoA lyase (Hmgcl), a ketogenesis and leucine catabolism enzyme, is significantly increased in skeletal muscle from old wildtype but not old Nrf2-/- mice (Fig. 4B).
3.5. Nrf2 deficiency impairs skeletal muscle redox status
Because Nrf2 deficiency is associated with increased ROS production, decreased antioxidant abundance (including the glutathione system), and increased oxidative stress, we hypothesized it would also result in dysregulated cellular redox status. We found a significant increase in the quantity of oxidized glutathione (GSSG) and decrease in the GSH:GSSG ratio (Fig. 5A) with age in wildtype and Nrf2-/- mice. The level of reduced (GSH) glutathione is decreased by Nrf2 deficiency compared to old wildtype suggesting a potentially diminished ability to respond to excess ROS (Fig. 5A). In addition, the oxidized form of NAD (NAD+) is significantly increased in old Nrf2-/- mice compared to old wildtype mice, while no change is observed in NADH (Fig. 5B). An increased NAD+/NADH ratio occurs in old Nrf2-/- skeletal muscle compared to both young and old wildtype mice (Fig. 5B). We observe no change in NADP+, but NADPH is dramatically increased in old Nrf2-/- compared to both young and old wildtype, and the NADP+/NADPH ratio is significantly decreased in old Nrf2-/- compared to young wildtype (Fig. 5C). Thus Nrf2-/- mice have decreased GSH and GSH:GSSG, increased NAD+ and NAD+/NADH, and increased NADPH but decreased NADP+/NADPH.
Fig. 5.

Redox potential and recycling of antioxidant enzymes. A) Levels of reduced (GSH) and oxidized glutathione (GSSG) and their ratio (GSH:GSSG) in gastrocnemius muscle. n = 6–8. B) Levels of oxidized (NAD+) and reduced (NADH) coenzymes and their ratio (NAD/NADH) in gastrocnemius muscle. n = 7–8. C) Levels of of oxidized (NADP+) and reduced (NADPH) cofactors and their ratio (NADP+/NADPH) in gastrocnemius muscle. n = 7–8. Data are mean ± SEM. *p < 0.05.
3.6. Nrf2 deficiency reduces mRNA and protein abundance of the acetylcholine receptor
We measured the expression of acetylcholine receptor (AChR) subunits by qRT-PCR and AChR-α abundance by western blot in gastrocnemius muscle. The mRNA levels of the α (Chrna1), δ (Chrnd), and ε (Chrne) AChR subunits increases with age (Fig. 6A). While the ε subunit also increases in old Nrf2-/- mice, neither of the α or δ AChR subunits are significantly increased in the old Nrf2-/- mice compared to young, and Chrna1 is significantly reduced compared to aged wildtype (Fig. 6A). The impaired age-related increase of AChR-α protein abundance is confirmed in Nrf2-/- mice by western blot (Fig. 6B). Interestingly, mRNA expression of Gadd45a, a stress-induced pro-atrophy protein [40], is increased in old wildtype but not Nrf2-/- mice (Fig. 6A). This is likely due to its induction being Nrf2-dependent, as described in other tissues [41]. Reduced grip strength of old Nrf2-/- mice compared to age-matched wildtype is likely due in part to reduced neuromuscular innervation (Fig. 1D).
Fig. 6.

mRNA and protein levels of acetylcholine receptor subunits and atrophy associated genes. A) mRNA levels of acetylcholine receptor (AChR) subunits and denervation genes normalized to 18S rRNA (Rn18s). n = 4–8. Data are presented as log2 fold change relative to young. Old wildtype (grey); old-Nrf2-/- (black). B) Protein abundance of AChR-α normalized to total protein. n = 5–6. Image, Top: Representative membrane images showing the abundances of AChR-α determined by western blot. Bottom: Representative protein abundance at molecular weight ~ 37 kDa from total proteins stained by Ponceau. Data are mean ± SEM. *p < 0.05.
4. Discussion
The main findings of our work are that Nrf2 deficiency exacerbates atrophy and contractile dysfunction in aged skeletal muscle. Nrf2 deficiency abrogates the antioxidant defense response induced in aged skeletal muscle, impairs mitochondrial respiration, and increases ROS generation leading to a greater redox imbalance and oxidative and nitrosative stress. Nrf2 deficiency also abrogates the increased neuromuscular junction proteins in old skeletal muscle, suggesting a role of Nrf2 in neuromuscular function.
4.1. Nrf2 deficiency exacerbates atrophy in aged skeletal muscle
Sarcopenia is a function of both skeletal muscle atrophy and contractile dysfunction. The loss of muscle mass we measured is ~ 15–20% at ~24 months of age compared to young mice, which is consistent with previous reports [3], [7], [42], [43]. Nrf2 deficiency exacerbated age-related atrophy by ~ 15–20% especially in large muscle groups for a total reduction of ~ 35–50% compared to young mice. Why and how Nrf2 deficiency might affect larger muscles more than small muscles is unclear. A potential mediator of age-associated muscle atrophy is excess oxidants that activate degradation pathways in skeletal muscles, including proteasome, autophagy, calpain and caspase [44]. Indeed, Miller et al. [17] have previously shown that ubiquitinated proteins and apoptotic pathways are significantly elevated in aged Nrf2-/- skeletal muscles, including AIF, Bax, Bad, and Caspase-3. We also find that Nrf2 deficiency reduces age-related chaperones, including levels of heat shock protein 90 β family member 1 (Hsp90b1), which can affect muscle mass via muscle IGF-1 production [45]. Nrf2 deficiency in old mice also impairs regenerative capacity in response to stress induced by exercise [18], suggesting a failure of compensatory responses to overcome apoptosis and degeneration.
4.2. Nrf2 deficiency exacerbates contractile dysfunction of aged skeletal muscle
Aged Nrf2-/- mice demonstrate a more pronounced functional deficit in grid hanging time than grip strength, which suggests the difference may be due to increased age-related weakness of hind limb muscles. Murine hindlimb muscles undergo atrophy with age compared to forelimb muscles likely due to increased loss of neuromuscular innervation [46]. Thus the underlying cause for the difference might be neurogenic, since continuous muscle contractions and sustained neuronal inputs are required for grid hanging. To determine the component of force deficit intrinsic to the hindlimb muscle, isolated EDL muscle was tested for contractile properties. Isometric maximum specific force is reduced by aging, and this reduction is exacerbated by Nrf2 deficiency. Underlying causes of specific force deficit involve selective loss of myosin heavy chain [47] and/or other post-translational modifications of sarcomeric proteins, including thiol oxidation and protein carbonylation [48]. Twitch characteristics demonstrate increased time to peak twitch and half relaxation time for aged Nrf2-/-, suggesting impaired kinetics of calcium release and reuptake. Although a previous report published age-related increase in half relaxation time, the animals in that report were 28 month old rats compared to our 24 month old mice [36].
4.3. Nrf2 deficiency increases mitochondrial ROS generation and decreases respiration
The rate of ROS generation is unchanged by aging in wildtype mice, which is consistent with previous observations using the same preparation (permeabilized fibers) in older mouse (~32 months old) gastrocnemius muscle [49]. We and others have demonstrated increased rate of mitochondrial ROS generation in aged mice isolated mitochondria [5], [50], [51], but the discrepancies might be caused by damage and selection bias during isolation step of mitochondria [52]. Indeed, endogenous antioxidants might still remain in the cytoplasm of permeabilized fibers, while not in isolated mitochondria. Deficiency in Nrf2 induces a significant increase in ROS generation in the fibers from old Nrf2-/- mice. Consistent with excess ROS, nitration of tyrosine residues is upregulated in aged Nrf2-/- gastrocnemius, suggesting nitrosative stress by Nrf2 deficiency. Although we fail to detect the increase in lipid peroxidation, previous studies reported that oxidative stress in aged Nrf2-/- mice induced lipid peroxidation [17], [18]. We assessed lipid peroxidation in quadriceps using F2-isoprostane, while previous studies used immunoblot in gastrocnemius, which might contribute to the discrepancy. Mitochondrial OCR is unchanged by aging, but Nrf2 deficiency significantly decreases the respiration of the mitochondrial complex I. This is associated with an increased NAD+/NADH ratio in Nrf2-/-, which could explain reduced complex-I linked OCR via reduced concentration of substrate (NADH). This finding is associated with a decrease in NADH dehydrogenase iron-sulfur protein 3 (Ndufs3) mRNA expression by Nrf2 deficiency, which plays an important role in electron transportation from complex I to other complexes in the respiratory system.
4.4. Aging-induced upregulation of antioxidant enzymes is abrogated in aged Nrf2-/- muscle
Rate of mtROS generation increases with advancing age in isolated mitochondria from 28- and 32-month old mouse gastrocnemius, and antioxidant enzymes (superoxide dismutase, catalase, prdx, and gpx) are correspondingly elevated in old mice [5] as a defense mechanism against excess ROS. A number of antioxidant enzymes are transcriptionally regulated by the Nrf2-ARE pathway [53]. These enzymes include superoxide dismutase 1 and 2, catalase, peroxiredoxins, thioredoxins, and glutathione peroxidase systems, GSH synthesis and metabolism systems, quinone recycling (Nqo1), and iron homeostasis (heme oxygenase 1). In aged Nrf2 deficient skeletal muscle, we observe that CuZnSOD and MnSOD protein abundances are unchanged by aging or Nrf2 deficiency. Miller et al. [17] previously reported a significant increase in MnSOD with no change in CuZnSOD protein levels in aged Nrf2 deficient skeletal muscles, while Narasimhan et al. [18] demonstrated decreased CuZnSOD with no change in MnSOD protein levels. The variability in results between these two studies and ours may be related to differences in the assays or may reflect biologic variability. Previous studies used immunoblot assays while our study determines protein levels using multiple sets of amino acid sequences coupled with HPLC. Although protein abundance of Nqo1 was significantly decreased regardless of age, its impact on redox balance appears to be minimal in skeletal muscle [17] unlike neuronal tissues [54].
Our findings demonstrate that enzymes involved with detoxification of hydrogen peroxide are diminished by Nrf2 deficiency in aged skeletal muscle. Glutathione peroxidase (GPX) systems catalyze the detoxification of hydrogen peroxide [55]. Six antioxidant enzymes in the glutathione peroxidase systems are significantly elevated by aging, suggesting a protective response against excess oxidants in aged muscle. However, the protective mechanism was abrogated in aged Nrf2-/- mice in addition to a significant reduction in the total quantity of oxidized and reduced glutathione. GPX is an enzyme that catalyzes the antioxidant reaction of GSH, which leads to its oxidation (i.e. GSSG). Regeneration of GSH is facilitated by the enzyme glutathione reductase, which requires oxidation of NADPH to NADP+. It is possible that the significant upregulation of NADPH might be related to the reduced glutathione reductase in the old Nrf2-/- mice, although other organelles from the cell can oxidize NADPH independent of redox regulation [56]. Glutathione is a small antioxidant molecule whose biosynthesis is tightly regulated by its rate-limiting enzyme, γ-glutamyl cysteine ligase (γ-GCL). Existing literature has demonstrated that γ-GCL is transcriptionally regulated by Nrf2 [57], [58]. GSH is responsible for releasing disulfide bond formation (i.e. thiol oxidation). Thiol oxidation in skeletal muscle is of particular importance since contractile apparatus (i.e. myosin, actin, troponin) and calcium handling proteins (i.e. sarcoplasmic reticulum, SERCA pump) contain multiple thiol groups, excess oxidation of which can impair cross-bridge cycling interactions [59].
It is interesting to note that impaired muscle mitochondrial respiration, increased mitochondrial ROS production, and fatigue resistance in three month old Nrf2-/- mice was previously shown to be restored by an Nrf2-independent pathway following exercise training [16]. This response includes increased expression of mitochondrial proteins including COXIV and Tfam but does not involve an increase in assayed Nrf2 target antioxidant enzymes. Crilly et al. did not identify the nature of this Nrf2 independent exercise pathway. The effect of exercise on the increase in respiration independent of Nrf2 likely involves proliferation of mitochondria electron transport chain enzymes and increased mitochondrial density independent of antioxidant signaling [66]. Consistent with this, Narasimhan et al. demonstrated an induction of PGC1- in response to acute endurance exercise stress [18]. The impact of an Nrf2 independent increase in antioxidants may have contributed to the findings we report here.
4.5. Nrf2 signaling is involved with oxidative stress-induced neurological diseases
Age-related loss of neuromuscular innervation plays a major role in sarcopenia pathogenesis and is associated with increased expression of acetylcholine receptor (AChR) subunits. The mRNA and protein levels of AChR-α are elevated in aged gastrocnemius muscle. This finding is consistent with previous observations using aged laryngeal muscles, which exhibited decreased NMJ density with increases in mRNA and protein abundances of AChR [60]. It is predictable that a compensatory response is activated in aged skeletal muscle due to functional decline (i.e. failure in neural transmission) and morphological disruption of NMJ. However, the increase in mRNA and protein levels of AChR is attenuated in old Nrf2 deficient mice, suggesting a role of oxidative stress on skeletal muscle NMJ. We have previously demonstrated that mice lacking Sod1, a model of oxidative stress with accelerated sarcopenia, exhibits decreases in AChR mRNA and protein expressions that are associated with upregulation of the calcium-activated protease calpain [43], [61]. Nrf2 and antioxidant response element (ARE) pathway has been demonstrated to be decreased in multiple neurological diseases [62]. Indeed, activation of the pathways by Nrf2 activators ameliorated symptoms of neurodegenerative diseases, including Alzheimer's [63], Parkinson's diseases [64], and amyotrophic lateral sclerosis [65].
5. Conclusions
Nrf2 deficiency significantly reduces antioxidant defense enzymes and is associated with excess ROS from aged skeletal muscle, which causes redox imbalance and increases in markers of oxidative modifications. Abrogation of Nrf2 exacerbates age-related loss of skeletal muscle mass and contractile dysfunction, which are associated with diminished independence and quality of life. Our findings suggest that Nrf2/ARE pathway is a potential therapeutic target of sarcopenia to extend healthspan.
Acknowledgements
The authors would like to thank the Integrative Redox Biology and Multiplex Protein Quantification Cores at Nathan Shock Center for sharing their expertise. We would like to thank Kendra Plafker and Scott Plafker for sharing their expert opinions and Nrf2 antibody.
Funding sources
This work was supported by the National Institute of Health and National Institute of Aging [P01AG051442, R01-AG047879, R01-AG055395, P30AG050911, T32AG052363] and the United States Department of Veterans Affairs [1I01BX002595-01].
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2018.04.004.
Appendix A. Supplementary material
Supplementary material Supplemental Figure 1. Immunoblot and protein gel images show the abundance of Nrf2 proteins in young, old and old-Nrf2-/-. Retinal pigment epithelial cells were treated with MG132 for 0 and 4 h for identification of Nrf2. n = 4. *p < 0.05 old vs. old-Nrf2-/-. Supplemental Table 1. Gastrocnemius muscle protein abundances of all proteins assessed by targeted mass spectrometry and SRM. Proteins identified as Gene ID. Data are represented as mean ± SEM standard deviation (pmol assayed protein/100 µg total protein). n = 6. *p < 0.05 young vs. old or old-Nrf2-/-. # p < 0.05 old vs. old-Nrf2-/-. Supplemental Table 2. Information of primary antibodies for immunoblots and primers for qRT-PCR. Supplemental Table 3. Gastrocnemius mitochondrial enzyme mRNA expression determined by qRT-PCR. Data are represented as percent RQ relative to young ± SEM normalized to 18S rRNA (Rn18s) expression. n = 8. *p < 0.05 young vs. old or old-Nrf2-/-. # p < 0.05 old vs. old-Nrf2-/-. Supplemental Table 4. qRT-PCR inflammation gene panel. Data are presented as percent RQ relative to young ± SEM normalized to 18S rRNA. N = 5–10 per group. *p < 0.05 young vs. old or old-Nrf2-/-. # p < 0.05 old vs. old-Nrf2-/-.
References
- 1.Karakelides H., Sreekumaran Nair K. Current Topics in Developmental Biology. 2005. Sarcopenia of aging and its metabolic impact; pp. 123–148. [DOI] [PubMed] [Google Scholar]
- 2.Chin S.O., Rhee S.Y., Chon S., Hwang Y.-C., Jeong I.-K., Oh S., Ahn K.J., Chung H.Y., Woo J.-T., Kim S.-W., Kim J.-W., Kim Y.S., Ahn H.-Y. Sarcopenia is independently associated with cardiovascular disease in older Korean adults: the Korea National Health and Nutrition Examination Survey (KNHANES) from 2009. PLoS One. 2013;8:e60119. doi: 10.1371/journal.pone.0060119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muller F.L., Song W., Liu Y., Chaudhuri A., Pieke-Dahl S., Strong R., Huang T.-T., Epstein C.J., Roberts L.J., 2nd, Csete M., Faulkner J.A., Van Remmen H. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radic. Biol. Med. 2006;40:1993–2004. doi: 10.1016/j.freeradbiomed.2006.01.036. [DOI] [PubMed] [Google Scholar]
- 4.Sakellariou G.K., Davis C.S., Shi Y., Ivannikov M.V., Zhang Y., Vasilaki A., Macleod G.T., Richardson A., Van Remmen H., Jackson M.J., McArdle A., Brooks S.V. Neuron-specific expression of CuZnSOD prevents the loss of muscle mass and function that occurs in homozygous CuZnSOD-knockout mice. FASEB J. 2014;28:1666–1681. doi: 10.1096/fj.13-240390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muller F.L., Song W., Jang Y.C., Liu Y., Sabia M., Richardson A., Van Remmen H. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007;293:R1159–R1168. doi: 10.1152/ajpregu.00767.2006. [DOI] [PubMed] [Google Scholar]
- 6.Jang Y.C., Liu Y., Hayworth C.R., Bhattacharya A., Lustgarten M.S., Muller F.L., Chaudhuri A., Qi W., Li Y., Huang J.-Y., Verdin E., Richardson A., Van Remmen H. Dietary restriction attenuates age-associated muscle atrophy by lowering oxidative stress in mice even in complete absence of CuZnSOD. Aging Cell. 2012;11:770–782. doi: 10.1111/j.1474-9726.2012.00843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Larkin L.M., Davis C.S., Sims-Robinson C., Kostrominova T.Y., Van Remmen H., Richardson A., Feldman E.L., Brooks S.V. Skeletal muscle weakness due to deficiency of CuZn-superoxide dismutase is associated with loss of functional innervation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011;301:R1400–R1407. doi: 10.1152/ajpregu.00093.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vasilaki A., van der Meulen J.H., Larkin L., Harrison D.C., Pearson T., Van Remmen H., Richardson A., Brooks S.V., Jackson M.J., McArdle A. The age-related failure of adaptive responses to contractile activity in skeletal muscle is mimicked in young mice by deletion of Cu, Zn superoxide dismutase. Aging Cell. 2010;9:979–990. doi: 10.1111/j.1474-9726.2010.00635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Remmen H., Salvador C., Yang H., Huang T.T., Epstein C.J., Richardson A. Characterization of the antioxidant status of the heterozygous manganese superoxide dismutase knockout mouse. Arch. Biochem. Biophys. 1999;363:91–97. doi: 10.1006/abbi.1998.1060. [DOI] [PubMed] [Google Scholar]
- 10.Lustgarten M.S., Jang Y.C., Liu Y., Qi W., Qin Y., Dahia P.L., Shi Y., Bhattacharya A., Muller F.L., Shimizu T., Shirasawa T., Richardson A., Van Remmen H. MnSOD deficiency results in elevated oxidative stress and decreased mitochondrial function but does not lead to muscle atrophy during aging. Aging Cell. 2011;10:493–505. doi: 10.1111/j.1474-9726.2011.00695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y., Ikeno Y., Qi W., Chaudhuri A., Li Y., Bokov A., Thorpe S.R., Baynes J.W., Epstein C., Richardson A., Van Remmen H. Mice deficient in both Mn superoxide dismutase and glutathione peroxidase-1 have increased oxidative damage and a greater incidence of pathology but no reduction in longevity. J. Gerontol. A Biol. Sci. Med. Sci. 2009;64A:1212–1220. doi: 10.1093/gerona/glp132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jang Y.C., Perez V.I., Song W., Lustgarten M.S., Salmon A.B., Mele J., Qi W., Liu Y., Liang H., Chaudhuri A., Ikeno Y., Epstein C.J., Van Remmen H., Richardson A. Overexpression of Mn superoxide dismutase does not increase life span in mice. J. Gerontol. A Biol. Sci. Med. Sci. 2009;64A:1114–1125. doi: 10.1093/gerona/glp100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Itoh K., Chiba T., Takahashi S., Ishii T., Igarashi K., Katoh Y., Oyake T., Hayashi N., Satoh K., Hatayama I., Yamamoto M., Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. 〈https://www.ncbi.nlm.nih.gov/pubmed/9240432〉 [DOI] [PubMed] [Google Scholar]
- 14.Safdar A., deBeer J., Tarnopolsky M.A. Dysfunctional Nrf2-Keap1 redox signaling in skeletal muscle of the sedentary old. Free Radic. Biol. Med. 2010;49:1487–1493. doi: 10.1016/j.freeradbiomed.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 15.Kitaoka Y., Takeda K., Tamura Y., Fujimaki S., Takemasa T., Hatta H. Nrf2 deficiency does not affect denervation‐induced alterations in mitochondrial fission and fusion proteins in skeletal muscle. Physiol. Rep. 2016;4:e13064. doi: 10.14814/phy2.13064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crilly M.J., Tryon L.D., Erlich A.T., Hood D.A. The role of Nrf2 in skeletal muscle contractile and mitochondrial function. J. Appl. Physiol. 2016;121:730–740. doi: 10.1152/japplphysiol.00042.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller C.J., Gounder S.S., Kannan S., Goutam K., Muthusamy V.R., Firpo M.A., Symons J.D., Paine R., 3rd, Hoidal J.R., Rajasekaran N.S. Disruption of Nrf2/ARE signaling impairs antioxidant mechanisms and promotes cell degradation pathways in aged skeletal muscle. Biochim. Biophys. Acta. 1822;2012:1038–1050. doi: 10.1016/j.bbadis.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 18.Narasimhan M., Hong J., Atieno N., Muthusamy V.R., Davidson C.J., Abu-Rmaileh N., Richardson R.S., Gomes A.V., Hoidal J.R., Rajasekaran N.S. Nrf2 deficiency promotes apoptosis and impairs PAX7/MyoD expression in aging skeletal muscle cells. Free Radic. Biol. Med. 2014;71:402–414. doi: 10.1016/j.freeradbiomed.2014.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roberts B.M., Frye G.S., Ahn B., Ferreira L.F., Judge A.R. Cancer cachexia decreases specific force and accelerates fatigue in limb muscle. Biochem. Biophys. Res. Commun. 2013;435:488–492. doi: 10.1016/j.bbrc.2013.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuznetsov A.V., Veksler V., Gellerich F.N., Saks V., Margreiter R., Kunz W.S. Analysis of mitochondrial function in situ in permeabilized muscle fibers, tissues and cells. Nat. Protoc. 2008;3:965–976. doi: 10.1038/nprot.2008.61. [DOI] [PubMed] [Google Scholar]
- 21.Anderson E.J., Darrell Neufer P. Type II skeletal myofibers possess unique properties that potentiate mitochondrial H2O2 generation. Am. J. Physiol.-Cell Physiol. 2006;290:C844–C851. doi: 10.1152/ajpcell.00402.2005. [DOI] [PubMed] [Google Scholar]
- 22.Krumschnabel G., Fontana-Ayoub M., Sumbalova Z., Heidler J., Gauper K., Fasching M., Gnaiger E. Simultaneous high-resolution measurement of mitochondrial respiration and hydrogen peroxide production. Methods Mol. Biol. 2015;1264:245–261. doi: 10.1007/978-1-4939-2257-4_22. [DOI] [PubMed] [Google Scholar]
- 23.Makrecka-Kuka M., Krumschnabel G., Gnaiger E. High-resolution respirometry for simultaneous measurement of oxygen and hydrogen peroxide fluxes in permeabilized cells, tissue homogenate and isolated mitochondria. Biomolecules. 2015;5:1319–1338. doi: 10.3390/biom5031319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Votion D.-M., Gnaiger E., Lemieux H., Mouithys-Mickalad A., Serteyn D. Physical fitness and mitochondrial respiratory capacity in horse skeletal muscle. PLoS One. 2012;7:e34890. doi: 10.1371/journal.pone.0034890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gnaiger E. Vol. 17. 2012. Mitochondrial Pathways and Respiratory Control: An Introduction to OXPHOS Analysis; Mitochondr Physiol Network; p. 18.〈https://books.google.com/books/about/Mitochondrial_Pathways_and_Respiratory_C.html?hl=&id=WfTbrQEACAAJ〉 [Google Scholar]
- 26.McLain A.L., Cormier P.J., Kinter M., Szweda L.I. Glutathionylation of α-ketoglutarate dehydrogenase: the chemical nature and relative susceptibility of the cofactor lipoic acid to modification. Free Radic. Biol. Med. 2013;61:161–169. doi: 10.1016/j.freeradbiomed.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roberts L.J., Morrow J.D. Measurement of F(2)-isoprostanes as an index of oxidative stress in vivo. Free Radic. Biol. Med. 2000;28:505–513. doi: 10.1016/s0891-5849(99)00264-6. 〈https://www.ncbi.nlm.nih.gov/pubmed/10719231〉 [DOI] [PubMed] [Google Scholar]
- 28.Lane R.S., Fu Y., Matsuzaki S., Kinter M., Humphries K.M., Griffin T.M. Mitochondrial respiration and redox coupling in articular chondrocytes. Arthritis Res. Ther. 2015;17:54. doi: 10.1186/s13075-015-0566-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rindler P.M., Plafker S.M., Szweda L.I., Kinter M. High dietary fat selectively increases catalase expression within cardiac mitochondria. J. Biol. Chem. 2013;288:1979–1990. doi: 10.1074/jbc.M112.412890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kinter C.S., Lundie J.M., Patel H., Rindler P.M., Szweda L.I., Kinter M. A quantitative proteomic profile of the Nrf2-mediated antioxidant response of macrophages to oxidized LDL determined by multiplexed selected reaction monitoring. PLoS One. 2012;7:e50016. doi: 10.1371/journal.pone.0050016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.MacLean B., Tomazela D.M., Shulman N., Chambers M., Finney G.L., Frewen B., Kern R., Tabb D.L., Liebler D.C., MacCoss M.J. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Mealey G.B., Plafker K.S., Berry W.L., Janknecht R., Chan J.Y., Plafker S.M. A PGAM5-KEAP1-Nrf2 complex is required for stress-induced mitochondrial retrograde trafficking. J. Cell Sci. 2017;130:3467–3480. doi: 10.1242/jcs.203216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pharaoh G., Pulliam D., Hill S., Sataranatarajan K. Ablation of the mitochondrial complex IV assembly protein Surf1 leads to increased expression of the UPRMT and increased resistance to oxidative stress in primary cultures of fibroblasts. Redox Biol. 2016 doi: 10.1016/j.redox.2016.05.001. 〈http://www.sciencedirect.com/science/article/pii/S2213231716300301〉 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sataranatarajan K., Qaisar R., Davis C., Sakellariou G.K., Vasilaki A., Zhang Y., Liu Y., Bhaskaran S., McArdle A., Jackson M., Brooks S.V., Richardson A., Van Remmen H. Neuron specific reduction in CuZnSOD is not sufficient to initiate a full sarcopenia phenotype. Redox Biol. 2015;5:140–148. doi: 10.1016/j.redox.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Toth P., Tarantini S., Ashpole N.M., Tucsek Z., Milne G.L., Valcarcel-Ares N.M., Menyhart A., Farkas E., Sonntag W.E., Csiszar A., Ungvari Z. IGF-1 deficiency impairs neurovascular coupling in mice: implications for cerebromicrovascular aging. Aging Cell. 2015;14:1034–1044. doi: 10.1111/acel.12372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fitts R.H., Troup J.P., Witzmann F.A., Holloszy J.O. The effect of ageing and exercise on skeletal muscle function. Mech. Ageing Dev. 1984;27:161–172. doi: 10.1016/0047-6374(84)90041-1. 〈https://www.ncbi.nlm.nih.gov/pubmed/6492893〉 [DOI] [PubMed] [Google Scholar]
- 37.Zhang H., Davies K.J.A., Forman H.J. Oxidative stress response and Nrf2 signaling in aging. Free Radic. Biol. Med. 2015;88:314–336. doi: 10.1016/j.freeradbiomed.2015.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peake J., Della Gatta P., Cameron-Smith D. Aging and its effects on inflammation in skeletal muscle at rest and following exercise-induced muscle injury. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010;298:R1485–R1495. doi: 10.1152/ajpregu.00467.2009. [DOI] [PubMed] [Google Scholar]
- 39.Altara R., Manca M., Hessel M.H., Gu Y., van Vark L.C., Akkerhuis K.M., Staessen J.A., Struijker-Boudier H.A.J., Booz G.W., Blankesteijn W.M. CXCL10 is a circulating inflammatory marker in patients with advanced heart failure: a pilot study. J. Cardiovasc. Transl. Res. 2016;9:302–314. doi: 10.1007/s12265-016-9703-3. [DOI] [PubMed] [Google Scholar]
- 40.Ebert S.M., Dyle M.C., Kunkel S.D., Bullard S.A., Bongers K.S., Fox D.K., Dierdorff J.M., Foster E.D., Adams C.M. Stress-induced skeletal muscle Gadd45a expression reprograms myonuclei and causes muscle atrophy. J. Biol. Chem. 2012;287:27290–27301. doi: 10.1074/jbc.M112.374777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cho H.-Y., Reddy S.P., Debiase A., Yamamoto M., Kleeberger S.R. Gene expression profiling of NRF2-mediated protection against oxidative injury. Free Radic. Biol. Med. 2005;38:325–343. doi: 10.1016/j.freeradbiomed.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 42.Brooks S.V., Faulkner J.A. Contractile properties of skeletal muscles from young, adult and aged mice. J. Physiol. 1988;404:71–82. doi: 10.1113/jphysiol.1988.sp017279. 〈https://www.ncbi.nlm.nih.gov/pubmed/3253447〉 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jang Y.C., Lustgarten M.S., Liu Y., Muller F.L., Bhattacharya A., Liang H., Salmon A.B., Brooks S.V., Larkin L., Hayworth C.R., Richardson A., Van Remmen H. Increased superoxide in vivo accelerates age-associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. FASEB J. 2010;24:1376–1390. doi: 10.1096/fj.09-146308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Powers S.K., Jackson M.J. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol. Rev. 2008;88:1243–1276. doi: 10.1152/physrev.00031.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barton E.R., Park S., James J.K., Makarewich C.A., Philippou A., Eletto D., Lei H., Brisson B., Ostrovsky O., Li Z., Argon Y. Deletion of muscle GRP94 impairs both muscle and body growth by inhibiting local IGF production. FASEB J. 2012;26:3691–3702. doi: 10.1096/fj.11-203026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hashizume K., Kanda K. Differential effects of aging on motoneurons and peripheral nerves innervating the hindlimb and forelimb muscles of rats. Neurosci. Res. 1995;22:189–196. doi: 10.1016/0168-0102(95)00889-3. 〈https://www.ncbi.nlm.nih.gov/pubmed/7566699〉 [DOI] [PubMed] [Google Scholar]
- 47.Shi Y., Ivannikov M.V., Walsh M.E., Liu Y., Zhang Y., Jaramillo C.A., Macleod G.T., Van Remmen H. The lack of CuZnSOD leads to impaired neurotransmitter release, neuromuscular junction destabilization and reduced muscle strength in mice. PLoS One. 2014;9:e100834. doi: 10.1371/journal.pone.0100834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wei L., Gregorich Z.R., Lin Z., Cai W., Jin Y., McKiernan S.H., McIlwain S., Aiken J.M., Moss R.L., Diffee G.M., Ge Y. Novel sarcopenia-related alterations in sarcomeric protein post-translational modifications in skeletal muscles Identified by top-down proteomics. Mol. Cell. Proteom. 2017 doi: 10.1074/mcp.RA117.000124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Picard M., Ritchie D., Wright K.J., Romestaing C., Thomas M.M., Rowan S.L., Taivassalo T., Hepple R.T. Mitochondrial functional impairment with aging is exaggerated in isolated mitochondria compared to permeabilized myofibers. Aging Cell. 2010;9:1032–1046. doi: 10.1111/j.1474-9726.2010.00628.x. [DOI] [PubMed] [Google Scholar]
- 50.Mansouri A., Muller F.L., Liu Y., Ng R., Faulkner J., Hamilton M., Richardson A., Huang T.-T., Epstein C.J., Van Remmen H. Alterations in mitochondrial function, hydrogen peroxide release and oxidative damage in mouse hind-limb skeletal muscle during aging. Mech. Ageing Dev. 2006;127:298–306. doi: 10.1016/j.mad.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 51.Capel F., Buffière C., Patureau Mirand P., Mosoni L. Differential variation of mitochondrial H2O2 release during aging in oxidative and glycolytic muscles in rats. Mech. Ageing Dev. 2004;125:367–373. doi: 10.1016/j.mad.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 52.Picard M., Taivassalo T., Ritchie D., Wright K.J., Thomas M.M., Romestaing C., Hepple R.T. Mitochondrial structure and function are disrupted by standard isolation methods. PLoS One. 2011;6:e18317. doi: 10.1371/journal.pone.0018317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nguyen T., Nioi P., Pickett C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009;284:13291–13295. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu S.-S., Zhao J., Lei S.-P., Lin X.-M., Wang L.-L., Zhao Y. 4-hydroxybenzyl alcohol ameliorates cerebral injury in rats by antioxidant action. Neurochem. Res. 2011;36:339–346. doi: 10.1007/s11064-010-0335-8. [DOI] [PubMed] [Google Scholar]
- 55.Tomanek L. Proteomic responses to environmentally induced oxidative stress. J. Exp. Biol. 2015;218:1867–1879. doi: 10.1242/jeb.116475. [DOI] [PubMed] [Google Scholar]
- 56.Ahn B., Beharry A.W., Frye G.S., Judge A.R., Ferreira L.F. NAD(P)H oxidase subunit p47phox is elevated, and p47phox knockout prevents diaphragm contractile dysfunction in heart failure. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015;309:L497–L505. doi: 10.1152/ajplung.00176.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chan J.Y., Kwong M. Impaired expression of glutathione synthetic enzyme genes in mice with targeted deletion of the Nrf2 basic-leucine zipper protein. Biochim. Biophys. Acta. 2000;1517:19–26. doi: 10.1016/s0167-4781(00)00238-4. 〈https://www.ncbi.nlm.nih.gov/pubmed/11118612〉 [DOI] [PubMed] [Google Scholar]
- 58.Wild A.C., Mulcahy R.T. Regulation of gamma-glutamylcysteine synthetase subunit gene expression: insights into transcriptional control of antioxidant defenses. Free Radic. Res. 2000;32:281–301. doi: 10.1080/10715760000300291. 〈https://www.ncbi.nlm.nih.gov/pubmed/10741850〉 [DOI] [PubMed] [Google Scholar]
- 59.Jackson M.J. Redox regulation of skeletal muscle. IUBMB Life. 2008;60:497–501. doi: 10.1002/iub.72. [DOI] [PubMed] [Google Scholar]
- 60.McMullen C.A., Andrade F.H. Functional and morphological evidence of age-related denervation in rat laryngeal muscles. J. Gerontol. A Biol. Sci. Med. Sci. 2009;64:435–442. doi: 10.1093/gerona/gln074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jang Y.C., Van Remmen H. Age-associated alterations of the neuromuscular junction. Exp. Gerontol. 2011;46:193–198. doi: 10.1016/j.exger.2010.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang M., An C., Gao Y., Leak R.K., Chen J., Zhang F. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog. Neurobiol. 2013;100:30–47. doi: 10.1016/j.pneurobio.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kanninen K., Malm T.M., Jyrkkänen H.-K., Goldsteins G., Keksa-Goldsteine V., Tanila H., Yamamoto M., Ylä-Herttuala S., Levonen A.-L., Koistinaho J. Nuclear factor erythroid 2-related factor 2 protects against beta amyloid. Mol. Cell. Neurosci. 2008;39:302–313. doi: 10.1016/j.mcn.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 64.Burton N.C., Kensler T.W., Guilarte T.R. In vivo modulation of the Parkinsonian phenotype by Nrf2. Neurotoxicology. 2006;27:1094–1100. doi: 10.1016/j.neuro.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 65.Neymotin A., Calingasan N.Y., Wille E., Naseri N., Petri S., Damiano M., Liby K.T., Risingsong R., Sporn M., Flint Beal M., Kiaei M. Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2011;51:88–96. doi: 10.1016/j.freeradbiomed.2011.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lundby C., Jacobs R.A. Adaptations of skeletal muscle mitochondria to exercise training. Exp. Physiol. 2016;101:17–22. doi: 10.1113/EP085319. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material Supplemental Figure 1. Immunoblot and protein gel images show the abundance of Nrf2 proteins in young, old and old-Nrf2-/-. Retinal pigment epithelial cells were treated with MG132 for 0 and 4 h for identification of Nrf2. n = 4. *p < 0.05 old vs. old-Nrf2-/-. Supplemental Table 1. Gastrocnemius muscle protein abundances of all proteins assessed by targeted mass spectrometry and SRM. Proteins identified as Gene ID. Data are represented as mean ± SEM standard deviation (pmol assayed protein/100 µg total protein). n = 6. *p < 0.05 young vs. old or old-Nrf2-/-. # p < 0.05 old vs. old-Nrf2-/-. Supplemental Table 2. Information of primary antibodies for immunoblots and primers for qRT-PCR. Supplemental Table 3. Gastrocnemius mitochondrial enzyme mRNA expression determined by qRT-PCR. Data are represented as percent RQ relative to young ± SEM normalized to 18S rRNA (Rn18s) expression. n = 8. *p < 0.05 young vs. old or old-Nrf2-/-. # p < 0.05 old vs. old-Nrf2-/-. Supplemental Table 4. qRT-PCR inflammation gene panel. Data are presented as percent RQ relative to young ± SEM normalized to 18S rRNA. N = 5–10 per group. *p < 0.05 young vs. old or old-Nrf2-/-. # p < 0.05 old vs. old-Nrf2-/-.