

Abstract
The clinical use of doxorubicin for cancer therapy is limited by its cardiotoxicity, which involves cardiomyocyte apoptosis and oxidative stress. Previously, we showed that general control nonderepressible 2 (GCN2), an eukaryotic initiation factor 2α (eIF2α) kinase, impairs the ventricular adaptation to chronic pressure overload by affecting cardiomyocyte apoptosis. However, the impact of GCN2 on Dox-induced cardiotoxicity has not been investigated. In the present study, we treated wild type (WT) and Gcn2−/− mice with four intraperitoneal injections (5 mg/kg/week) to induce cardiomyopathy. After Dox treatment, Gcn2−/− mice developed less contractile dysfunction, myocardial fibrosis, apoptosis, and oxidative stress compared with WT mice. In the hearts of the Dox-treated mice, GCN2 deficiency attenuated eIF2α phosphorylation and induction of its downstream targets, activating transcription factor 4 (ATF4) and C/EBP homologous protein (CHOP), and preserved the expression of anti-apoptotic factor Bcl-2 and mitochondrial uncoupling protein-2(UCP2). Furthermore, we found that GCN2 knockdown attenuated, whereas GCN2 overexpression exacerbated, Dox-induced cell death, oxidative stress and reduction of Bcl-2 and UCP2 expression through the eIF2α-CHOP-dependent pathway in H9C2 cells. Collectively, our data provide solid evidence that GCN2 has a marked effect on Dox induced myocardial apoptosis and oxidative stress. Our findings suggest that strategies to inhibit GCN2 activity in cardiomyocyte may provide a novel approach to attenuate Dox-related cardiotoxicity.
Abbreviations: ANP, atrial natriuretic peptide; ATF4, activating transcription factor 4; CHOP, C/EBP homologous protein; CRISPR, clustered regularly interspaced short palindromic repeats; DHE, dihydroethidium; DMEM, Dulbecco's Modified Eagle Medium; Dox, doxorubicin; Dusp1, dual specificity protein phosphatase 1; eIF2α, eukaryotic initiation factor 2α; ELISA, enzyme-linked immunosorbent assay; ER, endoplasmic reticulum; FBS, fetal bovine serum; GCN2, General control nonderepressible 2; Glrx, glutaredoxin; GSH, reduced glutathione; GSSG, oxidized glutathione; LV, left ventricular; MDA, malondialdehyde; Mt, metallothionein; NT, nitrotyrosine; PBS, phosphate-buffered saline; qPCR, quantitative real-time polymerase chain reaction; ROS, reactive oxygen species; SOD, superoxide dismutase; shRNA, short hairpin RNA; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling; UCP2, uncoupling protein-2; WT, wild type
Keywords: GCN2, Doxorubicin, Cardiotoxicity, Oxidative stress, CHOP, UCP2
Graphical abstract
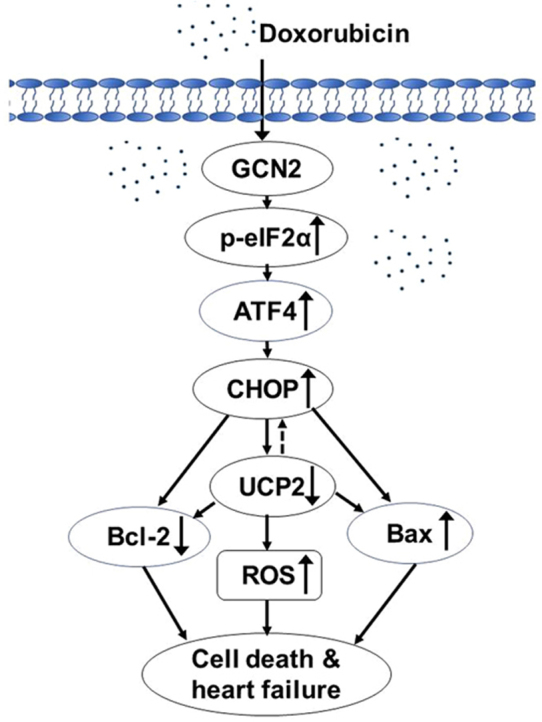
Highlights
-
•
GCN2 deficiency ameliorates doxorubicin-induced cardiac dysfunction.
-
•
GCN2 promotes doxorubicin-induced cardiomyocyte apoptosis and oxidative stress.
-
•
GCN2 decreases Bcl-2 and UCP2 expression via a CHOP dependent manner.
-
•
Knockdown of UCP2 exacerbated doxorubicin-induced cell death and oxidative stress.
1. Introduction
Doxorubicin (Dox) is a powerful anti-cancer chemotherapy drug that is widely used in the therapy of a broad range of hematological malignancies, carcinomas and solid sarcomas [1]. However, the clinical use of Dox can also trigger dilated cardiomyopathy and heart failure in a dose dependent manner [2], [3]. The molecular mechanism involved in the cardiotoxicity of Dox is multifactorial [4], including increased reactive oxygen species (ROS) production [5], [6], [7], DNA damage and apoptosis [8], [9], and autophagy dysregulation [10]. As the risk of Dox-induced cardiotoxicity in patients with cancer is increasing [11], there is an urgent need to explore new drugs and strategies to reduce Dox-induced cardiomyopathy.
General control nonderepressible 2 (GCN2) is a serine/threonine kinase that is activated by uncharged transfer RNA accumulation [12]. Under conditions of amino acid deprivation, GCN2 maintains amino acid homeostasis through phosphorylating eukaryotic initiation factor 2α at Ser51 (eIF2αSer51) and selectively stimulating the expression of amino acid biosynthetic genes [13], [14]. In addition to regulating amino acid starvation response, GCN2 is also involved in memory formation [15], immune response [16], [17] and muscle atrophy [18]. Interestingly, GCN2 was found to promote sodium salicylate- or histone deacetylase inhibitor-induced apoptosis [19], [20], and it has been suggested that GCN2 expression levels determine the sensitivity of cancer cells to Na+, K+-ATPase ligand-induced apoptosis [21]. We have previously demonstrated that GCN2 deficiency attenuated transverse aortic constriction (TAC)-induced cardiac dysfunction and cardiomyocyte apoptosis [22]. However, whether GCN2 can also affect Dox-induced cardiotoxicity remains unclear. To address the issues, we first examined the effect of GCN2 on Dox-induced cardiotoxicity using Gcn2 deletion (Gcn2-/-) mice and wild-type (WT) littermates. Then, we used the H9C2 rat cardiomyoblast cell line to investigate the role of GCN2 in Dox-induced cell death and oxidative stress.
2. Materials and methods
2.1. Antibodies and reagents
Antibodies against β-tubulin, C/EBP homologous protein (CHOP), activating transcription factor 4 (ATF4), Bcl-2 and uncoupling protein-2 (UCP2), and mouse 3′- nitrotyrosine (3′-NT) ELISA kit were acquired from Abcam PLC (#ab6046, #ab11419, #ab23760, #ab194583, #ab203244, #ab116691, Cambridge, UK); GCN2, eIF2α, phospho-eIF2αser51, cleaved caspase-3 and Bax antibodies were acquired from Cell Signaling Technology (#3302, #5324, #3398, #9661, #2772, Danvers, MA, USA). Dulbecco's modified Eagle's medium (DMEM),fetal bovine serum (FBS), lipofectamine 2000 and TRIzol reagent were obtained from Invitrogen (#11965175, #10099141, #11668019, #15596026, Grand Island, NY, USA). Dox, 3-(4,5-dimethylthiazol-2-yl)− 2,5-diphenyltetrazolium bromide (MTT), DCFH-DA (2′,7′-Dichlorodihydrofluorescein diacetate), dihydroethidium (DHE) were purchased from Sigma (#D1515,#M2128, #D6883, #D7008, St. Louise, MO, USA). Short hairpin RNA (ShRNA) sequences targeting GCN2, eIF2α or UCP2 were constructed into pLKO.1 lentiviral vectors [23] (Addgene Plasmid # 10878) for viral packaging. The forward oligo targeting sequences of shRNA hairpins were follows: shGCN2, TTAGCTTATATCCATGAGAAA; sheIF2α, GCTTGGAATCCTAGTTCTACG; shUCP2, CGGCTGCAGATCCAAGGAGAG. GCN2 cDNA clone and single guide RNA (sgRNA) targeting CHOP (antisense sequence TCAGCCAAGCTAGGGATGCA) were sub-cloned into pLVX-Tet3G plasmid (Takara, Otsu, Japan) and lentiCRISPRv2 [24] (Addgene Plasmid # 52961) for viral packaging, respectively. The GCN2 and CHOP adenovirus were purchased from Oubokang Co. Ltd. (customized products, Beijing, China). DIO (3,3΄-dioctadecyloxacarbocyanine perchlorate) and kits for TUNEL assay, malondialdehyde (MDA) and glutathione measurement were obtained from Beyotime Institute of Biotechnology (#C1038, #C1090, #S0131, #S0053, Shanghai, China). All other chemicals made in China were analytical grade.
2.2. Experimental animals
Gcn2–/– mice [25] were obtained from The Jackson Laboratory (Bar Harbor, ME, USA) and then crossed with C57BL/6 J strain (obtained from HFK Bioscience Co., Beijing). Adult male littermates (10–12 weeks of age) were used in this study. Animal studies were performed in accordance with the principles of laboratory animal care (NIH publication no. 85–23, revised 1985) and with approval of the University Of Chinese Academy of Science Animal Care and Use Committee. Mice were received intraperitoneal injections of Dox (5 mg/kg/week) for 4 weeks (cumulative dose: 20 mg/kg). Initial body weight and age matched mice were used as controls.
2.3. Echocardiography
Mice were anesthetized with 1.5% isoflurane and echocardiographic images were obtained with a Visualsonics high-resolution 2100 system as previously described [26], [27].
2.4. Cell culture
H9C2 cardiomyocytes derived from rat myocardium were obtained from China Infrastructure of Cell Line Resource (Beijing, China) and grown in DMEM medium supplemented with 10% (v/v) inactivated FBS and 1% penicillin and streptomycin at 37 °C with 5% CO2.
To generate a stable GCN2/eIF2α/UCP2 knockdown or doxycycline-inducible GCN2 overexpression cell line, 5 × 105 exponentially growing cells were transfected with the shRNA lentivirus or pLVX-Tet3G-GCN2 lentivirus for 24 h, followed by puromycin selection (1 μg/ml) for 3 weeks.
2.5. Measurement of intracellular ROS and superoxide levels
The levels of intracellular ROS and superoxide were determined by spectrophotometry using DCFH-DA and DHE respectively. The cells were washed with PBS and incubated with 5 μM fluorescence dyes (final concentration) for 30 mins at 37 °C in dark. Then cells were washed three times with PBS and the fluorescence intensity was determined using a Synergy H1 Hybrid Multi-Mode Microplate Reader (Biotek Instruments, Inc., Winooski, VT, USA).
2.6. Tissue processing
Heart paraffin sections (5 µm) were stained with a trichrome stain kit (Modified Masson', ScyTek Laboratories, Inc., UT, USA) and TUNEL apoptosis detection kit to assess fibrosis and apoptosis, respectively. At least 4 mice per group were used for these experiments.
2.7. Western blots and quantitative real-time polymerase chain reaction (qPCR) analysis
As previously reported [28], [29], protein was extracted from heart tissue or cells with buffer (50 mM Tris-Cl, 150 mM NaCl, 100 μg/ml phenylmethylsulfonyl fluoride (#P7626, Sigma), protease and phosphatase inhibitor cocktail (#04693124001, #4906837001, Roche, Basel, Switzerland) and 1%Triton X-100) on ice for 30 min. After centrifugation at 12,000 g and 4 °C for 20 min, the supernatant was used for Western blot.
TRIzol reagent was used to extract total RNA from heart tissue. Reverse transcriptional reactions were performed using the PrimeScript RT Reagent Kit (#RR036B, TaKaRa, Otsu, Japan). The mRNA levels were determined using the SYBR® Premix Ex Taq™ II Kit (#RR820DS, TaKaRa). The cycling conditions for qPCR were as follows: initial denaturation at 95 °C for 30 s, followed by 40 cycles of 5 s at 95 °C, 30 s at 60 °C, and 30 s at 72 °C. Primers are listed in Supplemental Table 1. The results were normalized to 18 S rRNA.
2.8. Data and statistical analysis
All values are expressed as the means ± standard error. Statistical significance was defined as p < .05. One- or two-way analysis of variance (ANOVA) was used to test each variable for differences among the treatment groups with StatView (SAS Institute Inc.). If ANOVA demonstrated a significant effect, pair wise post hoc comparisons were made with the Fisher's least significant difference test.
3. Results
3.1. Gcn2–/– attenuated Dox-induced cardiac dysfunction, fibrosis and apoptosis
As we previously demonstrated [22], GCN2 deficiency does not affect body weight, heart weight and left ventricular function. Exposure to Dox for 4 weeks significantly decreased body weight and increased the ratio of heart weight to body weight in WT and Gcn2-/- mice; however, Gcn2-/- mice exhibited more body weight than WT mice (Fig. 1A-B). qPCR results showed that the mRNA levels of atrial natriuretic peptide (ANP; a marker for cardiac stress) were significantly higher in WT hearts than in Gcn2-/- hearts after Dox treatment (Fig. 1C). Echocardigraphyic examination also revealed that Gcn2–/– mice developed significantly less left ventricular (LV) dysfunction in response to Dox treatment as indicated by significantly less reduction in LV ejection fraction (53.7 ± 2.1% in WT vs 62.3 ± 2.0% in Gcn2–/–; p < .05) (Fig. 1D, E).
Fig. 1.

General control nonderepressible 2 (GCN2) deficiency attenuated doxorubicin (Dox)-induced left venticular (LV) dysfunction. Male Gcn2–/– and wild-type (WT) mice received intraperitoneal injections (5 mg/kg/week) for 4 weeks. The body weight (A), the heart weight to body weight ratio (B), and the mRNA levels of atrial natrurietic peptide (ANP) (C) were measured. Echocardiography was used to measure LV ejection fraction (D, E). n = 7–8, * indicates p < .05, * * indicates p < .01.
LV fibrosis and cardiomyocyte death are the main mechanisms responsible for Dox-induced LV dysfunction. As demonstrated by Masson staining, we found that Dox treatment resulted in more LV fibrosis in WT than in Gcn2−/− hearts (Fig. 2A-B). Exposure to Dox also increased more collagen I mRNA expression in WT hearts than in Gcn2–/– hearts (2.29 ± .19 in WT vs 1.39 ± .11 in Gcn2–/–) (Supplemental Fig. 1). Next, we performed terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining to determine whether GCN2 deficiency affects Dox-induced cardiomyocyte apoptosis. As shown in Fig. 2A, Dox treatment significantly increased apoptotic cells in WT and Gcn2–/– hearts; however, the increase in apoptotic cells was significantly less in Gcn2–/– hearts than in WT hearts (Fig. 2A, C). Consistent with TUNEL staining results, WT hearts exhibited higher caspase-3 activity and greater activation (cleavage) of pro-apoptotic protein caspase-3 than Gcn2–/– hearts after Dox treatment (Fig. 2D, E). In addition, the expression of anti-apoptotic factor Bcl-2 was significantly decreased, whereas the expression of pro-apoptotic factor Bax was significantly increased in Dox-treated WT hearts; however, these changes were significantly attenuated by Gcn2 deletion (Fig. 2D, E).
Fig. 2.

GCN2 deficiency attenuated Dox-induced myocardial fibrosis and apoptosis. After exposure to Dox or control conditions for 4 weeks, representative heart sections from control and Dox-treated WT and Gcn2–/– mice were stained with Masson trichrome (upper panel, scale bar=50 µm), and DiO (green), DAPI (blue) and TUNEL assay kit (red) (lower panel, scale bar=10 µm, arrows point to TUNEL positive cells) (A). The fibrosis area (B) and TUNEL positive cells (C) were quantified. Heart tissue was collected from control and Dox-treated mice and lysates were subjected to casapse-3 activity assay (D) and Western blot for the expression of cleaved caspase-3, Bcl-2 and Bax. β-tubulin was used as a loading control (E). N = 3–5, * indicates p < .05, ** indicates p < .01, *** indicates p < .001.
3.2. Gcn2-/- attenuated Dox-induced oxidative stress and endoplasmic reticulum (ER) stress
Since Dox-induced cardiotoxicity is associated with increased oxidative stress, we measured oxidative stress markers such as MDA, 3′-NT and the ratio of reduced glutathione (GSH)-to-oxidized glutathione (GSSG), to evaluate whether Dox treatment differentially stimulated oxidative stress in WT and GCN2-/- hearts. Under basal conditions, myocardial MDA and 3-NT levels, as well as the GSH/GSSG ratio were not different in hearts of WT and Gcn2–/– mice. However, Dox treatment caused greater oxidative stress in hearts of WT mice than in hearts of Gcn2-/- mice, as indicated by higher MDA and 3′-NT levels, and lower GSH/GSSG ratios (Fig. 3A–C). To further explore the mechanism responsible for the attenuated oxidative stress in Dox-treated Gcn2-/- hearts, we performed qPCR to measure anti-oxidative stress response genes, including superoxide dismutase 1 (Sod1), Sod2, Sod3, uncoupling protein 2 (Ucp2), glutaredoxin (Glrx), metallothionein 1 (Mt1), Mt2, and dual specificity protein phosphatase 1 (Dusp1). Sod1 was not affected by Gcn2 deletion and Dox treatment. Sod2 and Sod3 were downregulated in Dox-treated Gcn2-/- hearts; however, there was no difference between WT and Gcn2–/– hearts. Dox treatment also significantly decreased mRNA levels of Ucp2, Glrx, Mt1, Mt2 and Dusp1 in WT hearts, while the decreases in Ucp2, Glrx and Mt1 were significantly attenuated by Gcn2 deletion (Fig. 3D). Western blot analysis also showed that GCN2 deficiency attenuated Dox-induced UCP2 reduction (Fig. 3E).
Fig. 3.

GCN2 deficiency attenuated Dox-induced myocardial oxidative stress and endoplasmic reticulum (ER) stress. Heart tissue was collected from WT and Gcn2−/− mice 4 weeks after Dox treatment or control conditions. Then, myocardial 3΄-nitrotyrosine (3΄-NT) (A), malondialdehyde (MDA) (B) levels, and the ratio of reduced glutathione to oxidized glutathione (GSH/GSSG) ratio (C) were determined. The mRNA levels of anti-oxidative stress response genes were examined using real-time polymerase chain reaction (PCR) (D). Heart lysates were examined by Western blotting for the expression of total and phosphorylated eIF2α, activating transcription factor 4 (ATF4), C/EBP homologous protein (CHOP) and uncoupling protein 2 (UCP2). β-tubulin was used as a loading control (E). N = 3–4, * indicates p < .05, ** indicates p < .01.
It is well known that ER stress is associated with phosphorylation of eIF2α and induction of its downstream targets, ATF4 and CHOP. Although GCN2 is an eIF2α kinase, loss of GCN2 has no significant effect on myocardial eIF2αSer51 phosphorylation under basal conditions. However, while eIF2α phosphorylation and myocardial ATF4 and CHOP levels were significantly increased in the hearts of WT mice after Dox treatment, these increases were significantly attenuated in hearts of Gcn2–/– mice (Fig. 3E).
3.3. GCN2 affects Dox-induced oxidative stress, ER stress and cell death in H9C2 cardiomyocytes
To confirm the pro-apoptotic role of GCN2 in cardiomyocytes, we stably transfected H9C2 cells with shRNA lentiviral vector targeting GCN2. Another cell line stably transfected with PLKO.1-scramble shRNA was used as a control (shScr). Compared with control cells, GCN2-depleted cells exhibited an ~16% increase in cell viability. Although Dox treatment decreased cell viability in both cell lines, the viability of GCN2-depleted cells was significantly higher than that of control cells after treatment with 1 or 2 μM Dox (Fig. 4A). Knockdown of GCN2 had no obvious effects on intracellular ROS or superoxide levels under basal conditions, but significantly attenuated the increase of ROS and superoxide levels in Dox-treated (1 μM, 24 h) H9C2 cells (Fig. 4B–C). Furthermore, GCN2 knockdown also significantly attenuated Dox-induced upregulation of p-eIF2α, ATF4, CHOP and Bax, as well as downregulation of Bcl-2 and UCP2 in H9C2 cells (Fig. 4D).
Fig. 4.

GCN2 affects Dox induced cell death, oxidative stress and ER stress in H9C2 cardiomyoblast cells. Scramble shRNA (shScr) and GCN2-specific shRNA (shGCN2) stably transfected cells were treated with 0–2 μM Dox for 24 h and the cell viability (A) was determined. After treatment with 1 μM Dox for 24 h, the intracellular reactive oxygen species (ROS) (B) and superoxide (C) levels in control and GCN2 knockdown cells were measured, and cell lysates were examined by Western blot (D). After treatment with 0–2 μM Dox for 24 h, the cell viability of control and GCN2 overexpressed cells was determined (E). Control and GCN2 overexpressed cells were treated with 1 μM Dox for 24 h, then the intracellular ROS (F) and superoxide (G) levels were measured, and cell lysates were examined by Western blot (H). N = 3–8, * indicates p < .05, ** indicates p < .01.
Then we generated a stable H9C2 cell line with tetracycline (Tet)-controlled expression of FLAG-tagged mouse GCN2 using pLVX-Tet3G-GCN2 lentivirus. Under basal conditions, GCN2 overexpression (Tet on) did not affect cell viability and cellular redox state. However, Dox treatment caused more cell death and higher levels of intracellular ROS and superoxide in GCN2 overexpressing cells than in control (Tet off) cells (Fig. 4E-G). After 1 μM Dox treatment for 24 h, GCN2-overexpressing cells also exhibited higher levels of p-eIF2α, ATF4, CHOP and Bax, as well as lower levels of Bcl-2 and UCP2 (Fig. 4H). Together, these data suggested that GCN2 affects Dox-induced oxidative stress, ER stress and cell death in cardiomyocytes.
3.4. Knockdown of eIF2α attenuated Dox-induced oxidative stress and cell death
Since eIF2α phosphorylation was increased in Dox-treated hearts and H9C2 cells, we also stably transfected H9C2 cells with shRNA lentiviral vectors targeting eIF2α to determine whether knockdown of eIF2α also protects against Dox-induced cell death. Compared to control cells, eIF2α-depleting cells exhibited higher viability and lower intracellular ROS levels in response to Dox treatment. In addition, Dox-induced upregulation of p-eIF2α, ATF4, CHOP and Bax, as well as downregulation of UCP2 were significantly attenuated by depleting eIF2α in H9C2 cells (Supplemental Fig. 2).
3.5. CHOP expression affects Dox-induced oxidative stress and cell death
To determine whether the GCN2/eIF2α pathway affects Dox-induced cell death through a CHOP-dependent pathway, we overexpressed FLAG-tagged CHOP in H9C2 cells by adenovirus transfection. Adenovirus with empty vector (Ad-Empty) was used as a control. Compared to control cells, CHOP-overexpressing cells exhibited lower levels of cell viability in response to Dox treatment (Fig. 5A), indicating that CHOP overexpression exacerbated Dox-induced cell death. Furthermore, CHOP overexpression significantly increased levels of intracellular ROS and superoxide in both control and Dox-treated H9C2 cells (Fig. 5B-C). More interestingly, CHOP overexpression resulted in a significant increase in Bax expression, as well as remarkable reductions in Bcl-2 and UCP2 expression under basal conditions. After Dox treatment, Bcl-2 and UCP2 were further decreased, while Bax was increased in CHOP-overexpressing cells (Fig. 5D).
Fig. 5.

CHOP affects Dox-induced cell death, oxidative stress and ER stress in H9C2 cells. Cells were infected with adenovirus with empty vector (Ad-Empty) or FLAG-tagged CHOP (Ad-CHOP), then treated with different concentrations (0–1 μM) of Dox for 24 h, and the cell viability (A) was determined. After treatment with 1 μM Dox for 24 h, the intracellular reactive oxygen species (ROS) (B) and superoxide (C) levels in Ad-Empty and Ad-CHOP transfected cells were measured, and cell lysates were examined by Western blot (D). After treatment with 0–2 μM Dox for 24 h, the cell viability of WT and Chop-/- cells was determined (E). WT and Chop-/- cells were treated with 1 μM Dox for 24 h; then, the intracellular ROS (F) and superoxide (G) levels were measured, and cell lysates were examined by Western blot (H). N = 3–8, * indicates p < .05, ** indicates p < .01.
Next, we knocked out Chop in H9C2 cells using clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 lentiviral vectors targeting CHOP. Under basal condition, Chop deletion had no obvious effect on cell viability and cell redox state. After Dox treatment for 24 h, Chop-/- cells exhibited higher cell viability and lower levels of intracellular ROS and superoxide levels than WT cells (Fig. 5E-G). Western blot results also showed that CHOP depletion significantly decreased Bax expression, and increased Bcl-2 and UCP2 expression in both control and Dox-treated cells (Fig. 5H).
To further confirm the role of CHOP in the GCN2/eIF2α pathway, we overexpressed CHOP in shScr, sheIF2α or shGCN2 stably transfected cells with Ad-CHOP adenovirus. After treatment with .5 μM Dox for 24 h, the viability of GCN2- or eIF2α-depleted cells was still significantly higher than that of control cells (Supplemental Fig. 3). However, overexpression of GCN2 using Ad-GCN2 adenovirus had no obvious effect on cell viability in Dox-treated Chop-/- cells (Supplemental Fig. 4), indicating that CHOP is required for GCN2 to regulate Dox-induced cell death.
3.6. Knockdown of UCP2 exacerbated Dox-induced oxidative stress and cell death
We noticed that UCP2 expression was reduced in Dox treated hearts and H9C2 cells, and this reduction was regulated by the GCN2/eIF2α/CHOP pathway. To determine whether UCP2 affects Dox-induced cell death and oxidative stress, we further depleted UCP2 in H9C2 cells by stably transfecting shUCP2 shRNA lentiviral vectors. Under basal conditions, UCP2 knockdown resulted in a significant decrease in cell viability and increases in intracellular ROS and superoxide levels. After Dox treatment, UCP2-depleted cells exhibited a significantly lower cell viability and higher levels of intracellular ROS and superoxide than those of shScr stably transfected cells (Fig. 6A-C). Western blot results revealed that UCP2 expression was reduced ~70% in UCP2-depleted cells, and the expression of UCP2 was further reduced after treatment with 1 μM Dox for 24 h (Fig. 6D). Interestingly, UCP2 knockdown significantly increased CHOP and Bax expression, whereas decreased Bcl-2 expression in both control and Dox-treated cells (Fig. 6D).
Fig. 6.

Knockdown of UCP2 exacerbates Dox-induced cell death and oxidative stress in H9C2 cells. ShScr and UCP2-specific shRNA (shUCP2) stably transfected cells were treated with 0–1 μM Dox for 24 h, and then the cell viability was determined (A). After treatment with 1 μM Dox for 24 h, the intracellular reactive oxygen species (ROS) (B) and superoxide (C) levels in control and UCP2 knockdown cells were measured, and cell lysates were examined by Western blot (D). Control or UCP2 knockdown cells were infected with Ad-Empty, Ad-GCN2, Ad-CHOP or Ad-GCN2 plus Ad-CHOP for 24 h, and the cell viability was determined (E). N = 3–8, * indicates p < .05, ** indicates p < .01, *** indicates p < .001, NS, not significant.
To further understand the role of GCN2/CHOP/UCP2 pathway in cell death, we overexpressed GCN2, CHOP and GCN2 plus CHOP in both shScr and shUCP2 stably transfected cells. In control cells, overexpression of GCN2 or CHOP by adenovirus transfection had no obvious effect on cell viability. However, when GCN2 and CHOP were co-overexpressed, there was an ~35% reduction in cell viability. In UCP2-depleted cells, overexpression of GCN2 and CHOP resulted in ~24% and ~45% reduction in cell viability, respectively. Co-overexpression of GCN2 and CHOP in UCP2-depleted cells decreased cell viability by ~64% (Fig. 6E), indicating that UCP2 depletion renders H9C2 cells more vulnerable to cell death induced by GCN2 activation and CHOP induction.
4. Discussion
In the present study, we demonstrated that GCN2 deficiency attenuates Dox-induced heart failure by inhibiting cell death and decreasing oxidative stress. The underlying mechanism for the pathological role of GCN2 in response to Dox was due to eIF2α-mediated CHOP induction, which results in upregulation of Bax and downregulation of Bcl-2 and UCP2. Our work implies that GCN2 not only acts as a sensor for amino acid availability but also controls myocardial redox state via regulation of the eIF2α/CHOP/UCP2 pathway.
Emerging evidence has suggested that cardiomyocyte apoptosis represents a critical process in the development and progression of doxorubicin-induced cardiotoxicity [8], [30]. Although GCN2 did not affect Dox-induced cell death in mouse embryonic fibroblasts [31], we found that Gcn2-/- mice exhibited preserved EF values and decreased cardiomyocyte apoptosis after Dox treatment. The regulation of GCN2 on cardiac cell apoptosis was further confirmed in H9C2 cells, where GCN2 knockdown attenuated while GCN2 overexpression exacerbated Dox-induced cell death. This is in accordance with previous studies demonstrating that GCN2 exerts a pro-apoptotic role of GCN2 in cardiomyocytes [22] and cancer cells [19], [20]. Consistent with our previous finding that GCN2 is a novel negative regulator of cardiac Bcl-2 expression [22], we found that Gcn2-/- hearts or GCN2-depleted H9C2 cells exhibited higher levels of Bcl-2, whereas GCN2 overexpression decreased Bcl-2 expression in Dox-treated cells. We also observed that GCN2 increases the pro-apoptotic factor Bax expression in Dox-treated hearts and cardiomyocytes. Since a decreased ratio of Bcl-2/Bax can lead to the formation of pores in the mitochondria, release of cytochrome c, and activation of the apoptotic pathway [32], [33], it is likely that GCN2 deficiency confers resistance to Dox-induced cardiomyocyte apoptosis by increasing the ratio of Bcl-2 and Bax.
It has been well established that increased ROS production and a marked reduction in expression of anti-oxidant enzymes are associated with the cardiotoxicity of Dox [7], [34], while decreased oxidative stress attenuates heart dysfunction [35]. In that regard, the lower levels of oxidative stress observed in the hearts of Dox-treated Gcn2–/– mice provide an important alternate mechanism for improved cardiac function. Dox treatment decreased the mRNA levels of a series of anti-oxidative stress response genes, while the reduction of Ucp2, Glrx and Mt1 was attenuated by GCN2 deletion. We also demonstrated that GCN2 knockdown increases, while GCN2 overexpression decreases, UCP2 expression in Dox-treated H9C2 cells. It has been proposed that Dox-induced UCP2 reduction contributes to the increased ROS production in cardiomyocytes [36], [37]. UCP2 overexpression can inhibit H2O2-induced mitochondrial death pathway in cardiomyocytes [38], while UCP2 knockdown exacerbated Dox-induced cell death and oxidative stress. In addition, we demonstrated that UCP2 knockdown directly decreases Bcl-2 expression and increases Bax expression in H9C2 cells. With KO or transgenic mice, Glrx and Mt1 have also been found to protect against Dox-induced cardiomyopathy and oxidative stress [39], [40]. Accordingly, the preserved expression of Ucp2, Glrx and Mt1 observed in hearts from Gcn2–/– mice after Dox treatment may partially contribute to reduced myocardial oxidative stress.
Since eIF2α is the only identified substrate of GCN2, it is reasonable to propose that GCN2 promotes cell death and oxidative stress through phosphorylation of eIF2α. In fact, we consistently observed that GCN2 expression affects eIF2α phosphorylation in Dox-treated hearts and cells. In addition, eIF2α knockdown had a similar effect to GCN2 knockdown on Dox-induced cell death and oxidative stress. In response to amino acid deprivation, GCN2-mediated eIF2α phosphorylation increases the translation of ATF4, which in turn increases the expression of CHOP and other stress response genes [14]. The regulation of the GCN2-eIF2α pathway on ATF4/CHOP expression has also been demonstrated in anti-cancer drug treated cells [20], [21]. Consistent with these reports, we found that GCN2 deletion or depletion attenuated, whereas GCN2 overexpression exacerbated, ATF4/CHOP induction in Dox-treated hearts or H9C2 cells, indicating that CHOP induction under stress conditions is partially GCN2 dependent. Furthermore, when Chop was deleted, GCN2 overexpression could not exacerbate Dox-induced cell death, suggesting a crucial role for CHOP in GCN2-eIF2α pathway. It should be noticed that other factors apart from CHOP may also be involved in GCN2-eIF2a pathway mediated cell death, as overexpressing CHOP could not diminish the protective effect of GCN2 or eIF2α knockdown on Dox-induced cell death. Therefore, more careful studies are still needed to address this point.
As an ER-resident transcription factor, CHOP is increased in the failing heart and plays a pivotal role in ER stress-mediated cardiomyocyte apoptosis [41], [42], [43]. There is some evidence that CHOP promotes apoptosis through inhibition of Bcl-2 and activation of Bax [44], [45]. In the present study, we found that the attenuated CHOP induction in Dox-treated Gcn2-/- hearts or GCN2-depleted H9C2 cells was associated with increased Bcl-2 expression and decreased Bax expression, whereas the enhanced CHOP induction had an opposite effect on Bcl-2 and Bax expression. In addition, using CHOP adenovirus vector and Chop-/- cells, we demonstrated that CHOP can directly regulate Bcl-2 and Bax expression in H9C2 cells. Therefore, it is likely that GCN2 regulates Bcl-2 and Bax expression under stress conditions via a CHOP dependent manner.
In addition to downregulating Bcl-2 expression, CHOP can also sensitize cells to endoplasmic reticulum stress by perturbing the cellular redox state [44], [46]. In the failing heart, CHOP induction was associated with increased oxidative stress [22], [47]. Conversely, Chop deletion reduces ROS levels of β cells under diabetic conditions [48], and attenuates renal ischemia-reperfusion induced oxidative stress [49]. Mechanistically, CHOP represses a series of anti-oxidative stress response genes in ER stress-mediated cell death [46]. In this study, we observed that both CHOP overexpression and UCP2 downregulation increased intracellular ROS and superoxide levels in both Dox treated and untreated cells. Overexpression of CHOP remarkably decreased UCP2 expression, whereas deletion of Chop significantly increased UCP2 expression in H9C2 cells. On the other hand, knockdown of UCP2 also increased CHOP expression, which forms a positive feedback loop. Our and previous studies demonstrated that forced expression of CHOP alone does not induce cell death [44], [46], [50]. However, overexpression of CHOP can induce cell death in UCP2-depleted cells and this effect could be further enhanced by GCN2 overexpression. Thus, GCN2-mediated interaction between CHOP and UCP2 might be critical for cardiomyocyte survival and oxidative stress in the failing heart.
In summary, our study demonstrates that GCN2 deletion protects against Dox-induced cardiomyopathy via inhibiting eIF2α-ATF4-CHOP signaling, which mediates the reduction of Bcl-2/Bax ratio and UCP2 expression. Our results suggest that inhibiting GCN2 activity may help in reducing Dox-related cardiotoxicity.
Acknowledgments
This study was supported by grants from National Natural Science Foundation of China (91743104, 91643206, and 81470520) and Chinese Academy of Sciences (KJRH2015-005, Hundred Talents Program and CAS/SAFEA International Partnership Program for Creative Research Teams). We would like to thank Fang Li, Shasha Zuo and Dandan Sun from University of Chinese Academy of Science for their kindly help in instrument operation.
Acknowledgments
Author disclosure statement
No competing financial interests exist.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2018.04.009.
Appendix A. Supplementary material
Supplementary material
References
- 1.Carvalho C., Santos R.X., Cardoso S., Correia S., Oliveira P.J., Santos M.S., Moreira P.I. Doxorubicin: the good, the bad and the ugly effect. Curr. Med. Chem. 2009;16:3267–3285. doi: 10.2174/092986709788803312. [DOI] [PubMed] [Google Scholar]
- 2.Von Hoff D.D., Layard M.W., Basa P., Davis H.L., Jr, Von Hoff A.L., Rozencweig M., Muggia F.M. Risk factors for doxorubicin-induced congestive heart failure. Ann. Intern. Med. 1979;91:710–717. doi: 10.7326/0003-4819-91-5-710. [DOI] [PubMed] [Google Scholar]
- 3.Swain S.M., Whaley F.S., Ewer M.S. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer. 2003;97:2869–2879. doi: 10.1002/cncr.11407. [DOI] [PubMed] [Google Scholar]
- 4.McGowan J.V., Chung R., Maulik A., Piotrowska I., Walker J.M., Yellon D.M. Anthracycline Chemotherapy and Cardiotoxicity. Cardiovasc. Drugs Ther. 2017;31:63–75. doi: 10.1007/s10557-016-6711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singal P.K., Deally C.M., Weinberg L.E. Subcellular effects of adriamycin in the heart: a concise review. J. Mol. Cell. Cardiol. 1987;19:817–828. doi: 10.1016/s0022-2828(87)80392-9. [DOI] [PubMed] [Google Scholar]
- 6.Nithipongvanitch R., Ittarat W., Cole M.P., Tangpong J., Clair D.K., Oberley T.D. Mitochondrial and nuclear p53 localization in cardiomyocytes: redox modulation by doxorubicin (adriamycin)? Antioxid. Redox Signal. 2007;9:1001–1008. doi: 10.1089/ars.2007.1632. [DOI] [PubMed] [Google Scholar]
- 7.Sterba M., Popelova O., Vavrova A., Jirkovsky E., Kovarikova P., Gersl V., Simunek T. Oxidative stress, redox signaling, and metal chelation in anthracycline cardiotoxicity and pharmacological cardioprotection. Antioxid. Redox Signal. 2013;18:899–929. doi: 10.1089/ars.2012.4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar D., Kirshenbaum L.A., Li T., Danelisen I., Singal P.K. Apoptosis in adriamycin cardiomyopathy and its modulation by probucol. Antioxid. Redox Signal. 2001;3:135–145. doi: 10.1089/152308601750100641. [DOI] [PubMed] [Google Scholar]
- 9.Asensio-Lopez M.C., Sanchez-Mas J., Pascual-Figal D.A., de Torre C., Valdes M., Lax A. Ferritin heavy chain as main mediator of preventive effect of metformin against mitochondrial damage induced by doxorubicin in cardiomyocytes. Free Radic. Biol. Med. 2014;67:19–29. doi: 10.1016/j.freeradbiomed.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 10.Bartlett J.J., Trivedi P.C., Pulinilkunnil T. Autophagic dysregulation in doxorubicin cardiomyopathy. J. Mol. Cell. Cardiol. 2017;104:1–8. doi: 10.1016/j.yjmcc.2017.01.007. [DOI] [PubMed] [Google Scholar]
- 11.Jones L.W., Haykowsky M.J., Swartz J.J., Douglas P.S., Mackey J.R. Early breast cancer therapy and cardiovascular injury. J. Am. Coll. Cardiol. 2007;50:1435–1441. doi: 10.1016/j.jacc.2007.06.037. [DOI] [PubMed] [Google Scholar]
- 12.Wek S.A., Zhu S., Wek R.C. The histidyl-tRNA synthetase-related sequence in the eIF-2 alpha protein kinase GCN2 interacts with tRNA and is required for activation in response to starvation for different amino acids. Mol. Cell. Biol. 1995;15:4497–4506. doi: 10.1128/mcb.15.8.4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sood R., Porter A.C., Olsen D.A., Cavener D.R., Wek R.C. A mammalian homologue of GCN2 protein kinase important for translational control by phosphorylation of eukaryotic initiation factor-2alpha. Genetics. 2000;154:787–801. doi: 10.1093/genetics/154.2.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harding H.P., Novoa I., Zhang Y., Zeng H., Wek R., Schapira M., Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell. 2000;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 15.Costa-Mattioli M., Gobert D., Harding H., Herdy B., Azzi M., Bruno M., Bidinosti M., Ben Mamou C., Marcinkiewicz E., Yoshida M., Imataka H., Cuello A.C., Seidah N., Sossin W., Lacaille J.C., Ron D., Nader K., Sonenberg N. Translational control of hippocampal synaptic plasticity and memory by the eIF2alpha kinase GCN2. Nature. 2005;436:1166–1173. doi: 10.1038/nature03897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ravindran R., Loebbermann J., Nakaya H.I., Khan N., Ma H., Gama L., Machiah D.K., Lawson B., Hakimpour P., Wang Y.C., Li S., Sharma P., Kaufman R.J., Martinez J., Pulendran B. The amino acid sensor GCN2 controls gut inflammation by inhibiting inflammasome activation. Nature. 2016;531:523–527. doi: 10.1038/nature17186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ravishankar B., Liu H., Shinde R., Chaudhary K., Xiao W., Bradley J., Koritzinsky M., Madaio M.P., McGaha T.L. The amino acid sensor GCN2 inhibits inflammatory responses to apoptotic cells promoting tolerance and suppressing systemic autoimmunity. Proc. Natl. Acad. Sci. USA. 2015;112:10774–10779. doi: 10.1073/pnas.1504276112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo Y., Wang H., Tang Y., Wang Y., Zhang M., Yang Z., Nyirimigabo E., Wei B., Lu Z., Ji G. GCN2 deficiency protects mice from denervation-induced skeletal muscle atrophy via inhibiting FoxO3a nuclear translocation. Protein Cell. 2018 doi: 10.1007/s13238-018-0504-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peidis P., Papadakis A.I., Rajesh K., Koromilas A.E. HDAC pharmacological inhibition promotes cell death through the eIF2alpha kinases PKR and GCN2. Aging. 2010;2:669–677. doi: 10.18632/aging.100216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gentz S.H., Bertollo C.M., Souza-Fagundes E.M., da Silva A.M. Implication of eIF2alpha kinase GCN2 in induction of apoptosis and endoplasmic reticulum stress-responsive genes by sodium salicylate. J. Pharm. Pharmacol. 2013;65:430–440. doi: 10.1111/jphp.12002. [DOI] [PubMed] [Google Scholar]
- 21.Wei C., Lin M., Jinjun B., Su F., Dan C., Yan C., Jie Y., Jin Z., Zi-Chun H., Wu Y. Involvement of general control nonderepressible kinase 2 in cancer cell apoptosis by posttranslational mechanisms. Mol. Biol. Cell. 2015;26:1044–1057. doi: 10.1091/mbc.E14-10-1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu Z., Xu X., Fassett J., Kwak D., Liu X., Hu X., Wang H., Guo H., Xu D., Yan S., McFalls E.O., Lu F., Bache R.J., Chen Y. Loss of the eukaryotic initiation factor 2alpha kinase general control nonderepressible 2 protects mice from pressure overload-induced congestive heart failure without affecting ventricular hypertrophy. Hypertens. (Dallas, Tex.: 1979) 2014;63:128–135. doi: 10.1161/HYPERTENSIONAHA.113.02313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moffat J., Grueneberg D.A., Yang X., Kim S.Y., Kloepfer A.M., Hinkle G., Piqani B., Eisenhaure T.M., Luo B., Grenier J.K., Carpenter A.E., Foo S.Y., Stewart S.A., Stockwell B.R., Hacohen N., Hahn W.C., Lander E.S., Sabatini D.M., Root D.E. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124:1283–1298. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 24.Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Munn D.H., Sharma M.D., Baban B., Harding H.P., Zhang Y., Ron D., Mellor A.L. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. 2005;22:633–642. doi: 10.1016/j.immuni.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 26.Lu Z., Xu X., Hu X., Lee S., Traverse J.H., Zhu G., Fassett J., Tao Y., Zhang P., dos Remedios C., Pritzker M., Hall J.L., Garry D.J., Chen Y. Oxidative stress regulates left ventricular PDE5 expression in the failing heart. Circulation. 2010;121:1474–1483. doi: 10.1161/CIRCULATIONAHA.109.906818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu Z., Xu X., Hu X., Fassett J., Zhu G., Tao Y., Li J., Huang Y., Zhang P., Zhao B., Chen Y. PGC-1 alpha regulates expression of myocardial mitochondrial antioxidants and myocardial oxidative stress after chronic systolic overload. Antioxid. Redox Signal. 2010;13:1011–1022. doi: 10.1089/ars.2009.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi L., Zhao C., Wang H., Lei T., Liu S., Cao J., Lu Z. Dimethylarginine Dimethylaminohydrolase 1 Deficiency Induces the Epithelial to Mesenchymal Transition in Renal Proximal Tubular Epithelial Cells and Exacerbates Kidney Damage in Aged and Diabetic Mice. Antioxid. Redox Signal. 2017;27:1347–1360. doi: 10.1089/ars.2017.7022. [DOI] [PubMed] [Google Scholar]
- 29.Li T., Feng R., Zhao C., Wang Y., Wang J., Liu S., Cao J., Wang H., Wang T., Guo Y., Lu Z. Dimethylarginine Dimethylaminohydrolase 1 protects against high-fat diet-induced hepatic steatosis and insulin resistance in mice. Antioxid. Redox Signal. 2017;26:598–609. doi: 10.1089/ars.2016.6742. [DOI] [PubMed] [Google Scholar]
- 30.Arola O.J., Saraste A., Pulkki K., Kallajoki M., Parvinen M., Voipio-Pulkki L.M. Acute doxorubicin cardiotoxicity involves cardiomyocyte apoptosis. Cancer Res. 2000;60:1789–1792. [PubMed] [Google Scholar]
- 31.Peidis P., Papadakis A.I., Muaddi H., Richard S., Koromilas A.E. Doxorubicin bypasses the cytoprotective effects of eIF2alpha phosphorylation and promotes PKR-mediated cell death. Cell death Differ. 2011;18:145–154. doi: 10.1038/cdd.2010.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kumar D., Jugdutt B.I. Apoptosis and oxidants in the heart. J. Lab. Clin. Med. 2003;142:288–297. doi: 10.1016/S0022-2143(03)00148-3. [DOI] [PubMed] [Google Scholar]
- 33.Wolter K.G., Hsu Y.T., Smith C.L., Nechushtan A., Xi X.G., Youle R.J. Movement of Bax from the cytosol to mitochondria during apoptosis. J. Cell Biol. 1997;139:1281–1292. doi: 10.1083/jcb.139.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sterba M., Popelova O., Lenco J., Fucikova A., Brcakova E., Mazurova Y., Jirkovsky E., Simunek T., Adamcova M., Micuda S., Stulik J., Gersl V. Proteomic insights into chronic anthracycline cardiotoxicity. J. Mol. Cell. Cardiol. 2011;50:849–862. doi: 10.1016/j.yjmcc.2011.01.018. [DOI] [PubMed] [Google Scholar]
- 35.Octavia Y., Tocchetti C.G., Gabrielson K.L., Janssens S., Crijns H.J., Moens A.L. Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J. Mol. Cell. Cardiol. 2012;52:1213–1225. doi: 10.1016/j.yjmcc.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 36.Bugger H., Guzman C., Zechner C., Palmeri M., Russell K.S., Russell R.R., 3rd Uncoupling protein downregulation in doxorubicin-induced heart failure improves mitochondrial coupling but increases reactive oxygen species generation. Cancer Chemother. Pharmacol. 2011;67:1381–1388. doi: 10.1007/s00280-010-1441-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hsu H.C., Chen C.Y., Chen M.F. N-3 polyunsaturated fatty acids decrease levels of doxorubicin-induced reactive oxygen species in cardiomyocytes -- involvement of uncoupling protein UCP2. J. Biomed. Sci. 2014;21:101. doi: 10.1186/s12929-014-0101-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Teshima Y., Akao M., Jones S.P., Marban E. Uncoupling protein-2 overexpression inhibits mitochondrial death pathway in cardiomyocytes. Circ. Res. 2003;93:192–200. doi: 10.1161/01.RES.0000085581.60197.4D. [DOI] [PubMed] [Google Scholar]
- 39.Guo J., Guo Q., Fang H., Lei L., Zhang T., Zhao J., Peng S. Cardioprotection against doxorubicin by metallothionein Is associated with preservation of mitochondrial biogenesis involving PGC-1alpha pathway. Eur. J. Pharmacol. 2014;737:117–124. doi: 10.1016/j.ejphar.2014.05.017. [DOI] [PubMed] [Google Scholar]
- 40.Diotte N.M., Xiong Y., Gao J., Chua B.H., Ho Y.S. Attenuation of doxorubicin-induced cardiac injury by mitochondrial glutaredoxin 2. Biochim. Et. Biophys. Acta. 2009;1793:427–438. doi: 10.1016/j.bbamcr.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 41.Fu H.Y., Okada K., Liao Y., Tsukamoto O., Isomura T., Asai M., Sawada T., Okuda K., Asano Y., Sanada S., Asanuma H., Asakura M., Takashima S., Komuro I., Kitakaze M., Minamino T. Ablation of C/EBP homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation. 2010;122:361–369. doi: 10.1161/CIRCULATIONAHA.109.917914. [DOI] [PubMed] [Google Scholar]
- 42.Miyazaki Y., Kaikita K., Endo M., Horio E., Miura M., Tsujita K., Hokimoto S., Yamamuro M., Iwawaki T., Gotoh T., Ogawa H., Oike Y. C/EBP homologous protein deficiency attenuates myocardial reperfusion injury by inhibiting myocardial apoptosis and inflammation. Arterioscler., Thromb., Vasc. Biol. 2011;31:1124–1132. doi: 10.1161/ATVBAHA.111.224519. [DOI] [PubMed] [Google Scholar]
- 43.Okada K., Minamino T., Tsukamoto Y., Liao Y., Tsukamoto O., Takashima S., Hirata A., Fujita M., Nagamachi Y., Nakatani T., Yutani C., Ozawa K., Ogawa S., Tomoike H., Hori M., Kitakaze M. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation. 2004;110:705–712. doi: 10.1161/01.CIR.0000137836.95625.D4. [DOI] [PubMed] [Google Scholar]
- 44.McCullough K.D., Martindale J.L., Klotz L.O., Aw T.Y., Holbrook N.J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 2001;21:1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oyadomari S., Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell death Differ. 2004;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 46.Han J., Back S.H., Hur J., Lin Y.H., Gildersleeve R., Shan J., Yuan C.L., Krokowski D., Wang S., Hatzoglou M., Kilberg M.S., Sartor M.A., Kaufman R.J. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013;15:481–490. doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu X., Kwak D., Lu Z., Xu X., Fassett J., Wang H., Wei Y., Cavener D.R., Hu X., Hall J., Bache R.J., Chen Y. Endoplasmic reticulum stress sensor protein kinase R-like endoplasmic reticulum kinase (PERK) protects against pressure overload-induced heart failure and lung remodeling. Hypertens. (Dallas, Tex.: 1979) 2014;64:738–744. doi: 10.1161/HYPERTENSIONAHA.114.03811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Song B., Scheuner D., Ron D., Pennathur S., Kaufman R.J. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J. Clin. Investig. 2008;118:3378–3389. doi: 10.1172/JCI34587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen B.L., Sheu M.L., Tsai K.S., Lan K.C., Guan S.S., Wu C.T., Chen L.P., Hung K.Y., Huang J.W., Chiang C.K., Liu S.H. CCAAT-enhancer-binding protein homologous protein deficiency attenuates oxidative stress and renal ischemia-reperfusion injury. Antioxid. Redox Signal. 2015;23:1233–1245. doi: 10.1089/ars.2013.5768. [DOI] [PubMed] [Google Scholar]
- 50.Chikka M.R., McCabe D.D., Tyra H.M., Rutkowski D.T. C/EBP homologous protein (CHOP) contributes to suppression of metabolic genes during endoplasmic reticulum stress in the liver. J. Biol. Chem. 2013;288:4405–4415. doi: 10.1074/jbc.M112.432344. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material