

Abstract
Background:
LMNA-related muscular dystrophy can manifest in a wide variety of disorders, including Emery-Dreifuss muscular dystrophy (EDMD), limb-girdle muscular dystrophy (LGMD), and LMNA-associated congenital muscular dystrophy (L-CMD). Muscle magnetic resonance imaging (MRI) has become a useful tool in the diagnostic workup of patients with muscle dystrophies. This study aimed to investigate whether there is a consistent pattern of MRI changes in patients with LMNA mutations in various muscle subtypes.
Methods:
Twenty-two patients with LMNA-related muscular dystrophies were enrolled in this study. MRI of the thigh and/or calf muscles was performed in them. The muscle MRI features of the three subtypes were compared by the Mann-Whitney U-test. The relationship between the clinical and MRI findings was also investigated by Spearman's rank analyses.
Results:
The present study included five EDMD, nine LGMD, and eight L-CMD patients. The thigh muscle MRI revealed that the fatty infiltration of the adductor magnus, semimembranosus, long and short heads of the biceps femoris, and vasti muscles, with relative sparing of the rectus femoris, was the predominant change observed in the EDMD, LGMD, and advanced-stage L-CMD phenotypes, although the involvement of the vasti muscles was not prominent in the early stage of L-CMD. At the level of the calf, six patients (one EDMD, four LGMD, and one L-CMD) also showed a similar pattern, in which the soleus and the medial and lateral gastrocnemius muscles were most frequently observed to have fatty infiltration. The fatty infiltration severity demonstrated higher scores associated with disease progression, with a corresponding rate of 1.483 + 0.075 × disease duration (X) (r = 0.444, P = 0.026). It was noteworthy that in six L-CMD patients with massive inflammatory cell infiltration in muscle pathology, no remarkable edema-like signals were observed in muscle MRI.
Conclusions:
EDMD, LGMD and advanced-staged L-CMD subtypes showed similar pattern of muscle MRI changes, while early-staged L-CMD showed somewhat different changes. Muscle MRI of L-CMD with a muscular dystrophy pattern in MRI provided important clues for differentiating it from childhood inflammatory myopathy. The fatty infiltration score could be used as a reliable biomarker for outcome measure of disease progression.
Keywords: Emery-Dreifuss Muscular Dystrophy, Limb-Girdle Muscular Dystrophy, Congenital Muscular Dystrophy, LMNA, Muscle Magnetic Resonance Imaging
摘要
背景:
LMNA相关肌营养不良是LMNA基因突变导致的遗传性骨骼肌疾病,具有显著的临床异质性,常见的临床表型包 Emery-Dreifus肌营养不良(EDMD)、肢带型肌营养不良(LGMD1B)、婴儿期起病的先天性肌营养不良(L-CMD)。既 往研究表明骨骼肌MRI在骨骼肌疾病诊断中具有重要作用。本文报道一组LMNA相关肌营养不良的临床表现、肌肉病理及骨 骼肌MRI改变特点。
方法:
通过临床表现、病理改变、基因筛查,对符合LMNA相关肌营养不良诊断标准的病例进行临床诊断并纳入研究,共收集 22例。对上述22位患者行大腿和/或小腿骨骼肌MRI检查。回顾性分析不同临床表型之间的临床表现、肌肉病理改变及骨骼肌 MRI改变特点,探讨临床表型及骨骼肌MRI表现之间的相关性。
结果:
根据不同的临床表现,22例患者分别为5例EDMD、9例LGMD、8例L-CMD。大腿骨骼肌MRI检查显示EDMD、LGMD和 晚期L-CMD患者大收肌、半膜肌、股二头肌长头及短头、股四头肌肌群出现明显脂肪化,其中股直肌相对保留。早期L-CMD 患者股四头肌肌群脂肪化不明显。6例患者(1例EDMD、4例LGMD、1例L-CMD)行小腿骨骼肌MRI检查,所有患者均出现 明显的比目鱼肌及腓肠肌脂肪化改变。所有患者股四头肌群脂肪化评分与病程的相关性有统计学意义,股四头肌群脂肪化评 分(Y) =1.483+0.075×病程 (X) (r = 0.444, P = 0.026).。6例L-CMD患者骨骼肌病理可见大量的炎症细胞浸润,骨骼肌MRI检查 未见明显水肿改变。
结论:
EDMD、LGMD和晚期L-CMD患者在大腿骨骼肌MRI上有相似的脂肪化浸润模式,而早期L-CMD患者在股四头肌群的 受累程度则有不同。LMNA相关肌营养不良与婴儿期炎性肌肉病在病理改变上有一定相似,骨骼肌MRI检查有助于鉴别两者。 此外,股四头肌群脂肪化增加与疾病的进程有一定的相关性。
INTRODUCTION
The LMNA gene, located on chromosome 1q21.1–21.3, encodes lamin A and C, which are nuclear envelope intermediate filament proteins found on the inner nuclear membrane. These proteins play an important role in many cellular processes, such as in providing a nuclear scaffold for protein complexes, maintaining nuclear structure, regulating gene expression, and playing roles in signaling pathways.[1,2,3,4] Mutations in the LMNA gene cause a series of rare diseases known as laminopathies, which involve skeletal muscles, cardiac muscles, peripheral nerves and fat tissue.[5,6] Skeletal muscle disorders caused by mutations in the LMNA gene can manifest as diverse phenotypes, such as LMNA-associated congenital muscular dystrophy (L-CMD), Emery-Dreifuss muscular dystrophy (EDMD), and limb-girdle muscular dystrophy type 1B (LGMD1B).
EDMD was the first described myopathic phenotype of laminopathies. This disorder is characterized by weakness in a scapulo-humero-peroneal distribution; prominent contractures of the ankle, elbow, and spine; and heart issues. The cardiomyopathy and conduction defects may result in a high risk of cardiac sudden death.[7,8,9] LGMD1B is characterized by predominant scapular and pelvic girdle muscle involvement, and the onset age is later than that of EDMD. Joint contractures and restrictive neck flexions are not prominent in the early stage of the disease, in contrast to EDMD.[10,11] L-CMD is described in patients whose symptoms manifest in the first few months of life or even during the prenatal period.[12,13] Patients with L-CMD have delayed motor development, hypotonia, and proximal limb and cervical-axial muscle weakness.[14] In addition, strikingly, some patients present with dropped head syndrome.[15] Most patients are unable to walk or sit without assistant during childhood, and some even require early respiratory support or die due to cardiac involvement.[16,17] The pathological diagnosis of L-CMD is difficult because the histological evidence of inflammation in laminopathies makes it difficult to distinguish from juvenile idiopathic inflammatory myopathy.[18]
As reported in a previous articles, though these clinical features may be determined by the same LMNA mutation, it is not easy to distinguish according to the genotype.[11,19] Muscle imaging has been reported as an important part of the diagnostic workup of myopathies in recent years.[20,21,22,23] Magnetic resonance imaging (MRI) has excellent soft-tissue contrast and resolution, it is a safe and noninvasive method for studying and evaluating myopathy.[20,24,25,26] It is, therefore, the method of detection of the changes, such as fatty infiltration into the muscle, that might serve as indications that differential diagnosis or the evaluation of the severity of the disease should be performed. It has been demonstrated that patients have a similar pattern of fatty infiltration regardless of whether their phenotype is EDMD or LGMD1B.[26,27,28,29] Our objective was to investigate whether there is a consistent pattern of muscle MRI changes in patients with LMNA mutations related to EDMD, LGMD1B, and L-CMD. Moreover, we were dedicated to assessing the possibility of MRI scanning in distinguishing L-CMD from idiopathic inflammatory myopathy.
METHODS
Ethical approval
The study was approved by the Ethics Committee of Peking University First Hospital, and written informed consent was obtained from all patients or their parents.
Patients
Twenty-two patients from twenty-one unrelated families with LMNA mutations were recruited from the Department of Neurology, Peking University First Hospital, between March 2012 and October 2017. Their clinical data were recorded, including age of onset; presenting features; joint contractures; muscle weakness involving cervical, proximal, and distal upper and lower limbs; and serum creatine kinase (CK) level [Table 1]. Cardiac function was assessed via electrocardiograph (ECG), Holter or echocardiogram.
Table 1.
Clinical characteristics of all patients with various clinical subtypes of LMNA-related muscular dystrophy
| Patient number/sex/age at last review (year) | Presenting feature/age at onset (year) | Motor function | Contractures | CK (U/L) | Heart | EMG | Muscle biopsy |
|---|---|---|---|---|---|---|---|
| 1/female/24 | Limb weakness and gait disorder/10 | Proximal LL weakness | Left ankle | 223 | Normal | Myogenic change | Myodystrophic change |
| 2/male/19 | Limb weakness and myopathic gait/1 | Proximal UL and LL weakness | Both sides of elbow and ankle | 929 | Normal | Myogenic change | Myodystrophic change |
| 3/female/29 | Limb weakness/3 | Proximal and distal LL weakness | Both sides of elbow and wrist, rigid spine | 902 | Atrioventricular junctional escape rhythm | Myogenic change | Myodystrophic change |
| 4/male/4 | Limb weakness/2 | Proximal LL weakness | Both sides of ankle | 1361 | Normal | Myogenic change | Myodystrophic change |
| 5/female/14 | Limb weakness/4 | Proximal and distal LL weakness | Spine, both sides of ankle | 820 | Tachycardia, ultrasonic cardiogram is normal | Myogenic change | Myodystrophic change |
| 6/male/3.5 | Limb weakness/2 | Proximal LL weakness | No | 215 | Normal | Myogenic change | Necrotic myopathic change |
| 7/male/57 | Limb weakness/56 | Proximal LL and UL weakness | No | 900 | Normal | Myogenic change | Nonspecific change |
| 8/female/3 years | Limb weakness, myopathic gait/1.3 | Proximal LL and UL weakness | No | 2623 | Normal | Myogenic change | Myodystrophic change |
| 9/male/2.5 years | Myopathic gait/1 | LL weakness | No | 413 | Normal | Myogenic change | Necrotic myopathic change |
| 10/female/16 | Limb weakness/11 | Proximal and distal LL weakness | No | 710 | Bradycardia | Myogenic change | Myodystrophic change |
| 11/female/3 | Limb weakness/1 | Proximal and distal LL weakness | No | 350 | Normal | Myogenic change | Myodystrophic change |
| 12/female/3 | Limb weakness/1.2 | Proximal LL weakness | Both sides of ankle | 1005 | Normal | Myogenic change | Myodystrophic change |
| 13/male/38 | Limb weakness/5 | LL and UL weakness | Both sides of ankle | 800 | Normal | Myogenic change | Myodystrophic change |
| 14/male/3 | Limb weakness/1.2 | Proximal LL weakness | No | 904 | Normal | Myogenic change | Myodystrophic change |
| 15/male/6 | Limb weakness, myopathic gait, hold head poorly/2 | Neck, proximal LL and UL weakness | Both sides of ankle | 568 | Normal | Myogenic change | Myodystrophic change with massive inflammation |
| 16/male/2.5 | Motor development delay/0.9 | Neck, proximal LL and UL weakness | Both sides of ankle | 1046 | Normal | Myogenic change | Myodystrophic change |
| 17/female/2.3 | Motor development delay/0.4 | Weakness and hypotonia | Both sides of ankle | 486 | Normal | Myogenic change | Myodystrophic change with inflammation |
| 18/male/1.5 | Motor development delay, hold head poorly/0.2 | Weakness and hypotonia | Both sides of ankle and wrist | 445 | Normal | Myogenic change | Myodystrophic change with inflammation |
| 19/female/4.5 | Motor development delay, hold head poorly/1 | Proximal UL and LL weakness; hold head poorly | No | 673 | Normal | Myogenic change | End-stage change |
| 20/female/8 | Motor development delay/0.7 | Proximal UL and LL weakness; hold head poorly | Both sides of knee and ankle | 372 | Normal | Myogenic change | Myodystrophic change with inflammation |
| 21/female/3 | Motor development delay/0.5 | Weakness and hypotonia, hold head poorly | No | 898 | Sinus arrhythmia | Myogenic change | Myodystrophic change with inflammation |
| 22/male/5 | Motor development delay/1 | Could not stand and walk | No | 444 | Normal | Myogenic change | Myodystrophic change with inflammation |
Patient number 13 and number 14 were a father and a son. EMG: Electromyography; CK: Creatine kinase; UL: Upper limbs; LL: Lower limbs.
Muscle pathology
We also analyzed the musculoskeletal system via electromyography and muscle biopsy. All biopsied samples were taken from the biceps brachii or quadriceps. These fresh-frozen muscle specimens were fixed in liquid nitrogen precooled isopentane and were processed for histochemical staining, enzyme histochemical staining, and immunohistochemical staining with first antibodies against major histocompatibility complex 1, complement membrane attack complex, CD3, CD4, CD8, CD20, and CD68.
Genetic test
Genomic DNA was extracted from the blood of patients and their parents. For patients No. 2–4, 8, and 15–18, LMNA gene mutations were screened via direct sequencing of polymerase chain reaction (PCR) products. All the exons and intron/exons of the LMNA gene were PCR-amplified using oligonucleotide primer designed by Primer Premier 5.0 online software (Premier Biosoft International, Palo Alto, CA, USA). For other patients, targeted next-generation sequencing of a panel including muscular dystrophy and congenital myopathy was performed initially, followed by Sanger sequencing of suspicious mutations.
Muscle imaging
MRI examinations were performed on a 3.0T (GE Signa, USA or Philips Achieva, NED) scanner at the level of the thigh in all patients, as previously described.[23] Lower leg muscle MRI was performed in six patients, including one with EDMD, four with LGMD, and one with L-CMD. Follow-up MRI scans were performed for three patients (patients No. 15, 19, and 22) 1–3 years after the first MRI. The following sequences were used: (1) axial T1-weighted fast spin-echo series for fatty infiltration and (2) axial T2-weighted short-time inversion recovery series with fat suppression series for edema. The parameters used for the MRI sequences were repetition time (TR) = 450–480 ms, echo time (TE) = 20 ms, thickness = 5 mm, section interval = 1 mm, and field of view = 28–32 cm. Two independent observers (Y.Z and H.L), blind to clinical data, quantified the fatty muscle infiltration on the muscle MRI scans using the modified version of the Mercuri score [Tables 2 and 3].[29,30]
Table 2.
Thigh muscle MRI scores of the 21 patients with various clinical subtypes of LMNA-related muscular dystrophy
| Patient number/duration of MRI | Diagnosis | Fatty infiltration | Edema | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GM | RF | VL | VI | VM | SA | AL | AM | GR | SM | ST | BFL | BFS | |||
| 1/14 years | EDMD | 2 | 1 | 3 | 4 | 3 | 2 | 1 | 3 | 0 | 2 | 1 | 2 | 4 | None |
| 2/18 years | EDMD | 2 | 0 | 3 | 3 | 3 | 1 | 2 | 3 | 0 | 1 | 0 | 2 | 2 | None |
| 3/26 years | EDMD | 2 | 0 | 3 | 4 | 3 | 1 | 1 | 3 | 0 | 2 | 1 | 3 | 4 | None |
| 4/2 years | EDMD | 1 | 0 | 1 | 1 | 1 | 0 | 2 | 2 | 0 | 2 | 2 | 2 | 0 | None |
| 6/1.5 years | LGMD | 3 | 1 | 4 | 4 | 4 | 1 | 1 | 4 | 1 | 2 | 1 | 4 | 4 | None |
| 7/1 years | LGMD | 0 | 1 | 2 | 2 | 2 | 3 | 0 | 1 | 3 | 2 | 2 | 1 | 1 | None |
| 8/1.7 years | LGMD | 2 | 0 | 4 | 4 | 4 | 0 | 4 | 4 | 0 | 4 | 2 | 4 | 4 | None |
| 9/1.5 years | LGMD | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | None |
| 10/5 years | LGMD | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 0 | None |
| 11/2 years | LGMD | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | None |
| 12/1.8 years | LGMD | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | None |
| 13/33 years | LGMD | 4 | 2 | 3 | 4 | 3 | 2 | 4 | 4 | 2 | 4 | 4 | 4 | 3 | None |
| 14/1.8 years | LGMD | 2 | 0 | 2 | 2 | 2 | 0 | 2 | 2 | 0 | 2 | 1 | 2 | 2 | None |
| 15/4 years | CMD | 1 | 0 | 1 | 1 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 2 | 2 | None |
| 15/5 years | CMD | 1 | 0 | 3 | 3 | 3 | 0 | 2 | 3 | 0 | 1 | 0 | 1 | 1 | None |
| 15/8 years | CMD | 1 | 0 | 3 | 3 | 3 | 0 | 2 | 3 | 0 | 1 | 0 | 1 | 1 | None |
| 16/1.6 years | CMD | 1 | 0 | 1 | 1 | 1 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | None |
| 17/2 years | CMD | 3 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 4 | 4 | 4 | 4 | None |
| 18/1.3 years | CMD | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | None |
| 19/3.5 years | CMD | 2 | 1 | 1 | 1 | 1 | 1 | 3 | 2 | 1 | 2 | 1 | 2 | 1 | Mild |
| 19/5 years | CMD | 2 | 1 | 2 | 1 | 1 | 1 | 3 | 2 | 1 | 3 | 2 | 3 | 1 | None |
| 20/8 years | CMD | 2 | 1 | 3 | 2 | 3 | 1 | 4 | 4 | 1 | 4 | 3 | 3 | 2 | None |
| 21/2.5 years | CMD | 2 | 0 | 1 | 1 | 1 | 0 | 2 | 3 | 1 | 2 | 2 | 3 | 1 | Mild |
| 22/4 years | CMD | 2 | 1 | 2 | 2 | 2 | 1 | 0 | 1 | 0 | 2 | 1 | 2 | 2 | None |
| 22/5 years | CMD | 2 | 3 | 3 | 3 | 3 | 2 | 3 | 3 | 2 | 3 | 2 | 3 | 3 | None |
GM: Gluteus maximus; RF: Rectus femoris; VL: Vastus lateralis; VI: Vastus intermedius; VM: Vastus medialis; SA: Sartorius; AM: Adductor magnus; AL: Adductor longus; GR: Gracilis femoris; SM: Semimembranosus; ST: Semitendinosus; BFL: Long heads of biceps femoris; BFS: Short heads of biceps femoris; EDMD: Emery-Dreifuss muscular dystrophy; LGMD: Limb-girdle muscular dystrophy; CMD: Congenital muscular dystrophy; MRI: Magnetic resonance imaging.
Table 3.
Lower leg muscle MRI scores of the six patients with various clinical subtypes of LMNA-related muscular dystrophy
| Patients number | Diagnosis | Fatty infiltration scores | Edema | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TA | ED | PB | PL | TP | FHL | FDL | SO | MHG | LHG | |||
| 5 | EDMD | 1 | 2 | 2 | 2 | 0 | 0 | 0 | 4 | 4 | 1 | None |
| 10 | LGMD | 1 | 1 | 2 | 2 | 1 | 1 | 1 | 3 | 3 | 3 | None |
| 11 | LGMD | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 2 | 1 | None |
| 13 | LGMD | 1 | 1 | 4 | 3 | 1 | 1 | 1 | 4 | 3 | 3 | None |
| 14 | LGMD | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | None |
| 19 | CMD | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 2 | 2 | 2 | None |
TA: Tibialis anterior; ED: Extensor digitorum; PB: Peroneus brevis; PL: Peroneus longus; TP: Tibialis posterior; FHL: Flexor hallucis longus; FDL: Flexor digitorum longus; SO: Soleus; MHG: Medial head of the gastrocnemius; LHG: Lateral head of the gastrocnemius; EDMD: Emery-Dreifuss muscular dystrophy; LGMD: Limb-girdle muscular dystrophy; CMD: Congenital muscular dystrophy; MRI: Magnetic resonance imaging.
Statistical analysis
Data were analyzed using SPSS version 22.0 (SPSS Inc., Chicago, IL, USA). Mann-Whitney U-tests were conducted to assess the significance of the differences in the MRI fatty infiltration scores among the three phenotypes. Differences were considered statistically significant at P < 0.05 (two-tailed). Correlations between the summed scores from the MRI findings and clinical assessments (including disease duration and age of onset) and correlations between the summed scores of fatty infiltration and genotypes were analyzed with nonparametric Spearman's rank analyses.
RESULTS
Clinical features
The study included 22 patients (11 males and 11 females) [Table 1]. The average age at onset was 4.8 ± 11.8 years (0.2–56.0 years). The average disease duration was 2.3 ± 6.8 years (1.0–33.0 years). CK was usually increased, ranging from 223 to 2623 U/L (normal limits: 70–170 U/L); most of the patients (16/22) had less than 5 times the upper limit of CK. The electromyographs showed myogenic changes. The three groups could be distinguished according to their phenotype.
Group I: Emery-Dreifuss muscular dystrophy
Patients No. 1–5 belonged to the EDMD group and their ages range from 4.0 to 29.0 years. These patients demonstrated normal developmental motor milestones and presented with slowly progressing muscle weakness, involving both the humeral and peroneal muscles and early onset of knee and elbow contractures. They had difficulties in running, jumping, standing up from a squatting position, or going up and down stairs. Heart function assessed by ECG showed tachycardia in patient No. 5. Holter monitoring of patient No. 3 showed no sinus P-wave or F-wave; atrioventricular junctional escape rhythm; her average heart rate was 46 beats/min; and her echocardiography showed mild tricuspid reflux and pulmonary arterial hypertension. The suggestion of a cardiologist was to implant a pacemaker.
Group II: Limb-girdle muscular dystrophy type 1B
Patients No. 6–14 belonged to the LGMD1B group, with ages ranging from 2.5 to 57.0 years. They showed scapular and girdle weakness at the onset of the disease and no evidence of joint contractures until the last interview, except for patients No. 12 and 13. Patient No. 10 had bradycardia, though her echocardiogram was normal. Patient No. 14 was a 3-year-old boy, who was the son of patient No. 13, and had muscle weakness in all his limbs.
Group III: LMNA-associated congenital muscular dystrophy
Patients No. 15–22 belonged to the L-CMD group, with ages ranging from 1.5 to 8.0 years. They all had abnormal developmental motor milestones and presented with motor development retardation. Most of them could not control their heads or sit or stand unaided. Patient No. 21 had sinus arrhythmia. None of them needed ventilation at the last visit.
Myopathological findings
The histological findings from the muscle biopsies were not uniform and included muscular dystrophic changes in 18 patients, necrotizing myopathic changes in two patients, a nonspecific change in one patient, and end-stage changes in one patient. Notably, there were six L-CMD patients who showed muscular dystrophic change with multifocal inflammatory cell infiltration in the endomysium or in the perivascular region of the perimysium (patients No. 15, 17, 18, 20, 21, and 22) [Figure 1].
Figure 1.
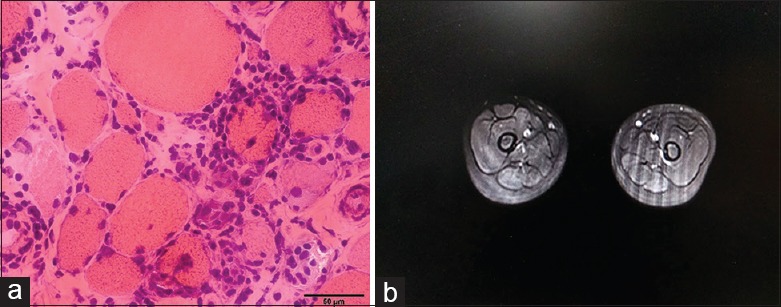
Representative image of the L-CMD patient. Muscle biopsy of L-CMD patient showed myodystrophic changes with massive inflammation (hematoxylin and eosin, original magnification ×400) (a). Bilateral thigh MRI of L-CMD patient showed no sign of edema during the T2-STIR sequence (b). MRI: Magnetic resonance imaging; STIR: Short tau inversion recovery; L-CMD: LMNA-associated congenital muscular dystrophy.
Mutation analysis
The genetic and phenotypic data are summarized in Supplementary Table 1. Ten of the heterozygous mutations (c.1357C>T, c.746G>A, c.1124C>G, c.695G>A, c.1558T>C, c.745C>T, c.736G>A, c.1151A>G, c.1118T>A, and c.1580G>C) had been reported previously, while the other eight were novel mutations (c.754G>A, c.1152G>C, c.1681C>G, c.827G>C, c.92_94del, c.770T>C, c.823C>G, and c.1478A>C) (according to http://www.dmd.nl/, the UMD-LMNA database and the HGMD pro-database). None of the mutations were found in their parents or immediate family members, except for patients No. 7, 13 and 14, which indicated that they occurred de novo.
Supplementary Table 1.
Phenotypes and genotypes of the 22 patients with various clinical subtypes of LMNA-related muscular dystrophy
| Patient number | Phenotype | Mutation | LMNA exon/intron | Protein domain | Previously, reported/novel | Inheritance |
|---|---|---|---|---|---|---|
| 1 | EDMD | c.754G>A | exon4 | Coil region 2 | Novel | De novo |
| 2 | EDMD | c.1152G>C | exon6 | Coil region 2 | Novel | De novo |
| 3 | EDMD | c.1478A>C | exon8 | Conserved IF-tail | Novel | De novo |
| 4 | EDMD | c.1357C>T | exon7 | Conserved IF-tail | Previously reported | De novo |
| 5 | EDMD | c.1357C>T | exon7 | Conserved IF-tail | Previously reported | De novo |
| 6 | LGMD | c.746G>A | exon4 | Coil region 2 | Previously reported | De novo |
| 7 | LGMD | c.1681C>G | exon10 | Linker | Novel | / |
| 8 | LGMD | c.695G>A | exon4 | Linker | Previously reported | De novo |
| 9 | LGMD | c.827G>C | exon5 | Coil region 2 | Novel | De novo |
| 10 | LGMD | c.770T>C | exon4 | Coil region 2 | Novel | De novo |
| 11 | LGMD | c.823C>G | exon5 | Coil region 2 | Novel | De novo |
| 12 | LGMD | c.1357C>T | exon7 | Conserved IF-tail | Previously reported | De novo |
| 13 | LGMD | c.1580G>C | exon9 | Conserved IF-tail | Previously reported | / |
| 14 | LGMD | c.1580G>C | exon9 | Conserved IF-tail | Previously reported | Paternal |
| 15 | L-CMD | c.1124C>G | exon 6 | Coil region 2 | Previously reported | De novo |
| 16 | L-CMD | c.1558T>C | exon9 | Conserved IF-tail | Previously reported | De novo |
| 17 | L-CMD | c.1118T>A | exon6 | Coil region 2 | Previously reported | De novo |
| 18 | L-CMD | c.745C>T | exon4 | Coil region 2 | Previously reported | De novo |
| 19 | L-CMD | c.92_94del | exon1 | Head region | Novel | De novo |
| 20 | L-CMD | c.745C>T | exon4 | Coil region 2 | Previously reported | De novo |
| 21 | L-CMD | c.1151A>G | exon6 | Coil region 2 | Previously reported | De novo |
| 22 | L-CMD | c.736G>A | exon4 | Coil region 2 | Previously reported | De novo |
EDMD: Emery-Dreifuss muscular dystrophy; LGMD: Limb-girdle muscular dystrophy; L-CMD: LMNA-associated congenital muscular dystrophy; IF: Intermediate filament. "/": Not done family genetic analysis.
Muscle magnetic resonance imaging findings
In total, 21 patients (four with EDMD, nine with LGMD1B, and eight with L-CMD) underwent thigh muscle MRI [Figures 1, 2 and Tables 2, 3]. The mean fatty scores varied greatly among the different thigh muscles across the different phenotypes. The results showed that the adductor magnus, the semimembranosus, and the long and short heads of the biceps femoris were the most frequently involved muscles across the three phenotypes, while the gracilis and sartorius were less involved. The involvement of the vasti muscles (vastus lateralis, vastus intermedius, and vastus medialis), while sparing the rectus femoris, was remarkable. Statistically, we did not find any significant differences in the severity of the fatty muscle infiltration between the EDMD and LGMD1B phenotypes.
Figure 2.

Muscle MRI of patients with three different clinical phenotypes caused by LMNA gene mutations of bilateral thigh (section on one third part of thigh). (a-d) The most prominently involved muscles of these three phenotypes were the adductor magnus (AM), the semimembranosus (SM), and the long and short heads of the biceps femoris (BF). The gracilis (GR) and sartorius (SA) exhibited less infiltration. The involvement of the vasti muscles (VM), with a relative sparing of the rectus femoris (RF), is prone to be observed in EDMD, LGMD, and advanced-stage CMD; (e and f): Follow-up MRI of CMD patient. MRI: Magnetic resonance imaging; EDMD: Emery-Dreifuss muscular dystrophy; LGMD: Limb-girdle muscular dystrophy; CMD: Congenital muscular dystrophy.
In the L-CMD patients, the results revealed that the adductor magnus, the semimembranosus, and the long and short heads of the biceps femoris were the most frequently involved muscles, while the gracilis and sartorius were less involved. Unlike EDMD and LGMD, we found infiltration of the vasti muscles in the early-stage L-CMD phenotype to a lesser degree [Figures 1 and 3]. This difference was significantly different (vastus lateralis: Z = −2.691, P = 0.007; vastus intermedius: Z = −2.520, P = 0.014; and vastus medialis: Z = −2.579, P = 0.011). However, in our follow-up research of some CMD patients, we observed fatty infiltration in the vasti muscle, with relative sparing of the rectus femoris during the course of the disease progression. The MRI demonstrated a pattern in which advanced-stage L-CMD presented in a similar manner as EDMD or LGMD. Hence, we speculated that the fatty infiltration scores of the vasti muscles were correlated with disease duration. The correlation between the mean score of the fatty infiltration of the vasti muscles and duration was analyzed with nonparametric Spearman's rank analyses. A significant difference was detected between the disease duration and mean scores of fatty infiltrations of the vasti muscles (r = 0.444, P = 0.026,). We used the linear regression method to determine the linear equation of the score of the vasti muscles and disease duration; the result revealed that the muscle score of the vasti muscle as (Y) = 1.483 + 0.075 × disease duration (X) [Figure 4].
Figure 3.
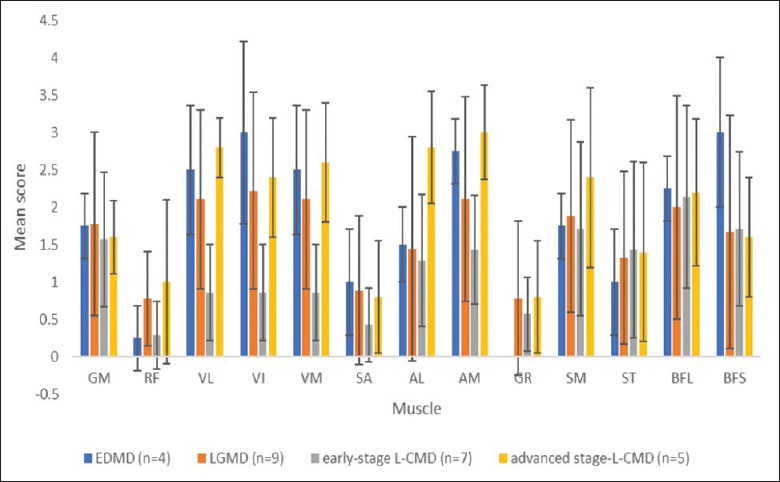
Mean scores of the fatty infiltration in individual thigh muscles. GM: Gluteus maximus; RF: Rectus femoris; VL: Vastus lateralis; VI: Vastus intermedius; VM: Vastus medialis; SA: Sartorius; AM: Adductor magnus; AL: Adductor longus; GR: Gracilis femoris; SM: Semimembranosus; ST: Semitendinosus; BFL: Long heads of the biceps femoris; BFS: Short heads of the biceps femoris.
Figure 4.
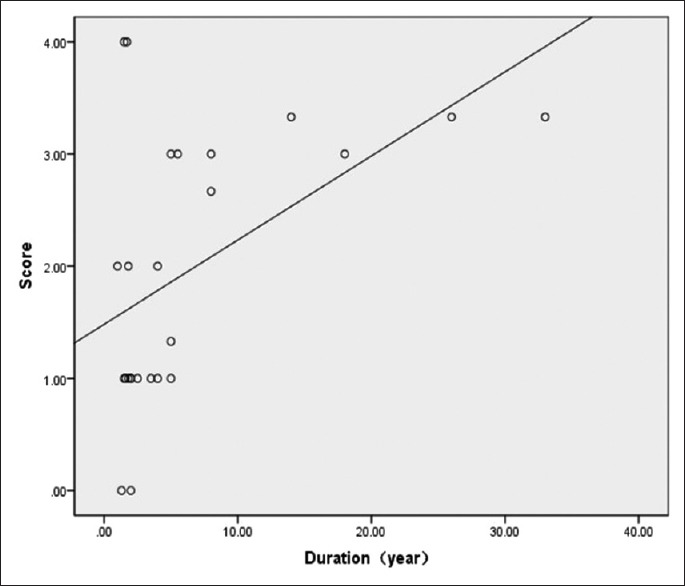
Correlation between the disease duration and mean scores of fatty infiltration in the vasti muscles (except the rectus femoris): the mean score of the fatty infiltration correlated positively with disease duration (n = 25; r = 0.444, P = 0.026).
Of the six patients who exhibited massive inflammatory cell infiltration in their muscle biopsies, only one patients had a mild edema-like condition in the thigh [Figure 1].
There were only six patients who underwent lower leg muscle MRI, including one with EDMD, four with LGMD1B, and one with L-CMD. We found that the soleus and the gastrocnemius medialis and lateralis were the muscles with the most severe fatty infiltration [Figure 5].
Figure 5.

Muscle MRI of patients with three different clinical phenotypes caused by LMNA gene mutations in the leg area. MRI: Magnetic resonance imaging. EDMD: Emery-Dreifuss muscular dystrophy; LGMD: Limb-girdle muscular dystrophy ; L-CMD: LMNA-associated congenital muscular dystrophy.
DISCUSSION
The present study contained different phenotypes of laminopathies.[31] The muscular dystrophies are a large group of genetic muscle disorders.[14,27,30] In general, different conditions often share similar clinical and histopathological phenotypes, so they are difficult to diagnose. The reliable method involves gene sequencing, but this method is expensive and time consuming. In recent years, there has been a great deal of encouraging data regarding the possible role of muscle imaging in identifying different muscular dystrophies. Some reported articles demonstrated a way to identify specific patterns in some muscular dystrophies, such as Ullrich congenital muscular dystrophy,[28,32] Duchenne muscular dystrophy,[33,34,37] and other congenital muscular dystrophies.[34] This study provided additional useful information regarding the differential diagnosis of laminopathies from other muscle diseases.
The present study showed, via MRI, that patients with LMNA gene mutations share a characteristic pattern of muscle involvement. We concluded that the adductor magnus, the semimembranosus, and the long and short heads of the biceps femoris were the most frequently involved muscles across the three phenotypes. The rectus femoris, gracilis, and sartorius either mildly involved or not involved completely. This result was in accordance with previous reports.[27,29,35] In L-CMD, although the muscle MRI scans showed less fatty infiltration in the vasti muscles in the early stage of the disease, the follow-up MRI revealed fatty infiltration in the vasti muscle, while the rectus femoris was relatively spared, indicating the same muscle involvement pattern as that in EDMD or LGMD1B over the course of disease progression. Some previous researchs have suggested that L-CMD may progress to EDMD or LGMD1B.[11,17] At the level of the lower leg, we also found that L-CMD, EDMD, and LGMD displayed similar changes. The soleus and the medial and lateral gastrocnemius muscles are the muscles most frequently found to have fatty infiltration. This result was similar to those of other reported imaging studies.[26,27] The study supported this hypothesis by revealing dynamic muscle MRI changes. L-CMD, EDMD, and LGMD1B are the phenotypes of a continuous entity at different stages; they share the same muscle involvement pattern throughout the disease progression. This information will lead to a way of helping us to further understand this disease and will guide follow-up researches of laminopathies.
Fatty infiltration of the vasti muscles was more frequently found in all phenotypes of the laminopathies, which identified the vasti muscles as a biomarker for evaluating the disease progress. We found that the fatty infiltration score of the vasti muscles is 1.483 + 0.075 × disease duration (X). The infiltration occurred more slowly in patients with DMD, in which the standard deviation of the fatty infiltration scores ranged from 2.41 to 4.87 before 5 years old and from 6.84 to 11.66 between 6 and 10 years old.[34]
The study provided important clues for differential diagnosis, especially between L-CMD and juvenile inflammatory myopathy. Myopathologically, most of the L-CMD patients in this study displayed massive inflammatory cell infiltration, as observed in the muscle biopsy, which were easily misdiagnosed as inflammatory myopathy. Previous studies also reported histological findings of inflammation in patients with laminopathies, especially in infants and children.[18,19,36,37] Similar inflammatory changes are not uncommon in other muscular dystrophy diseases including dysferlinopathy, LGMD2I, and facioscapulohumeral muscular dystrophy.[38,39,40,41] Up to now, the exact mechanism how mutant LMNA inducing inflammatory process in skeletal muscle has not been clarified, although one study revealed that nuclear lamina defects can cause the systemic inflammatory response through activating nuclear factor-kappa B signal pathways in HGPS (progeria caused by mutant LMNA) mouse model.[42] In this study, despite the inflammatory changes on muscle pathology, muscle MRI scans did not show any remarkable muscle edema, as is expected in inflammatory myopathies. Muscle MRI in inflammatory myopathies usually showed comprehensive edema as demonstrated by our previous studies and the others.[43,44] The follow-up studies using muscle imaging in the study also showed more severe fatty infiltration in L-CMD patients instead of edema changes. The reason for the mismatch between the biopsy and muscle imaging findings remains unknown, but it could imply that MRI would be a useful tool for distinguishing laminopathies from myositis.
In conclusion, the present study shows that patients with laminopathies share similar muscle imaging patterns across different phenotypes. We find that fatty infiltration of the vasti muscles is more frequently found in EDMD and LGMD1B patients compared to L-CMD patients, indicating that the vasti muscles are involved in the advanced stage of the disease. Our results can help guide diagnosis in patients with LMNA mutations and will lead the way for further research.
Supplementary information is linked to the online version of the paper on the Chinese Medical Journal website.
Financial support and sponsorship
This work was financially supported by grants from the National Nature Science Foundation of China (No. 81571219) and the Beijing Municipal Science and Technology Commission (No. Z151100003915126).
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
The authors extend their sincere appreciation to the patients and their parents for their participation and enthusiastic support.
Footnotes
Edited by: Ning-Ning Wang
REFERENCES
- 1.Dittmer TA, Misteli T. The lamin protein family. Genome Biol. 2011;12:222. doi: 10.1186/gb-2011-12-5-222. doi: 101186/gb-2011-12-5-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Puckelwartz MJ, Depreux FF, McNally EM. Gene expression, chromosome position and lamin A/C mutations. Nucleus. 2011;2:162–7. doi: 10.4161/nucl.2.3.16003. doi: 10.4161/nucl.2.3.16003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maraldi NM, Capanni C, Cenni V, Fini M, Lattanzi G. Laminopathies and lamin-associated signaling pathways. J Cell Biochem. 2011;112:979–92. doi: 10.1002/jcb.22992. doi: 10.1002/jcb.22992. [DOI] [PubMed] [Google Scholar]
- 4.Davidson PM Lammerding, J. Broken nuclei—Lamins, nuclear mechanics, and disease. Trends Cell Biol. 2014;24:247–56. doi: 10.1016/j.tcb.2013.11.004. doi: 10.1016/j.tcb.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Worman HJ. Nuclear lamins and laminopathies. J Pathol. 2012;226:316–25. doi: 10.1002/path.2999. doi: 10.1002/path.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Worman HJ, Bonne G. “Laminopathies”: A wide spectrum of human diseases. Exp Cell Res. 2007;313:2121–33. doi: 10.1016/j.yexcr.2007.03.028. doi: 10.1016/j.yexcr.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Emery AEH. Emery-Dreifuss muscular dystrophy: A 40-year retrospective. Neuromuscul Disord. 2000;10:228–32. doi: 10.1016/s0960-8966(00)00105-x. doi: 10.1016/S0960-8966(00)00105-X. [DOI] [PubMed] [Google Scholar]
- 8.Meune C, Van Berlo JH, Anselme F, Bonne G, Pinto YM, Duboc D, et al. Primary prevention of sudden death in patients with lamin A/C gene mutations. N Engl J Med. 2006;354:209–10. doi: 10.1056/NEJMc052632. doi: 10.1056/NEJMc052632. [DOI] [PubMed] [Google Scholar]
- 9.Bonne G, Di BM, Varnous S, Bécane HM, Hammouda EH, Merlini L, et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet. 1999;21:285–8. doi: 10.1038/6799. doi: 10.1038/6799. [DOI] [PubMed] [Google Scholar]
- 10.Muchir A, Bonne G, van der Kooi AJ, van Meegen M, Baas F, Bolhuis PA, et al. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B) Hum Mol Genet. 2000;9:1453–9. doi: 10.1093/hmg/9.9.1453. doi: 10.1093/hmg/9.9.1453. [DOI] [PubMed] [Google Scholar]
- 11.Maggi L, D'Amico A, Pini A, Sivo S, Pane M, Ricci G, et al. LMNA-associated myopathies: The Italian experience in a large cohort of patients. Neurology. 2014;83:1634–44. doi: 10.1212/WNL.0000000000000934. doi:10.1186/1750-1172-10-S2-O19. [DOI] [PubMed] [Google Scholar]
- 12.Flanigan KM, Kerr L, Bromberg MB, Leonard C, Tsuruda J, Zhang P, et al. Congenital muscular dystrophy with rigid spine syndrome: a clinical, pathological, radiological, and genetic study. Ann Neurol. 2000;47:152–61. doi: 10.1002/1531-8249(200002)47:2<152::AID-ANA4>3.3.CO;2-L. [PubMed] [Google Scholar]
- 13.Fu XN, Xiong H. Genetic and Clinical Advances of Congenital Muscular Dystrophy. Chin Med J. 2017;130:2624–31. doi: 10.4103/0366-6999.217091. doi: 10.4103/0366-6999.217091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iannaccone S T, Castro D. Congenital muscular dystrophies and congenital myopathies. Continuum. 2013;19:1509. doi: 10.1212/01.CON.0000440658.03557.f1. doi: 10.1212/01.CON.0000440658.03557.f1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hattori A, Komaki H, Kawatani M, Sakuma H, Saito Y, Nakagawa E, et al. A novel mutation in the LMNA gene causes congenital muscular dystrophy with dropped head and brain involvement. Neuromuscul Disord. 2012;22:149–51. doi: 10.1016/j.nmd.2011.08.009. doi: 10.1016/j.nmd.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 16.Menezes MP, Waddell LB, Evesson FJ, Cooper S, Webster R, Jones K, et al. Importance and challenge of making an early diagnosis in LMNA-related muscular dystrophy. Neurology. 2012;78:1258–63. doi: 10.1212/WNL.0b013e318250d839. doi: 10.1212/WNL.0b013e318250d839. [DOI] [PubMed] [Google Scholar]
- 17.Quijano-Roy S, Mbieleu B, Bönnemann CG, Jeannet PY, Colomer J, Clarke NF, et al. De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann Neurol. 2008;64:177–86. doi: 10.1002/ana.21417. doi: 10.1002/ana.21417. [DOI] [PubMed] [Google Scholar]
- 18.Komaki H, Hayashi YK, Tsuburaya R, Sugie K, Kato M, Nagai T, et al. Inflammatory changes in infantile-onset LMNA-associated myopathy. Neuromuscul Disord. 2011;21:563–8. doi: 10.1016/j.nmd.2011.04.010. doi: 10.1016/j.nmd.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 19.Mercuri E, Poppe M, Quinlivan R, Messina S, Kinali M, Demay L, et al. Extreme variability of phenotype in patients with an identical missense mutation in the lamin A/C gene: From congenital onset with severe phenotype to milder classic Emery-Dreifuss variant. Arch Neurol. 2004;61:690–4. doi: 10.1001/archneur.61.5.690. doi: 10.1001/archneur.61.5.690. [DOI] [PubMed] [Google Scholar]
- 20.Carlier PG, Mercuri E, Straub V. Applications of MRI in muscle diseases. Neuromuscul Disord. 2012;22(Suppl 2):S41–S41. doi: 10.1016/j.nmd.2012.08.001. doi: 10.1016/j.nmd.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 21.Mercuri E, Jungbluth H, Muntoni F. Muscle imaging in clinical practice: Diagnostic value of muscle magnetic resonance imaging in inherited neuromuscular disorders. Curr Opin Neurol. 2005;18:526–37. doi: 10.1097/01.wco.0000183947.01362.fe. doi: 10.1097/01.wco.0000183947.01362.fe. [DOI] [PubMed] [Google Scholar]
- 22.Quijano-Roy S, Avila-Smirnow D, Carlier RY. Whole body muscle MRI protocol: pattern recognition in early onset NM disorders. Neuromuscul Disord. 2012;22(Suppl 2):S68–84. doi: 10.1016/j.nmd.2012.08.003. doi: 101016/jnmd201208003. [DOI] [PubMed] [Google Scholar]
- 23.Fu J, Zheng YM, Jin SQ, Yi JF, Liu XJ, Lyn H, et al. “Target” and “Sandwich” Signs in Thigh Muscles have High Diagnostic Values for Collagen VI-related Myopathies. Chin Med J. 2016;129:1811–6. doi: 10.4103/0366-6999.186638. doi: 10.4103/0366-6999.186638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mercuri E, Pichiecchio A, Allsop J, Messina S, Pane M, Muntoni F. Muscle MRI in inherited neuromuscular disorders: past, present, and future. J Magnet Reson Imaging. 2007;25:433. doi: 10.1002/jmri.20804. doi: 10.1002/jmri.20804. [DOI] [PubMed] [Google Scholar]
- 25.Mercuri E, Pichiecchio A, Counsell S, Allsop J, Cini C, Jungbluth H, et al. A short protocol for muscle MRI in children with muscular dystrophies. Eur J Paediatr Neurol. 2002;6:305. doi: 10.1016/s1090-3798(02)90617-3. doi: 10.1016/S1090-3798(02)90617-3. [DOI] [PubMed] [Google Scholar]
- 26.Carboni N, Mura M, Marrosu G, Cocco E, Marini S, Solla E, et al. Muscle imaging analogies in a cohort of patients with different phenotypes caused by LMNA gene mutations. Muscle Nerve. 2010;41:458–63. doi: 10.1002/mus.21514. doi: 10.1002/mus.21514. [DOI] [PubMed] [Google Scholar]
- 27.Díaz-Manera J, Alejaldre A, González L, Olivé M, Gómez-Andrés D, Muelas N, et al. Muscle imaging in muscle dystrophies produced by mutations in the EMD and LMNA genes. Neuromuscul Disord. 2016;26:33–40. doi: 10.1016/j.nmd.2015.10.001. doi: 10.1016/j.nmd.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 28.Mercuri E, Clements E, Offiah A, Pichiecchio A, Vasco G, Bianco F, et al. Muscle magnetic resonance imaging involvement in muscular dystrophies with rigidity of the spine. Ann Neurol. 2010;67:201–8. doi: 10.1002/ana.21846. doi: 10.1002/ana.21846. [DOI] [PubMed] [Google Scholar]
- 29.Mercuri E, Counsell S, Allsop J, Jungbluth H, Kinali M, Bonne G, et al. Selective muscle involvement on magnetic resonance imaging in autosomal dominant Emery-Dreifuss muscular dystrophy. Neuropediatrics. 2002;33:10–4. doi: 10.1055/s-2002-23593. doi: 10.1055/s-2002-23593. [DOI] [PubMed] [Google Scholar]
- 30.Stramare R, Beltrame V, Dal BR, Gallimberti L, Frigo AC, Pegoraro E, et al. MRI in the assessment of muscular pathology: a comparison between limb-girdle muscular dystrophies, hyaline body myopathies and myotonic dystrophies[J] Radiologia Medica. 2010;115:585–99. doi: 10.1007/s11547-010-0531-2. doi: 10.1007/s11547-010-0531-2. [DOI] [PubMed] [Google Scholar]
- 31.Maggi L, Carboni N, Bernasconi P. Skeletal Muscle Laminopathies: A Review of Clinical and Molecular Features. Cells. 2016;5:33. doi: 10.3390/cells5030033. doi: 10.3390/cells50300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mercuri E, Lampe A, Allsop J, Knight R, Pane M, Kinali M, et al. Muscle MRI in Ullrich congenital muscular dystrophy and Bethlem myopathy. Neuromuscul Disord. 2005;15:303–10. doi: 10.1016/j.nmd.2005.01.004. doi: 10.1016/j.nmd.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 33.Zheng Y, Li W, Du J, Jin S, Li S, Zhao Y, et al. The trefoil with single fruit sign in muscle magnetic resonance imaging is highly specific for dystrophinopathies. Eur J Radiol. 2015;84:1992–8. doi: 10.1016/j.ejrad.2015.06.011. doi: 10.1016/j.ejrad.2015.06.011. [DOI] [PubMed] [Google Scholar]
- 34.Li W, Zheng Y, Zhang W, Wang Z, Xiao J, Yuan Y. Progression and variation of fatty infiltration of the thigh muscles in Duchenne muscular dystrophy, a muscle magnetic resonance imaging study. Neuromuscul Disord. 2015;25:375–80. doi: 10.1016/j.nmd.2015.01.003. doi: 10.1016/j.nmd.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 35.Gómez-Andrés D, Dabaj I, Mompoint D, Hankiewicz K, Azzi V, Ioos C, et al. Pediatric laminopathies: Whole-body magnetic resonance imaging fingerprint and comparison with Sepn1 myopathy. Muscle Nerve. 2016;54:192–202. doi: 10.1002/mus.25018. doi: 10.1002/mus.25018. [DOI] [PubMed] [Google Scholar]
- 36.Tan D, Yang H, Yuan Y, Bonnemann C, Chang X, Wang S, et al. Phenotype–Genotype Analysis of Chinese Patients with Early-Onset LMNA-Related Muscular Dystrophy. Plos One. 2015;10:e0129699. doi: 10.1371/journal.pone.0129699. doi: 10.1371/journal.pone.0129699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moraitis E, Foley AR, Pilkington CA, Manzur AY, Quinlivan R, Jacques TS, et al. Infantile-onset LMNA-associated Muscular Dystrophy Mimicking Juvenile Idiopathic Inflammatory Myopathy. J Rheumatol. 2015;42:1064–6. doi: 10.3899/jrheum.140554. doi: 10.3899/jrheum.140554. [DOI] [PubMed] [Google Scholar]
- 38.Pegoraro E, Mancias P, Swerdlow SH, Raikow RB, Cianno C, Marks H, et al. Congenital muscular dystrophy with primary laminin alpha2 (merosin) deficiency presenting as inflammatory myopathy. Ann Neurol. 1996;40:782–91. doi: 10.1002/ana.410400515. doi: 10.1002/ana.410400515. [DOI] [PubMed] [Google Scholar]
- 39.Hauerslev S, Ørngreen MC, Hertz J M, Vissing J, Krag TO. Muscle regeneration and inflammation in patients with facioscapulohumeral muscular dystrophy. Acta Neurol Scand. 2013;128:194–201. doi: 10.1111/ane.12109. doi: 10.1111/ane.12109. [DOI] [PubMed] [Google Scholar]
- 40.Gallardo E, Rojas-Garcã¬A R, De L N, Pou A, Jr B R, Illa I. Inflammation in dysferlin myopathy: immunohistochemical characterization of 13 patients. Neurology. 2001;57:2136–8. doi: 10.1212/wnl.57.11.2136. doi: 10.1212/WNL.57.11.2136. [DOI] [PubMed] [Google Scholar]
- 41.Darin N, Kroksmark A K, Åhlander A C, Moslemi A R, Oldfors A, Tulinius M. Inflammation and response to steroid treatment in limb-girdle muscular dystrophy 2I. Eur J Paediatr Neurol. 2007;11:353–7. doi: 10.1016/j.ejpn.2007.02.018. doi: 10.1016/j.ejpn.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 42.Osorio FG, Bárcena C, Soria-Valles C, Ramsay AJ, de Carlos F, Cobo J, et al. Nuclear lamina defects cause ATM-dependent NF-κB activation and link accelerated aging to a systemic inflammatory response. Genes Dev. 2012;26:2311–24. doi: 10.1101/gad.197954.112. doi: 10.1101/gad.197954.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng Y, Liu L, Wang L, Xiao J, Wang Z, Lv H, et al. Magnetic resonance imaging changes of thigh muscles in myopathy with antibodies to signal recognition particle. Rheumatology (Oxford) 2015;54:1017–24. doi: 10.1093/rheumatology/keu422. doi: 10.1093/rheumatology/keu422. [DOI] [PubMed] [Google Scholar]
- 44.Barsotti S, Zampa V, Talarico R, Minichilli F, Ortori S, Iacopetti V, et al. Thigh magnetic resonance imaging for the evaluation of disease activity in patients with idiopathic inflammatory myopathies followed in a single center. Muscle Nerve. 2016;54:666–72. doi: 10.1002/mus.25099. doi: 10.1002/mus.25099. [DOI] [PubMed] [Google Scholar]