

Abstract
Glucose, chief metabolic support for cancer cell survival and growth, is mainly imported into cells by facilitated glucose transporters (GLUTs). The increase in glucose uptake along with tumor progression is due to an increment of facilitative glucose transporters as GLUT1. GLUT1 prevents cell death of cancer cells caused by growth factors deprivation, but there is scarce information about its role on the damage caused by glucose deprivation, which usually occurs within the core of a growing tumor. In prostate cancer (PCa), GLUT1 is found in the most aggressive tumors, and it is regulated by androgens. To study the response of androgen-sensitive and insensitive PCa cells to glucose deprivation and the role of GLUT1 on survival mechanisms, androgen-sensitive LNCaP and castration-resistant LNCaP-R cells were employed. Results demonstrated that glucose deprivation induced a necrotic type of cell death which is prevented by antioxidants. Androgen-sensitive cells show a higher resistance to cell death triggered by glucose deprivation than castration-resistant cells. Glucose removal causes an increment of H2O2, an activation of androgen receptor (AR) and a stimulation of AMP-activated protein kinase activity. In addition, glucose removal increases GLUT1 production in androgen sensitive PCa cells. GLUT1 ectopic overexpression makes PCa cells more resistant to glucose deprivation and oxidative stress-induced cell death. Under glucose deprivation, GLUT1 overexpressing PCa cells sustains mitochondrial SOD2 activity, compromised after glucose removal, and significantly increases reduced glutathione (GSH). In conclusion, androgen-sensitive PCa cells are more resistant to glucose deprivation-induced cell death by a GLUT1 upregulation through an enhancement of reduced glutathione levels.
Abbreviations: 2DG, 2-deoxyglcuose; AMPK, AMPK-activated protein kinase; AR, Androgen Receptor; CAT, Catalase; DHA, Dehydroascorbic Acid; G6PG, Glucose-6-phosphate dehydrogenase; GPX, Glutathione peroxidase; GSH, Reduced glutathione; GSSG, Oxidized glutathione; HTLV, Human lymphotrophic virus; NAC, n-Acetylcysteine; NAMPT, Nicotinamide phosphoribosyltransferase; PCa, Prostate Cancer; PI3K, phosphoinositide 3-kinase; PSA, Prostate Specific Antigen; OXPHOS, Oxidative phosphorylation; RIPK1, Receptor-interacting serine/threonine-protein kinase 1; SOD, Superoxide dismutase; TRX, Thioredoxin; TXNIP, Thioredoxin interacting protein
Keywords: Glut1, Prostate cancer, Glucose deprivation, Androgen receptor, Glutathione, Oxidative stress
Graphical abstract
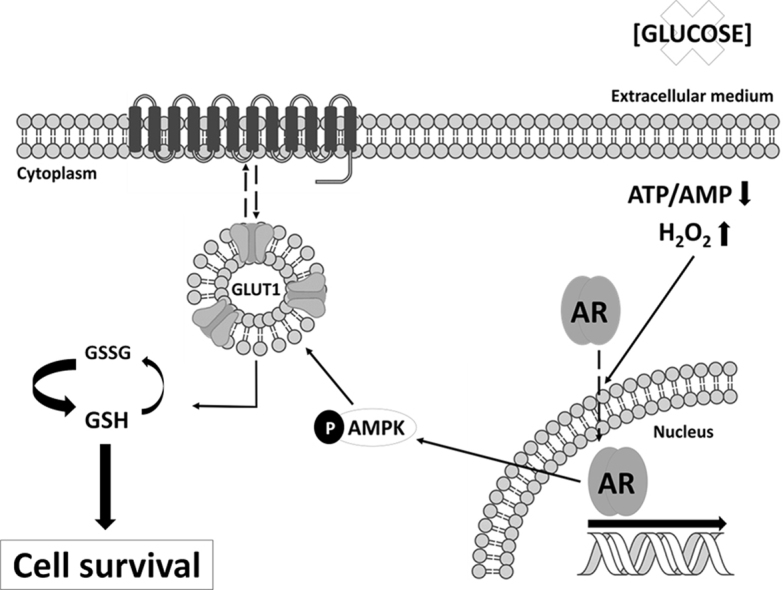
Highlights
-
•
Glucose deprivation carries an increase of free radicals in prostate cancer cells.
-
•
The androgen receptor activation after glucose deprivation proceeds with GLUT1 overproduction.
-
•
Treatment with hydrogen peroxide mimics the response to glucose deprivation.
-
•
GLUT1-overexpressing prostate cancer cells are more resistant to glucose deprivation.
-
•
Glutathione is more reduced in GLUT1-overexpressing cells under glucose deprivation.
1. Introduction
Cancer cells usually show different metabolic requirements than healthy cells to satisfy the higher demand aimed for proliferation. This evidence was first described by Otto Warburg in the 20's, and it is already considered as one of the hallmarks of cancer [1], [2]. Tumor cells usually increase their nutrients uptake, mainly glucose and glutamine. The increase in glycolytic flux, independent of oxygen concentration (i.e. “aerobic glycolysis”), is employed to obtain NADPH as well as the required precursors for biosynthesis [3].
Besides the promotion of cell proliferation, glucose metabolism protects from cell death. Thus, an increase in glycolytic flux turn cells into a more resistant phenotype to apoptosis [4]. The inhibition of cell metabolism by nutrient deprivation has been proposed as an effective approach to kill tumor cells [5]. After glucose withdrawal, reactive oxygen species (ROS) are generated by mitochondrial dysfunction [6], but sometimes cancer cells evolved to avoid this metabolic stress by mechanisms still unknown.
Glucose uptake is higher in tumor cells by an increase in protein production and membrane translocation of facilitative glucose transporters (GLUTs). Glucose uptake in cancer cells has been mainly associated with GLUT1, which is overexpressed by growth factors and usually correlates with cancer malignancy [7]. However, it may also involve other GLUTs [8], [9], [10]. In addition to a drop in oxygen levels, the core of a growing tumor also displays glucose starvation [11], and nutrient removal alters expression or location of GLUT transporters. It has been described that at least one of the GLUT isoforms is upregulated in response to glucose deprivation in cancer cells [12]. Moreover, when non-tumor cells are under growth factors deprivation, GLUT1 is usually internalized and degraded in lysosomes. Consequently, glucose uptake and metabolism dramatically decrease before cell death. Cancer cells often overexpress GLUT1 in response to the absence of growth factors, maintaining glucose metabolism and becoming resistant to apoptosis [13].
In prostate cancer (PCa), glucose metabolism plays a major role in progression [14]. Androgens can activate the metabolic regulator AMP-activated kinase (AMPK), promoting oxidative phosphorylation (OXPHOS) [15]. Besides, androgens increase GLUT1 [16], which overproduction has been described in the most aggressive tumors [17], [18]. Recently, our group described that this transporter is regulated by compounds that block glucose uptake in PCa cells and prevent cancer progression [19].
This work aimed to study the sensitivity of androgen-sensitive and insensitive prostate cancer cells to nutrients deprivation and the possible role of GLUT1 on survival mechanisms of PCa cells after glucose deprivation, a situation that occurs in the core of tumors.
2. Material and methods
2.1. Cell culture, transfection, and reagents
Human androgen-sensitive LNCaP cells were purchased from European Collection of Cell Cultures (Catalog number 89110211, Salisbury, UK) and were cultured in RPMI 1640 medium supplemented with 10% FBS, 2mM L-glutamine, 15 mM HEPES, 100 μg/ml ampicillin and 100 μg/ml kanamycin. PC-3 cells, a human androgen-insensitive cell line, were purchased from American Type Culture Collection (Catalog number CRL-1435, Manassas, VA, USA) and were grown in DMEM/F12 medium supplemented with 10% FBS, 2% L-glutamine and 1% antibiotic-antimycotic cocktail (100 U/ml penicillin, 10 μg/ml streptomycin and 0.25 μg/ml amphotericin B). Both cell lines were authenticated by short tandem repeat (STR) profiling. Jurkat cells were employed as a positive control in the study of GLUT1 surface levels [20]. Cells were maintained in complete RPMI-1640 and DMEM/F12, respectively. Cell lines were grown at 37 °C in a humidified 5% CO2 environment.
LNCaP-R cells, a clone of castration-resistant LNCaP were achieved as previously described [21]. Briefly, LNCaP cells were grown in RPMI-1640 supplemented with foetal bovine serum FBS was stripped from small molecular weight molecules including steroids by incubation with charcoal/dextran. For this purpose, FBS was incubated with a mixture of 2.5% charcoal-activated plus 0.025% Dextran T70 overnight at 4 °C. Charcoal/dextran-stripped FBS (FBSchst) was added to the culture medium and filtered through a 0.22-µm sterile filter. Media plus FBSchst were changed every other day. After 2 weeks, a massive cell death was observed and only a small amount of cells survived. After a month, isolated colonies were selected, subcultured in small 24-well plates and left to grow all the time in the presence of FBSchst culture media. Androgen-resistant colonies were maintained and grew continuously for at least 6 months. AR production in LNCaP-R was confirmed (Supplementary Fig. S1A). AR cytosolic location and androgen-insensitivity of LNCaP-R cells was proved (data not shown)
LNCaP or PC-3 GLUT1-overexpressing cells were obtained by using FuGENE®HD (Promega Biotech Iberica S.L., Alcobendas, Madrid), following manufacturer's instructions. Plasmid pcDNA3.2/v5-DEST hGLUT1 catalog number 18,085 (Addgene Europe, Teddington, UK) nicely deposited by Wolf Frommer [22]. Cells were selected by incubation with 300 μg/ml G418 (Sigma-Aldrich Quimica SL, Madrid, Spain). GLUT1 overproduction in LNCaPGLUT1 and PC-3GLUT1 cells were confirmed (Supplementary Fig. S1B). The increase in glucose uptake of GLUT1-overexpressing cells was also confirmed (data not shown).
For all treatments, cells were left to attach for 48 h. Glucose concentration was always set at 2 g/L (11 mM). For glucose/glutamine deprivation assays, complete medium without glucose or glutamine was purchased (Lonza Biologics Porriño SL, Barcelona, Spain). Fructose or mannose (11 mM) were added in the absence of glucose, and 2-deoxyglucose (2DG) (10 mM) (Alfa Aesar-Thermo Fisher, Karlsruhe, Germany) was employed to disable glycolysis in the presence of glucose.
Cell death was assayed by using 10 μM of the inhibitor of caspases q-VD-OPH (Sigma-Aldrich Quimica SL, Madrid, Spain) was used to study apoptosis, 100 μM necrostatin-1 to Receptor-Interacting serine/threonine-Protein Kinase 1 (RIPK1)-dependent necroptosis, 25 μM necrostatin-7 to RIPK1-independent necroptosis, 2 μM ferrostatin-1 to ferroptosis and 2 mM 3-methyladenine (3MA) to inhibit autophagy (Cayman Chemical Hamburg, Germany). N-acetyl-cysteine (NAC) (Alfa Aesar-Thermo Fisher, Karlsruhe, Germany), catalase, dehydroascorbic acid (DHA) or (±)-6-Hydroxy-2,5,7,8-tetramethyl-chromane-2-carboxylic acid (Trolox) (Sigma-Aldrich Quimica SL, Madrid, Spain) were employed as antioxidants. Hydrogen peroxide (H2O2) was used as oxidant agent (Sigma-Aldrich Quimica SL, Madrid, Spain) and phloretin was employed to inhibit GLUT1 transporter. All vehicles were adjusted to the same concentration in each experimental group.
2.2. Trypan Blue exclusion
Cells were seeded in 6-well plates. After treatments, cells were collected by scraping, centrifuged at 300×g for 5 min and then suspended in PBS. Total cells and dead cells were counted in 0.2% Trypan Blue solution after 5 min at RT.
2.3. Flow cytometry
Cell cycle was studied by staining with 50 μg/ml propidium iodide (PI) and analyzed by flow cytometry. Apoptosis was quantified by Annexin-V staining. Cells were reacted with annexin-V-fluorescein and PI for 10 min at RT following manufacturer's instructions (Immunostep SL, Salamanca, Spain) and then, cells were analyzed. The percentage of death cells corresponds to PI-positive cells and Annexin V/PI double positive cells.
The production of GLUT1 and AR was also studied by flow cytometry. For that, cells were fixed and permeability was performed by incubation in methanol for 10 min at RT. Cells were reacted against anti-GLUT1 and anti-AR at RT for 30 min (antibodies properties are collected in Sup. Table 1) and then, they were stained with rabbit secondary antibodies conjugated with Alexa Fluor 488 dye (Thermo Fisher Scientific, Madrid, Spain) [23]. To detect nuclear AR, cells were suspended in ice cold extraction buffer (320 mM sucrose, 5 mM MgCl2, 10 mM HEPES, 1% Triton X-100, pH 7.4) for 15 min to isolate nuclei. GLUT1 surface was monitored as a function of binding to the envelope glycoprotein of the human lymphotropic virus (HTVL) HTLV was fused to EGFP (HRBDEGFP, Metafora Biosystems, Montpellier, France), and it was added to cells at a concentration 1:10 in PBS with 2%FBS as previously reported [20].
All samples were analyzed in a Cytomics FC500 flow cytometer (Beckman Coulter), and data analysis was performed by using FlowJo and Kaluza software.
2.4. PAGE and immunoblotting
Total, cytoplasmic and nuclear proteins were obtained, separated and electrotransferred as previously described [24]. For total protein extraction, cells were lysed in RIPA lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% SDS, 1% Igepal C and 0.5% sodium deoxycholate) supplemented with 1 mM Dithiothreitol (DTT) and a protease inhibitor cocktail (10 μg/ml leupeptin, 2 μg/ml aprotinin A, 1 μg/ml pepstatin, 200 μM sodium orthovanadate, 1 mM sodium fluoride and 1 mM phenylmethylsulfonyl fluoride). To achieve cytoplasm and nuclear separation, cells were lysed in low-salt buffer (10 mM HEPES-KOH, 2 mM MgCl2, 15 mM NaCl, 0.1 mM EDTA) containing 1 mM DTT, protease inhibitors and 0.2% Igepal C for 20 min at 4 °C. Nuclear fractions were collected by centrifugation at 14,000×g 5 min at 4 °C and supernatants (cytosolic fraction) were transferred to clean tubes. Nuclear pellet was incubated with high-salt lysis buffer (420 mM NaCl, 20 mM HEPES-KOH, 10 mM KCl, 1 mM EDTA and 20% glycerol) supplemented with 1 mM DTT and protease inhibitors. After incubation on ice for 15 min, nuclear extracts were clarified by centrifugation at 13,000×g for 30 min at 4 °C. Protein concentration was estimated using Bradford protein assay (Bio-Rad Laboratories Inc., Madrid, Spain).
Antibodies (Sup. Table 1) were visualized by binding horseradish peroxidase-conjugated anti-rabbit or anti-mouse (Santa Cruz Biotechnology, Dallas, Tx, USA) secondary antibodies and detected with chemiluminescence substrate (Millipore, Merck Chemicals & Life Science SA, Madrid, Spain).
2.5. Glucose and glutamine uptake and glucose measurement in cell culture medium
Glucose and glutamine uptake were assessed as described previously [25], [26]. Briefly, 2DG uptake was initiated by the addition of labeled 2-deoxy-D[1–3H] glucose to a final concentration of 2 μCi (1Ci = 37 GBq). Glutamine uptake was initiated by the addition of labeled L-2,3,4-[3H] glutamine (0.5 μCi). Incubation was performed for 10 min at RT. For standardization, cells were counted using a Neubauer hemocytometer chamber before each experiment. 2.5×105 LNCaP or 1.5×105 PC3 cells were employed in each assay.
2.6. Immunocytochemistry
Cells were fixed in phosphate buffered 2% paraformaldehyde pH 7.4. Samples were blocked by incubation with TBS (Tris-HCl 20 mM pH 7.4, 150 mM NaCl) plus 1% Bovine Serum Albumin (BSA) or 3% goat serum and permeabilization was achieved by incubation with 0.15% Tween-20 (cytoplasmic) or 0.1% TritonX-100 (nuclear) for 20 min at RT. Samples were studied under a under Leica TCS SP8 microscope [19].
2.7. PSA levels
Prostate Specific Antigen (PSA) release was measured by ELISA following manufacturer's instructions (Human Diagnostics, Wiesbaden, Germany). Protein concentration was quantified by Bradford Assay for standardization (Bio-Rad Laboratories Inc, Madrid, Spain).
2.8. ATP/AMP determination by HPLC
Cells were seeded in 100 mm plate and harvested by scrapping at 70% confluence. After centrifugation at 500×g for 5 min 4 °C, cell pellets were dissolved in 0.3 M perchloric acid and incubated 5 min at 4 °C for protein precipitation. Samples were centrifuged at 9000×g for 5 min, and supernatants were neutralized with 1 M KOH. HPLC was performed after centrifugation at 9000×g for 10 min. Adenine nucleotides (ATP, ADP, and AMP) were separated in a 15 cm × 4.6 mm, 3 mm SUPELCOSIL LC-18-T column (SUPELCO, Sigma-Aldrich Quimica SL, Madrid, Spain) with a mobile phase composed of A mobile phase (KH2PO4/tetrabutylammonium hydrogen sulfate –TBHS-, pH 6.0) and B mobile phase (MeOH + TBHS, pH 5.5). Flow rate was set at 1.2 ml in a gradient mode 0 min (0%B), 2.5 min (0%B), 7 min (20%B), 14 min (40%B), 19 min (100%B), 24 min (100%B), 27 min (0%B). Nucleotides were detected by UV absorption at 260 nm and identified and quantified by the comparison of retention time with standards. Protein concentration was quantified by Bradford Assay for standardization (Bio-Rad Laboratories Inc, Madrid, Spain).
2.9. Detection of intracellular ROS levels
Cells were collected by trypsinization and washed twice with PBS. The number of cells was adjusted to 5×105/sample. Total ROS levels were measured incubating cells with 1 μM H2DCFDA (Bioquochem SL, Llanera, Spain) for 15 min at 37 °C. Mitochondrial O2.- was measured by using MitoSOX (Fisher Scientific, Madrid, Spain). Cells were stained with 5 μM MitoSOX at 37 °C for 15 min. Cells were analyzed using a Cytomics FC500 flow cytometer after staining. and data analysis was performed by using Kaluza software.
2.10. H2O2 determination
First, cells were seeded in 6-well plate, and cell culture medium was collected when cells reached at 70% confluence. Then, the medium was centrifuged to eliminate cell debris. An electro-oxidation and amperometric detection were employed for H2O2 determination as previously described [24]. Screen-printed electrodes with co-phthalocyanine were used as an electrochemical mediator (Bioquochem SL, Llanera, Spain). Intensity (mA) was registered for 200 s employing + 0.4 V as work potential. Each sample was measured before and after adding 25 units of catalase. Protein concentration was quantified by Bradford Assay for standardization (Bio-Rad Laboratories Inc, Madrid, Spain).
2.11. SOD activity assay
Cell lysis was accomplished by freeze-thaw cycles in PBS and sonication. Proteins were cleaned by centrifugation (12,000×g). Superoxide dismutase (SOD) activity was performed following manufacturer's instructions (Sigma-Aldrich Quimica SL, Madrid, Spain). Samples were treated with 5 mM potassium cyanide 30 min before the assay to inhibit the activity of cytosolic CuZnSOD (SOD1) in order to quantify mitochondrial SOD2 activity. Protein concentration was quantified by using Bradford Assay for standardization (Bio-Rad Laboratories Inc, Madrid, Spain).
2.12. Native gel for determination of H2O2 depuration by catalase
H2O2 depuration by catalase (CAT) was determined as previously described with minor modifications [27]. Briefly, protein samples were obtained by lysing cells using freeze-thaw cycles in PBS and sonication. Proteins were centrifuged (12,000×g) and quantified by Bradford Assay (Bio-Rad Laboratories Inc, Madrid, Spain). 30 μg of cell extracts were separated on a 6% native acrylamide gel, and CAT was determined by incubating with 0.003% H2O2 for 10 min and then adding 2% FeCl3 and 2% potassium ferrocianurum.
2.13. Thiol and disulfide assay
Thiol and disulfide assay was employed to measure total reduced and oxidized glutathione (GSH and GSSG). Cells were seeded in 6-well plates and collected at 70% confluence. Cells were harvested by scraping, pelleted, and suspended in 50 μl of deproteinization buffer (0.1% Triton X-100 in 5% sulfosalicylic). The assay was performed following manufacturer's instructions (Bioquochem, Llanera, Spain). Cell number was previously adjusted using a Neubauer counting chamber for standardization.
2.14. Statistical analysis
Data are presented as a mean ± standard error of the mean (SEM). Differences were assessed using one-way ANOVA, followed by a Student-Newman-Keuls (SNK) post-hoc ‘t′ test.
3. Results
3.1. Androgen-sensitive PCa cells are more resistant to cell death induced by glucose deprivation than androgen-insensitive cells
Tumor heterogeneity is usually reflected by metabolic differences within cancer cells, including the response to the limited source of nutrients. Here, the response to glucose deprivation of androgen-sensitive and insensitive cells was studied. LNCaP was employed as a model of androgen-sensitive cells. Castration-resistant LNCaP-R, established from LNCaP cells by growing in the absence of androgens for at least 6 months, were used as a model of androgen-insensitive cells.
After glucose deprivation, cells showed morphological features distinctive of cell death (Fig. 1A). Cell death was evaluated by dual staining with fluorescent Annexin V and propidium iodide (PI) after 48 h of glucose removal. The number of Annexin V/PI positive cells are shown in Fig. 1B-C. Androgen-insensitive LNCaP-R showed a higher sensitivity to glucose deprivation compared to androgen-sensitive LNCaP cells.
Fig. 1.

The response of LNCaP cells to glucose deprivation. (A-C) Annexin-V/PI assay of LNCaP and LNCaP-R cells after 48 h since glucose withdrawal. Micrographs (A) and one representative experiment (B) are shown. Original magnification 200×. Results are expressed as % PI-positive cells (dead cells) (C). (D) Trypan Blue results of LNCaP cells grown in absence of glucose and with 10 μM of the inhibitor of caspases q-VD-OPH, 100 μM of the inhibitor of necroptosis necrostatin-1, 25 μM of the inhibitor of RIPK1-independent necroptosis necrostatin-7, 2 μM of the inhibitor of ferroptosis ferrostatin-1 or 2 mM 3-methyladenine (3MA) to inhibit autophagy. DMSO was added as vehicle. (E) Trypan Blue assay was performed in LNCaP cells grown in the absence of glucose, with 10 mM 2DG or under both conditions for 48 h (left panel). Representative micrographs are shown (right panel). Original magnification 200×. (F) Cell counting and viability by trypan blue of LNCaP cells grown with 11 mM (2 g/L) fructose (fru) or mannose (man) without glucose for 48 h. Representative micrographs are shown (right panel). Original magnification 200×. Glucose concentration in control cells was always set at 2 g/L. Arbitrary 1.0 value was given to control cells. Results are expressed as mean ± SEM (n = 3). *p < 0.05 vs control; **p < 0.01 vs control; ***p < 0.001 vs control; #p < 0.05 vs w/o glu; ###p < 0.001 vs w/o glu. ΛΛΛp < 0.001 vs 2DG.
To determine the type of cell death caused by glucose withdrawal in LNCaP cells, several pharmacologic compounds that inhibit different types of cell death were tested in LNCaP cells (Fig. 1D). First, the pan-caspase inhibitor q-VD-OPH was employed, but it was found that it did not prevent cell death suggesting that, though annexin-V staining increased after glucose removal, a type of caspase-independent cell death is occurring. Necrostatin-1, an inhibitor of necroptosis via receptor-interacting serine/threonine-protein kinase 1 (RIPK1), and necrostatin-7, an inhibitor of RIPK1-independent necroptosis, did not prevent cell death. Ferrostatin-1, an inhibitor of ferroptosis, and 3-methyladenine, an inhibitor of autophagy, neither avoided cell death. Interestingly, blocking autophagy by 3MA, cell death was enhanced under the absence of glucose. These results suggested that glucose deprivation induces a non-apoptotic form of cell death as necrosis.
Once confirmed that cell death is driven by necrosis, it was studied whether this response might have been dependent on glucose availability or it was a general response to glycolysis interruption. Then, 2DG, an analog of glucose that does not follow glycolysis, was assayed in the presence of glucose. Fig. 1E shows that 2DG did not induce death in LNCaP cells. This result implies that the availability of glucose, and not a metabolite derived from glycolysis, is crucial for PCa survival.
Since glucose absence, but not the inhibition of glycolysis is responsible for cell death in LNCaP cells, it was studied whether another sugar source such as fructose or mannose would be able to prevent cell death. Cells cultivated in the presence of fructose and mannose did not die and still grew even under glucose deprivation (Fig. 1F), suggesting that it is sugar deprivation, the responsible for cell death.
In addition to glucose, glutamine is one of the essential nutrients of PCa cells. First, the balance in the uptake of glutamine in the absence of glucose was studied. After glucose removal, an increase in glutamine uptake was not found in LNCaP cells, and it was significantly reduced in LNCaP-R cells (Supplementary Fig. S2A), suggesting that the absence of glucose did not affect to glutamine uptake. Moreover, the removal of glutamine did not have any significant relevance in cell survival, since cells did not die in the absence of glutamine when glucose is present (Supplementary Fig. S2B).
3.2. Glucose withdrawal upregulates GLUT1 in androgen-sensitive prostate cancer cells
Since glucose uptake seemed to be essential for the survival of PCa cells, the role of glucose transportation in cell survival was studied. First, the rate of 2DG, an analog of glucose that enters the cell by using GLUT transporters and accumulates inside cells was examined. As shown, LNCaP cells significantly increased 2DG uptake after glucose removal (Fig. 2A) proposing an increment of GLUT transporters.
Fig. 2.

The regulation of GLUT1 under glucose deprivation in LNCaP cells. (A) 2DG uptake levels were evaluated in LNCaP and LNCaP-R cells grown in absence of glucose for 24 h. (B) GLUT1 protein expression was determined by western-blot under the same conditions. (C) GLUT1 surface expression was studied by flow cytometry. Levels were measured after 24 h since medium renewal. Jurkat cells were employed as a positive control. One representative experiment is shown. (D) TXNIP, phosphoAMPKThr172 and AMPK protein levels were also analyzed by western-blot in cells grown without glucose for 24 h. Glucose concentration in cell culture medium was always set at 2 g/L for control cells. In western-blot experiments, beta-actin was used as internal standard and one representative experiment is shown. Results are expressed as mean ± SEM (n = 3). *p < 0.05 vs control; **p < 0.01 vs control; ***p < 0.001 vs control.
Since GLUT1 is the major transporter of glucose in PCa cells, we investigated whether an increment of total protein levels or an increase in the surface location of GLUT1 occurred after glucose removal. GLUT1 production increased in androgen-sensitive LNCaP cells, but it was significantly down-regulated by glucose deprivation in androgen-insensitive LNCaP-R cells (Fig. 2B). To confirm if the uptake increment was related to surface GLUT1 levels, FACS analysis was performed. Interestingly, glucose deprivation increased total surface GLUT1 in both LNCaP and LNCaP-R cells (Fig. 2C).
The increment of GLUT1 production in androgen-sensitive LNCaP cells suggested the participation of androgen signaling in the mechanism of cell response to glucose deprivation. To confirm a possible role of androgens in response to glucose deprivation, GLUT1 protein levels were studied in LNCaP cells when they were cultured after glucose removal for 24 h in a steroid-depleted medium FBSchst. Interestingly, GLUT1 levels decreased by half in LNCaP cells cultured in androgen-depleted media, and they did not increase after glucose deprivation contrary to the findings observed in complete media in the presence of androgens (Supplementary Fig. S3). These results suggested that androgens have to be present to increase GLUT1 after glucose removal.
It has been previously described that AMP-activated protein kinase (AMPK) regulates production and membrane location of GLUT1 and it is activated by androgens. To explore whether AMPK would be related to the increment of GLUT1 production and membrane translocation after glucose withdrawal in androgen-sensitive cells, the phosphorylation of AMPK was studied in both androgen-sensitive and insensitive PCa cells. Phospho-AMPKThr172 levels were increased under glucose deprivation in androgen-sensitive LNCaP cells, but this increase was not significant in castration-resistant LNCaP-R cells (Fig. 2D). The production of Thioredoxin Interacting Protein (TXNIP), an AMPK-regulatory inhibitor described as a possible link between cellular redox state and metabolism, was also studied. Interestingly, TXNIP decreased under glucose deprivation in both cell types, being this reduction was much more significant in androgen-sensitive LNCaP cells.
3.3. Glucose removal increases AR activity
Since GLUT1 increased after glucose removal only in the presence of androgens, we assessed the role AR signaling on GLUT1 production. It was first found that glucose removal promoted an increment of AR mainly restricted to the nuclear compartment when observed under confocal microscopy (Fig. 3A) or analyzed by western-blot analysis (Fig. 3B). As expected, nuclear translocation was associated with a higher AR activity measured via the release of PSA (Fig. 3C).
Fig. 3.

The effect of glucose deprivation in AR signaling. (A) Immunocytochemical analysis of AR in LNCaP cells glucose-deprived for 24 h. Cells were incubated with a secondary antibody conjugated with phycoerythrin (red fluorescence) and then counterstained with DAPI (blue fluorescence). A representative picture is shown. (B) Nuclear (Nuc) and cytoplasmic (Cyto) AR protein levels were determined by western-blot using the same experimental conditions. beta-actin was used as cytoplasmic internal standard while HDAC2 was used as a nuclear internal standard. Nuclear/cytoplasmic AR ratio was determined. One representative experiment is shown. The experiment was performed 3 times (C) PSA levels in culture medium without glucose were determined by ELISA measurement. Results were standardized to protein concentration (ng PSA/μg protein). (D) GLUT1, total AR and nuclear AR protein production along cell cycle. Cells were incubated with a secondary antibody conjugated with AlexaFluor488 and then with Propidium iodide (PI) are shown in lower panels. Negative controls (C-), unstained cells, are shown in upper panels. Fluorescence was measured by flow cytometry. One representative of three different experiments is shown. Glucose concentration was always set at 2 g/L in control cells. Arbitrary 1.0 value was given to control cells and results are expressed as mean ± SEM (n = 3) in all experiments. ***p < 0.001 vs control.
Androgens and AR signaling are necessary for prostate maintenance and homeostasis, and they are the principal regulators of cancer cells proliferation in the prostate. Since AR activity was increased after glucose deprivation, GLUT1 and AR protein abundance across cell cycle were studied by flow cytometry (Fig. 3C). As expected total and nuclear AR increased in S and G2/M phases in LNCaP cells, but more importantly it correlated with GLUT1 because it is also enhanced in S and G2/M phases. These results suggested a concomitant overexpression of AR and GLUT1 along with cell cycle in androgen-sensitive prostate cancer cells.
3.4. Cell death driven by glucose deprivation is dependent on oxidative stress in PCa cells
Necrosis cell death is usually associated with a decrease of ATP production and an increase of free radicals. For this reason, the role of redox signaling in cell death caused by nutrients deprivation was considered. First, glucose deprivation significantly reduced ATP/AMP ratio as expected (Fig. 4A) and it also increased H2O2 production (Fig. 4B) and mitochondrial superoxide (Fig. 4C). To confirm that an oxidative pathway contributed to cell death induced by glucose deprivation in LNCaP cells, N-acetyl-L-cysteine (NAC), 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid (TROLOX), dehydroascorbic acid (DHA) or catalase (CAT) were added to cell culture medium after glucose removal. As shown in Fig. 4D, the incubation with NAC or catalase, significantly prevented cell death triggered by glucose deprivation. However, DHA or TROLOX did not have any effect. These results corroborate the involvement of oxidative pathways in nutrients deprivation induced cell death of androgen-sensitive prostate cancer cells.
Fig. 4.

The effect of glucose deprivation on redox balance. (A) ATP/AMP ratio was determined by measuring ATP and AMP levels by HPLC in LNCaP after 24 h since glucose withdrawal. (B) H2O2 was measured in cell culture medium (left panel) and (C) mitochondrial superoxide was determined by MitoSOX Red® staining (right panel) in glucose-deprived LNCaP cells after 24 h since medium renewal. (D) Trypan Blue results (left panel) and representative micrographs (right panel) of LNCaP grown in absence of glucose for 48 h and with 5 mM N-acetylcysteine (NAC), 100 μM dehydroascorbic acid (DHA), 100 μM Trolox or 50 U/ml catalase (CAT). DMSO was added as vehicle. Original magnification 200×. (E) Nuclear (Nuc) and cytoplasmic (Cyto) AR protein levels were determined by western-blot in LNCaP treated with 10 μM H2O2 for 24 h. Beta-actin was used as cytoplasmic internal standard while HDAC2 was used as a nuclear internal standard. Following the same conditions, phosphoAMPKThr172, AMPK and GLUT1 were also analyzed by western-blot. Beta-actin was used as internal standard. One representative experiment is shown. Glucose concentration in control cells was always set at 2 g/L. Arbitrary 1.0 value was given to untreated cells. Results are expressed as mean ± SEM (n = 3). *p < 0.05 vs control; ***p < 0.001 vs control; ###p < 0.001 vs. w/o glu.
Given that glucose deprivation caused the upregulation of GLUT1 in LNCaP cells, the relation between free radicals and GLUT1 production in PCa cells was studied. LNCaP cells were incubated with 10 µM H2O2 for 24 h. After treatment, translocation of AR, phosphorylation of AMPK and GLUT1 production was investigated. H2O2 increased nuclear AR translocation as well as the activation of phosphor-AMPKThr172 and, and then, it elevated GLUT1 production (Fig. 4E). Since after glucose removal H2O2 increased, H2O2 might be responsible for AR activation and in turn, of the increment of GLUT1 after glucose withdrawal.
3.5. GLUT1 overexpression protects from glucose deprivation-induced cell death in LNCaP cells
To assess the role of GLUT1 in cell death after glucose deprivation in LNCaP cells, the sensitivity to glucose removal and free radicals after the overexpression of GLUT1 in LNCaP cells was studied.
We studied the redox phenotype of GLUT1-overexpressing LNCaP cells since they should consume more glucose and then, they should have a more oxidative phenotype. Interestingly GLUT1 overexpression did not cause any increment of free radicals in LNCaP cells (Supplementary Fig. S4A, B). Moreover, it did not increase H2O2 or mitochondrial superoxide after glucose removal in LNCaP cells (Supplementary Fig. S4C, D).
LNCaP cells overexpressing GLUT1 showed a lower sensitivity to glucose removal then LNCaP cells, as showed in Fig. 5A. Moreover, GLUT1 overexpression protects cells from H2O2 toxicity. Thus, cells were incubated with 15 µM H2O2 for 48 h, LNCaPGLUT1 cells were more resistant to H2O2 toxicity than LNCaP cells (Fig. 5B).
Fig. 5.

The effect of GLUT1 overexpression in glucose-deprived LNCaP cells. LNCaPMock and LNCaPGLUT1 cells were employed for all experiments. (A) Representative micrographs after 48 h since glucose withdrawal (left panel). Original magnification 200×. Cell death was analyzed by PI staining. Results are expressed as % PI-positive cells (dead cells). Glucose concentration in control cells was always set at 2 g/L. Arbitrary 1.0 value was given to control cells. Results are expressed as mean ± SEM (n = 3). (B) Trypan Blue assay in cells treated with 15 μM H2O2 for 48 h (right panel). Representative micrographs are shown (left panel). Original magnification 200×. (C) Representative micrographs (left panel) and Trypan Blue assays of cells treated with 100 μM phl in cell culture medium without glucose for 48 h. *p < 0.05 vs control; **p < 0.01 vs control cells; ***p < 0.001 vs control cells; #p < 0.05 vs LNCaPMock cells; ###p < 0.001 vs LNCaPMock cells.
To confirm whether GLUT1 protection is due to an extracellular signal uptaken by the transporter or an intracellular signaling pathway promoted by GLUT1 overexpression, GLUT transporters were blocked by using phloretin, a natural product that has been used as a specific inhibitor of GLUT1 [19]. By blocking GLUT1 with phloretin cell death was enhanced in LNCaP and LNCaPGLUT1 cells after glucose deprivation (Fig. 5C). Furthermore, LNCaP and LNCaPGLUT1 cells were grown in low serum media to confirm whether a component of serum would participate in the role of glucose transporters in survival (Supplementary Fig. S5). Interestingly, as previously demonstrated, GLUT1 protected from cell death caused by depletion of growing factors induced by cultivation with 1% FBS even in the presence of glucose. However, under glucose removal in low serum incubation, ectopic overexpression of GLUT1 did no protect androgen-dependent prostate cancer cells.
3.6. GSH levels are stimulated in GLUT1-overexpressing LNCaP cells after glucose removal
Since the increment of GLUT1 makes cells more resistant to glucose deprivation and oxidative stress, the role of antioxidant pathways in survival after ectopic overexpression of GLUT1 was investigated.
First, total protein levels of the antioxidant enzymes SOD1, SOD2, CAT, glutathione peroxidase 1 (GPX1) and thioredoxin 1 (TRX1) were studied in LNCaP and LNCaPGLUT1 cells. After glucose removal, SOD2 protein levels diminished significantly, and its levels recover to controls by GLUT1 overexpression. Also, GPX protein production was increased in both LNCaP and LNCaPGLUT1 cells (Fig. 6A). However, no differences in SOD/SOD2 activity was found (Fig. 6C) and CAT activity increased in both LNCaP and LNCaPGLUT1 cells (Fig. 6A, C). Of all parameters investigated, GLUT1 overexpression significantly increased reducing power of cells by increasing GSH/GSSG ratio in LNCaPGLUT1 cells after glucose removal (Fig. 6D). This suggests that overexpression of GLUT1 might protect from glucose deprivation by increasing the reducing power inside cells.
Fig. 6.

The role of GLUT1 overexpression in the response of glucose deprivation-induced oxidative stress in LNCaP cells. LNCaPMock and LNCaPGLUT1 cells were employed for all experiments. Cell were glucose-starved for 24 h. (A) SOD1, SOD2, CAT, GPX1 and TRX1 protein production were analyzed by western-blot. Beta-actin was employed as internal standard and one representative experiment is shown. Only proteins with significant differences are graphically represented. (B) Total SOD and SOD2 activity were determined by enzymatic assay. (C) H2O2 depuration by CAT was determined by native acrylamide gel. One representative experiment is shown. (D) GSH/GSSG ratio was determined by enzymatic assay. Glucose concentration in control cells was set at 2 g/L. An arbitrary value of 1.0 was given to control cells. Results are expressed as mean ± SEM (n = 3). *p < 0.05 vs control cells; ***p < 0.001 vs control cells; #p < 0.05 vs LNCaPMock cells; ###p < 0.001 vs LNCaPMock cells.
3.7. GLUT1 also prevents cell death induced by glucose deprivation androgen-insensitive prostate cancer cells
To support that GLUT1 overexpression by itself has a role in the survival of PCa cells, we studied the answer to glucose deprivation of androgen-insensitive PC-3 cells, which do not produce AR or respond to androgens. First, it was found that PC-3 cells died after glucose deprivation (Fig. 7A). This effect was not dependent on glycolysis disruption since 2DG treatment did not alter cell viability, likewise androgen-sensitive LNCaP cells (Fig. 7B). Also, and also parallel to LNCaP, fructose and mannose supplementation in cell culture medium prevented cell death caused by glucose deprivation in PC-3 cells (Fig. 7C). However, contrary to LNCaP cells, GLUT1 protein levels did not increase in glucose-deprived cells (Fig. 7D). The fact that would confirm the necessity of an active form of AR to achieve an increment of GLUT1 production after glucose removal.
Fig. 7.

The response of PC-3 cells to glucose deprivation. (A) Annexin-V/PI assay of PC-3 cells after 48 h since glucose withdrawal. Micrographs and one representative experiment are shown (left panel). Original magnification 200×. Results are expressed as % PI-positive cells (dead cells). (A) Trypan Blue results of PC-3 cells grown in the absence of glucose, with 10 mM 2DG or under both conditions for 48 h (left panel). Representative micrographs are shown (right panel). Original magnification 200×. (B) Cell counting and viability by trypan blue of LNCaP and PC-3 cells grown with 11 mM (2 g/L) fructose (fru) or mannose (man) without glucose for 48 h. Representative micrographs are shown (right panel). Original magnification 200×. (D) GLUT1 protein levels were determined by western-blot in PC-3 cells grown without glucose for 24 h. beta-actin was used as internal standard and one representative experiment is shown. Glucose concentration in control cells was always set at 2 g/L. Arbitrary 1.0 value was given to control cells. Results are expressed as mean ± SEM (n = 3). *p < 0.05 vs control; ***p < 0.001 vs control; ###p < 0.001 vs w/o glu. ΛΛΛp < 0.001 vs 2DG.
When we ectopically overexpressed GLUT1 in PC-3 cells, the death induced by glucose deprivation (Fig. 8A) and the toxicity of H2O2 were also prevented. Besides, glucose removal also MitoSOX® staining in PC-3 cells confirming the increase of oxidative stress (data not shown).
Fig. 8.

The protective effect of GLUT1 overexpression in androgen-insensitive PC-3 cells. PC-3Mock and PC-3GLUT1 cells were employed for all experiments. (A) Representative micrographs of cells grown without glucose for 48 h are shown (original magnification 200×) (left panel). The percentage of death cells (PI positive) is compared (n = 3). (B) Trypan Blue assay in cells treated with 10 mM H2O2 for 48 h. Representative micrographs are shown (original magnification 200×) (right panel). (C) SOD2 and GPX1 protein production were analyzed by western-blot. Beta-actin was employed as internal standard and one representative experiment is shown. (D) Total SOD and SOD2 activity were determined by enzymatic assay. (E) H2O2 depuration by CAT was determined by native acrylamide gel. One representative experiment is shown. (F) GSH/GSSG ratio were determined by enzymatic assay. Glucose concentration in control cells was set at 2 g/L. An arbitrary value of 1.0 was given to control cells. Results are expressed as mean ± SEM (n = 3). ***p < 0.001 vs control cells; ###p < PC-3Mock cells.
In PC-3, after glucose removal there was an increased in GPX1 or SOD2 production in both native or overexpressing GLUT1 cells and a significant decrease in SOD2 activity was also found in PC-3 after glucose removal. Contrary to LNCaP, androgen insensitive PC-3 and PC-3GLUT1 cells showed a reduction of catalase activity after glucose removal.
Finally, after glucose deprivation, there was a significant reduction in GSH/GSSG balance in PC-3 cells that was recovered by GLUT1 overexpression (Fig. 8F).
Collectively, these data indicate that GLUT1 overexpression withstands glucose deprivation by a higher resistance to oxidative stress, events that might be directly related to a higher resistance to cell death.
4. Discussion
Cancer cells are characterized by an increase in glucose uptake and glycolysis. Therefore, targeting glucose metabolism is suggested as a promising approach in oncology. In this report, it is described for the first time that GLUT1 overproduction protects cells from cell death caused by glucose deprivation by an antioxidant mediated mechanism.
Glucose concentration inside the tumor core, away from blood vessels, vary from 0.25 to 2.5 mM [28], lower than in normal tissues. Together with hypoxia, glucose deprivation causes tumor cells to adapt their metabolism. Results shown here confirmed that cell death triggered by glucose deprivation does not follow the classical apoptotic response through caspase activation, necroptosis, ferroptosis or autophagy. We proved as previously demonstrated in other cell types that glucose removal activates necrosis [5]. Interestingly, we found that 2DG does not induce cell death in PCa cells, through glycolysis is not active. It would be possible that the treatment with 2DG could be promoting survival from autophagy in PCa, as it was previously described [29], [30].
It is well known that glucose deprivation cytotoxicity is mediated by oxidative stress in other cell types [6]. Some studies proposed that ROS induced by glucose deprivation are due to a promotion of mitochondrial metabolism in detriment of glycolysis [31], [32]. However, in some cell lines, cell death cannot be prevented with antioxidants [33], [34]. Here, glucose starvation increases both, H2O2 and mitochondrial superoxide and antioxidants such as NAC or catalase can prevent cell death. NAC maintains reduced glutathione levels and catalase depurates H2O2. However, DHA does not prevent cell death in PCa, and this might be due to the cost in GSH since GSH is required by cells to regenerate ascorbic acid [35].
Although glucose metabolism has not been considered as important as lipid or protein metabolism in PCa, it has been confirmed that glucose is essential for cell proliferation and survival [36]. In addition to glucose, glutamine is necessary for cell growth and proliferation of PCa cells [37], but surprisingly LNCaP cells do not die after glutamine deprivation. Interestingly, other sugars like fructose or mannose, also internalized by GLUT transporters, prevent cell death and diminish proliferation under glucose withdrawal perhaps because they replace glucose at any other point in the metabolic network.
Glucose metabolism in PCa is different when compared to other carcinomas. In non-pathological tissue, the prostate gland is primarily glycolytic because of a defect in tricarboxylic acid (TCA) cycle [38]. However, at the beginning of carcinogenesis, the gland becomes OXPHOS-dependent and then, in more aggressive stages, tumors become again to turn to glycolysis [39]. Therefore, resistance to glucose deprivation in PCa cells is different at the beginning or later stages of the disease. In fact, castration-resistant LNCaP-R cells are more sensitive to glucose starvation than parental LNCaP cells, indicating their higher dependence on glucose [40]. It was previously reported that cells deficient in upregulating OXPHOS are more sensitive to glucose deprivation [41], which it may be related to our results.
In the absence of glucose, cells usually overexpress an isoform of GLUT transporters [12]. Here it is shown that PCa cells with functional AR increase the levels of GLUT1 after glucose deprivation. It was previously described that GLUT1 regulation is dependent on AR activity [42]. Throughout carcinogenesis, the expression of this transporter is also differentially regulated. The healthy prostate produces GLUT1, decreasing its levels at early stages during tumor progression [43]. However, in most aggressive tumors, GLUT1 is found overexpressed, concomitant with a higher glycolytic activity and hypoxia [17].
Interestingly, when glucose is deleted in PCa cells, GLUT1 is increased. Also, AR is almost entirely located in the nucleus, correlating with the increment in GLUT1 levels. These results are in agreement with those reported by Vaz et al. [16], They described an increase in GLUT1 after DHT stimulation [16]. Furthermore, glucose deprivation fails to enhance GLUT1 expression when cells are grown in the absence of androgens. Hyperglycemia downregulates AR levels in Type 2 diabetes. Interestingly, diabetes type 2 is inversely related to prostate cancer incidence [44]. On the other hand, low glucose stimulates GLUT1 production, which might be concerned with the promotion of more aggressive tumors. Nuclear AR translocation, and consequently GLUT1 enhancement, are directly associated with cell proliferation. Since both proteins are found during S and G2/M cell cycle phases, they could act synergistically to promote cell proliferation under androgenic stimulation or suppressing cell cycle arrest in the absence of glucose. On the contrary, it is known that glycolysis only affects cell cycle distribution from G1 to S [45].
In PCa, AMPK is considered a key metabolic regulator involved in proliferation and cell survival [46]. In PCa, androgens stimulate AMPK activation leading to OXPHOS [15]. AMPK is also able to induce GLUT1 expression and membrane translocation by inhibiting TXNIP [47], indicating that GLUT1 overexpression via AMPK can protect from cell death caused by glucose deprivation. More interestingly, H2O2 stimulates glucose uptake in cells overexpressing GLUT1 [48]. Accordingly, with these results, H2O2 increases nuclear AR levels and GLUT1 production via AMPK in LNCaP cells. The protective activation of AMPK by H2O2 has been already described, and recently it was proposed as a regulator of mitochondrial ROS [49], [50].
In PCa cells that overexpress GLUT1, as expected, glucose consumption is higher than in parental cells. Recent studies showed that the overexpression of GLUT1 drives with an inflammatory response because of the increase of ROS in macrophages due to a higher glucose uptake [51], [52]. Here it is shown that oxidative stress is enhanced in GLUT1-overexpressing cells in the presence of glucose, but not under glucose deprivation.
To determine the mechanism by which GLUT1 protects cells from glucose removal, and given the oxidative signal caused by glucose removal, we analyzed several antioxidant pathways. The activity of mitochondrial SOD2, that fall after glucose removal in LNCaP cells, is sustained under glucose deprivation in GLUT1-overexpressing cells. Recently, it was described a role of this enzyme in the survival of renal carcinoma cells, as well as, in the promotion of cell proliferation of lung cancer cells via AMPK [53], [54].
Mainly GLUT1 overexpression increases reduced levels of glutathione. Since pentose phosphate pathway is not overstimulated after glucose withdrawal, the necessary NADPH to maintain GSH has to come from the mitochondria mainly. It was recently published that nicotinamide phosphoribosyltransferase (NAMPT), involved in NAD+ biosynthesis, protects cells from glucose deprivation-induced oxidative stress and that it is also regulated by AMPK [55], which confirms the role of mitochondria as the source of reducing power.
The protective role of GLUT1 in PCa cells may come from the uptake of some compound included in culture medium or by the activation/inhibition of an intracellular signaling pathway. Besides glucose uptake, it is well-known that GLUT1 can transport other compounds. The treatment with phloretin, which is well-established as a GLUT blocker [56] enhances cell death by glucose deprivation, implying that this response is not only a consequence of intracellular signaling. The increase of cell death by serum deprivation strengthens this idea. Moreover, it was recently described that ROS production is enhanced in L6 myoblasts when GLUT1 is inhibited by phloretin, which it is in agreement with our results [57].
5. Conclusions
GLUT1 acts as an oncoprotein in several tumors. However, it does not only promote proliferation because of increasing glucose uptake; it has a significant protective role against nutrients deprivation. Glucose starvation rises oxidative stress that in turn activates AR activity and, as a consequence, AMPK signaling and GLUT1 production. After GLUT1 increment, GSH levels rise protecting PCa cells from cell death. In PCa, GLUT1 is usually overexpressed in highly aggressive tumors so that it might protect cancer cells from low glucose microenvironments. Altogether, these results show the importance of glucose availability and glucose transporter for PCa survival and its relation to redox signaling.
Acknowledgements
We thank Marta Alonso-Guervos for her assistance with confocal microscopy. PGM thanks to European Association for Cancer Research for EACR travel fellowship. We also thank Dr. Cristina Muñoz-Pinedo for her help and advice in cell death studies.
Acknowledgments
Declaration of interest
The authors have nothing to disclose.
Funding
PGM thanks support to “Formación del Personal Universitario” grant from Ministerio de Educación, Cultura y Deporte, Gobierno de España (AP2012-4924) and IUOPA. DH is supported by Fundacion ONCE, SK by CNRS and NT by INSERM. This work was supported by Ministerio de Economía y Competitividad, Gobierno de España, co-funded by FEDER (MINECO-17-BFU2016-79139-R).
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2018.03.017.
Contributor Information
Juan C. Mayo, Email: mayojuan@uniovi.es.
Rosa M. Sainz, Email: sainzrosa@uniovi.es.
Appendix A. Supplementary material
Fig. S1.

AR production in LNCaP-R cells and GLUT1 production in LNCaPGLUT1and PC-3GLUT1cells. (A) AR protein production was determined by western-blot in LNCaP and LNCaP-R cells. (B) GLUT1 protein levels were determined in LNCaPMock, LNCaPGLUT1, PC-3Mock and PC-3GLUT1 cells. Beta-actin was employed as internal standard and one representative experiment is shown.
Fig. S2.

Glutamine deprivation in prostate cancer cells. (A) Glutamine uptake in LNCaP and LNCaP-R cells grown in absence of glucose for 24 h. An arbitrary value of 1.0 was given to control cells. (B) Representative micrographs of LNCaP cells under glucose or glutamine deprivation for 72 h. Original magnification 200x. Glucose concentration was always set at 2 g/L in control cells. Results are expressed as mean ± SEM (n = 3). ***p < 0.001 vs control; ##p < 0.01 vs LNCaP.
Fig. S3.

GLUT1 levels in androgen-deprived LNCaP cells. GLUT1 protein levels were analyzed by western-blot in LNCaP cells grown in absence of glucose in medium with FBSchst. LNCaP cells were previously grown 48 h in medium with FBSchst. An arbitrary value of 1.0 was given to control cells. Glucose concentration was always set at 2 g/L in control cells. Results are expressed as mean ± SEM (n = 3). Beta-actin was employed as internal standard and one representative experiment is shown. **p < 0.01,***p < 0.05.
Fig. S4.

ROS generation in LNCaPGLUT1cells after glucose withdrawal. LNCaPMock and LNCaPGLUT1 cells were employed for all experiments. (A) Determination of mitochondrial superoxide levels by MitoSOX Red® staining, and (B) H2O2 measurement in cell culture medium. (C) Determination of mitochondrial superoxide levels by MitoSOX Red® staining, and (D) H2O2 measurement in cell culture medium in glucose-starved cells for 24 h. Glucose concentration in control cells was always set at 2 g/L. Arbitrary 1.0 value was given to control cells. Results are expressed as mean ± SEM (n = 3). *p < 0.05 vs control cells; ***p < 0.001 vs control cells; ###p < 0.001 vs LNCaPMock cells.
Fig. S5.

Effect of serum deprivation in LNCaP cells. Representative micrographs (above panel) and Trypan Blue Assays of LNCaPMock and LNCaPGLUT1 cells grown in cell culture medium with 1% FBS and without glucose for 48 h. Glucose concentration was always set at 2 g/L in control cells. Results are expressed as mean ± SEM (n = 3). ***p < 0.001 vs control cells; ###p < 0.001 vs parental cells; ΛΛΛp < 0.001 vs LNCaP cells cultured with 1% FBS.
Supplementary material
References
- 1.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Pavlova N.N., Thompson C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muñoz-Pinedo C., El Mjiyad N., Ricci J.-E. Cancer metabolism: current perspectives and future directions. Cell Death Dis. 2012;3:e248. doi: 10.1038/cddis.2011.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.El Mjiyad N., Caro-Maldonado A., Ramírez-Peinado S., Muñoz-Pinedo C. Sugar-free approaches to cancer cell killing. Oncogene. 2011;30:253–264. doi: 10.1038/onc.2010.466. [DOI] [PubMed] [Google Scholar]
- 6.Liu Y., Song X.-D., Liu W., Zhang T.-Y., Zuo J. Glucose deprivation induces mitochondrial dysfunction and oxidative stress in PC12 cell line. J. Cell. Mol. Med. 2003;7:49–56. doi: 10.1111/j.1582-4934.2003.tb00202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Macheda M.L., Rogers S., Best J.D. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J. Cell. Physiol. 2005;202:654–662. doi: 10.1002/jcp.20166. [DOI] [PubMed] [Google Scholar]
- 8.Barron C.C., Bilan P.J., Tsakiridis T., Tsiani E. Facilitative glucose transporters: implications for cancer detection, prognosis and treatment. Metabolism. 2016;65:124–139. doi: 10.1016/j.metabol.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 9.Bryant N.J., Govers R., James D.E. Regulated transport of the glucose transporter GLUT4. Nat. Rev. Mol. Cell Biol. 2002;3:267–277. doi: 10.1038/nrm782. [DOI] [PubMed] [Google Scholar]
- 10.Chavez J.A., Roach W.G., Keller S.R., Lane W.S., Lienhard G.E. Inhibition of GLUT4 translocation by Tbc1d1, a Rab GTPase-activating protein abundant in skeletal muscle, is partially relieved by AMP-activated protein kinase activation. J. Biol. Chem. 2008;283:9187–9195. doi: 10.1074/jbc.M708934200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Saedeleer C.J., Porporato P.E., Copetti T., Pérez-Escuredo J., Payen V.L., Brisson L., Feron O., Sonveaux P. Glucose deprivation increases monocarboxylate transporter 1 (MCT1) expression and MCT1-dependent tumor cell migration. Oncogene. 2014;33:4060–4068. doi: 10.1038/onc.2013.454. [DOI] [PubMed] [Google Scholar]
- 12.Hu J., Locasale J.W., Bielas J.H., O’Sullivan J., Sheahan K., Cantley L.C., Vander Heiden M.G., Vitkup D. Heterogeneity of tumor-induced gene expression changes in the human metabolic network. Nat. Biotechnol. 2013;31:522–529. doi: 10.1038/nbt.2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rathmell J.C., Fox C.J., Plas D.R., Hammerman P.S., Cinalli R.M., Thompson C.B. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol. Cell. Biol. 2003;23:7315–7328. doi: 10.1128/MCB.23.20.7315-7328.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gonzalez-Menendez P., Hevia D., Mayo J.C., Sainz R.M. The dark side of glucose transporters in prostate cancer: are they a new feature to characterize carcinomas? Int. J. Cancer. 2017 doi: 10.1002/ijc.31165. [DOI] [PubMed] [Google Scholar]
- 15.Tennakoon J.B., Shi Y., Han J.J., Tsouko E., White M.A., Burns A.R., Zhang A., Xia X., Ilkayeva O.R., Xin L., Ittmann M.M., Rick F.G., Schally A.V., Frigo D.E. Androgens regulate prostate cancer cell growth via an AMPK-PGC-1α-mediated metabolic switch. Oncogene. 2014;33:5251–5261. doi: 10.1038/onc.2013.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vaz C.V., Marques R., Alves M.G., Oliveira P.F., Cavaco J.E., Maia C.J., Socorro S. Androgens enhance the glycolytic metabolism and lactate export in prostate cancer cells by modulating the expression of GLUT1, GLUT3, PFK, LDH and MCT4 genes. J. Cancer Res. Clin. Oncol. 2016;142:5–16. doi: 10.1007/s00432-015-1992-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stewart G.D., Gray K., Pennington C.J., Edwards D.R., Riddick A.C.P., Ross J.A., Habib F.K. Analysis of hypoxia-associated gene expression in prostate cancer: lysyl oxidase and glucose transporter-1 expression correlate with Gleason score. Oncol. Rep. 2008;20:1561–1567. [PubMed] [Google Scholar]
- 18.Xiao H., Wang J., Yan W., Cui Y., Chen Z., Gao X., Wen X., Chen J. GLUT1 regulates cell glycolysis and proliferation in prostate cancer. Prostate. 2018;78:86–94. doi: 10.1002/pros.23448. [DOI] [PubMed] [Google Scholar]
- 19.Gonzalez-Menendez P., Hevia D., Rodriguez-Garcia A., Mayo J.C., Sainz R.M. Regulation of GLUT transporters by flavonoids in androgen-sensitive and-insensitive prostate cancer cells. Endocrinology. 2014;155:3238–3250. doi: 10.1210/en.2014-1260. [DOI] [PubMed] [Google Scholar]
- 20.Manel N., Kim F.J., Kinet S., Taylor N., Sitbon M., Battini J.-L. The ubiquitous glucose transporter GLUT-1 is a receptor for HTLV. Cell. 2003;115:449–459. doi: 10.1016/s0092-8674(03)00881-x. [DOI] [PubMed] [Google Scholar]
- 21.Rodriguez-Garcia A., Mayo J.C., Hevia D., Quiros-Gonzalez I., Navarro M., Sainz R.M. Phenotypic changes caused by melatonin increased sensitivity of prostate cancer cells to cytokine-induced apoptosis. J. Pineal Res. 2013;54:33–45. doi: 10.1111/j.1600-079X.2012.01017.x. [DOI] [PubMed] [Google Scholar]
- 22.Takanaga H., Frommer W.B. Facilitative plasma membrane transporters function during ER transit. FASEB J. 2010;24:2849–2858. doi: 10.1096/fj.09-146472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosner M., Schipany K., Hengstschläger M. Merging high-quality biochemical fractionation with a refined flow cytometry approach to monitor nucleocytoplasmic protein expression throughout the unperturbed mammalian cell cycle. Nat. Protoc. 2013;8:602–626. doi: 10.1038/nprot.2013.011. [DOI] [PubMed] [Google Scholar]
- 24.Quiros-Gonzalez I., Sainz R.M., Hevia D., Mayo J.C. MnSOD drives neuroendocrine differentiation, androgen independence, and cell survival in prostate cancer cells. Free Radic. Biol. Med. 2011;50:525–536. doi: 10.1016/j.freeradbiomed.2010.10.715. [DOI] [PubMed] [Google Scholar]
- 25.Montel-Hagen A., Kinet S., Manel N., Mongellaz C., Prohaska R., Battini J.-L., Delaunay J., Sitbon M., Taylor N. Erythrocyte Glut1 triggers dehydroascorbic acid uptake in mammals unable to synthesize vitamin C. Cell. 2008;132:1039–1048. doi: 10.1016/j.cell.2008.01.042. [DOI] [PubMed] [Google Scholar]
- 26.Klysz D., Tai X., Robert P.A., Craveiro M., Cretenet G., Oburoglu L., Mongellaz C., Floess S., Fritz V., Matias M.I., Yong C., Surh N., Marie J.C., Huehn J., Zimmermann V., Kinet S., Dardalhon V., Taylor N. Glutamine-dependent α-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci. Signal. 2015;8 doi: 10.1126/scisignal.aab2610. [DOI] [PubMed] [Google Scholar]
- 27.Weydert C.J., Cullen J.J. Measurement of superoxide dismutase, catalase and glutathione peroxidase in cultured cells and tissue. Nat. Protoc. 2010;5:51–66. doi: 10.1038/nprot.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirayama A., Kami K., Sugimoto M., Sugawara M., Toki N., Onozuka H., Kinoshita T., Saito N., Ochiai A., Tomita M., Esumi H., Soga T. Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time-of-flight mass spectrometry. Cancer Res. 2009;69 doi: 10.1158/0008-5472.CAN-08-4806. [DOI] [PubMed] [Google Scholar]
- 29.Ben Sahra I., Laurent K., Giuliano S., Larbret F., Ponzio G., Gounon P., Le Marchand-Brustel Y., Giorgetti-Peraldi S., Cormont M., Bertolotto C., Deckert M., Auberger P., Tanti J.-F., Bost F. Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010;70:2465–2475. doi: 10.1158/0008-5472.CAN-09-2782. [DOI] [PubMed] [Google Scholar]
- 30.Stein M., Lin H., Jeyamohan C., Dvorzhinski D., Gounder M., Bray K., Eddy S., Goodin S., White E., DiPaola R.S. Targeting tumor metabolism with 2-deoxyglucose in patients with castrate-resistant prostate cancer and advanced malignancies. Prostate. 2010;70:1388–1394. doi: 10.1002/pros.21172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang S.-H., Garcia J., Melendez J.A., Kilberg M.S., Agarwal A. Haem oxygenase 1 gene induction by glucose deprivation is mediated by reactive oxygen species via the mitochondrial electron-transport chain. Biochem. J. 2003;371:877–885. doi: 10.1042/BJ20021731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schulz T.J., Zarse K., Voigt A., Urban N., Birringer M., Ristow M. Glucose restriction extends caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007;6:280–293. doi: 10.1016/j.cmet.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 33.Leon-Annicchiarico C.L., Ramirez-Peinado S., Dom?nguez-Villanueva D., Gonsberg A., Lampidis T.J., Muñoz-Pinedo C. ATF4 mediates necrosis induced by glucose deprivation and apoptosis induced by 2-deoxyglucose in the same cells. FEBS J. 2015;282:3647–3658. doi: 10.1111/febs.13369. [DOI] [PubMed] [Google Scholar]
- 34.Ding B., Parmigiani A., Divakaruni A.S., Archer K., Murphy A.N., Budanov A.V. Sestrin2 is induced by glucose starvation via the unfolded protein response and protects cells from non-canonical necroptotic cell death. Sci. Rep. 2016;6:22538. doi: 10.1038/srep22538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yun J., Mullarky E., Lu C., Bosch K.N., Kavalier A., Rivera K., Roper J., Chio I.I.C., Giannopoulou E.G., Rago C., Muley A., Asara J.M., Paik J., Elemento O., Chen Z., Pappin D.J., Dow L.E., Papadopoulos N., Gross S.S., Cantley L.C. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science. 2015;350:1391–1396. doi: 10.1126/science.aaa5004. (80-. ) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh G., Lakkis C.L., Laucirica R., Epner D.E. Regulation of prostate cancer cell division by glucose. J. Cell. Physiol. 1999;180:431–438. doi: 10.1002/(SICI)1097-4652(199909)180:3<431::AID-JCP14>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 37.Wang Q., Hardie R.-A., Hoy A.J., van Geldermalsen M., Gao D., Fazli L., Sadowski M.C., Balaban S., Schreuder M., Nagarajah R., Wong J.J.-L., Metierre C., Pinello N., Otte N.J., Lehman M.L., Gleave M., Nelson C.C., Bailey C.G., Ritchie W., Rasko J.E., Holst J. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J. Pathol. 2015;236:278–289. doi: 10.1002/path.4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singh K.K., Desouki M.M., Franklin R.B., Costello L.C. Mitochondrial aconitase and citrate metabolism in malignant and nonmalignant human prostate tissues. Mol. Cancer. 2006;5:14. doi: 10.1186/1476-4598-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vayalil P.K., Landar A. Mitochondrial oncobioenergetic index: a potential biomarker to predict progression from indolent to aggressive prostate cancer. Oncotarget. 2015;6:43065–43080. doi: 10.18632/oncotarget.5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vaz C.V., Alves M.G., Marques R., Moreira P.I., Oliveira P.F., Maia C.J., Socorro S. Androgen-responsive and nonresponsive prostate cancer cells present a distinct glycolytic metabolism profile. Int. J. Biochem. Cell Biol. 2012;44:2077–2084. doi: 10.1016/j.biocel.2012.08.013. [DOI] [PubMed] [Google Scholar]
- 41.Birsoy K., Possemato R., Lorbeer F.K., Bayraktar E.C., Thiru P., Yucel B., Wang T., Chen W.W., Clish C.B., Sabatini D.M. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature. 2014;508:108–112. doi: 10.1038/nature13110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Massie C.E., Lynch A., Ramos-Montoya A., Boren J., Stark R., Fazli L., Warren A., Scott H., Madhu B., Sharma N., Bon H., Zecchini V., Smith D.-M., DeNicola G.M., Mathews N., Osborne M., Hadfield J., MacArthur S., Adryan B., Lyons S.K., Brindle K.M., Griffiths J., Gleave M.E., Rennie P.S., Neal D.E., Mills I.G. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011;30:2719–2733. doi: 10.1038/emboj.2011.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reinicke K., Sotomayor P., Cisterna P., Delgado C., Nualart F., Godoy A. Cellular distribution of Glut-1 and Glut-5 in benign and malignant human prostate tissue. J. Cell. Biochem. 2012;113:553–562. doi: 10.1002/jcb.23379. [DOI] [PubMed] [Google Scholar]
- 44.Grossmann M., Wittert G. Androgens, diabetes and prostate cancer. Endocr. Relat. Cancer. 2012;19:F47–F62. doi: 10.1530/ERC-12-0067. [DOI] [PubMed] [Google Scholar]
- 45.Estévez-García I.O., Cordoba-Gonzalez V., Lara-Padilla E., Fuentes-Toledo A., Falfán-Valencia R., Campos-Rodríguez R., Abarca-Rojano E. Glucose and glutamine metabolism control by APC and SCF during the G1-to-S phase transition of the cell cycle. J. Physiol. Biochem. 2014;70:569–581. doi: 10.1007/s13105-014-0328-1. [DOI] [PubMed] [Google Scholar]
- 46.Park H.U., Suy S., Danner M., Dailey V., Zhang Y., Li H., Hyduke D.R., Collins B.T., Gagnon G., Kallakury B., Kumar D., Brown M.L., Fornace A., Dritschilo A., Collins S.P. AMP-activated protein kinase promotes human prostate cancer cell growth and survival. Mol. Cancer Ther. 2009;8:733–741. doi: 10.1158/1535-7163.MCT-08-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu N., Zheng B., Shaywitz A., Dagon Y., Tower C., Bellinger G., Shen C.-H., Wen J., Asara J., McGraw T.E., Kahn B.B., Cantley L.C. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol. Cell. 2013;49:1167–1175. doi: 10.1016/j.molcel.2013.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prasad R.K., Ismail-Beigi F. Mechanism of stimulation of glucose transport by H2O2: role of phospholipase C. Arch. Biochem. Biophys. 1999;362:113–122. doi: 10.1006/abbi.1998.1026. [DOI] [PubMed] [Google Scholar]
- 49.Zmijewski J.W., Banerjee S., Bae H., Friggeri A., Lazarowski E.R., Abraham E. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J. Biol. Chem. 2010;285:33154–33164. doi: 10.1074/jbc.M110.143685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rabinovitch R.C., Samborska B., Faubert B., Ma E.H., Gravel S.-P., Andrzejewski S., Raissi T.C., Pause A., Pierre J. St, Jones R.G. AMPK maintains cellular metabolic homeostasis through regulation of mitochondrial reactive oxygen species. Cell Rep. 2017;21:1–9. doi: 10.1016/j.celrep.2017.09.026. [DOI] [PubMed] [Google Scholar]
- 51.Freemerman A.J., Johnson A.R., Sacks G.N., Milner J.J., Kirk E.L., Troester M.A., Macintyre A.N., Goraksha-Hicks P., Rathmell J.C., Makowski L. Metabolic reprogramming of macrophages. J. Biol. Chem. 2014;289:7884–7896. doi: 10.1074/jbc.M113.522037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Long D., Wu H., Tsang A.W., Poole L.B., Yoza B.K., Wang X., Vachharajani V., Furdui C.M., McCall C.E. The oxidative state of cysteine thiol 144 regulates the SIRT6 glucose homeostat. Sci. Rep. 2017;7:11005. doi: 10.1038/s41598-017-11388-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Isono T., Chano T., Yonese J., Yuasa T. Therapeutic inhibition of mitochondrial function induces cell death in starvation-resistant renal cell carcinomas. Sci. Rep. 2016;6:25669. doi: 10.1038/srep25669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hart P.C., Mao M., de Abreu A.L.P., Ansenberger-Fricano K., Ekoue D.N., Ganini D., Kajdacsy-Balla A., Diamond A.M., Minshall R.D., Consolaro M.E.L., Santos J.H., Bonini M.G. MnSOD upregulation sustains the Warburg effect via mitochondrial ROS and AMPK-dependent signalling in cancer. Nat. Commun. 2015;6:6053. doi: 10.1038/ncomms7053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hong S.M., Park C.W., Kim S.W., Nam Y.J., Yu J.H., Shin J.H., Yun C.H., Im S.-H., Kim K.-T., Sung Y.C., Choi K.Y. NAMPT suppresses glucose deprivation-induced oxidative stress by increasing NADPH levels in breast cancer. Oncogene. 2016;35:3544–3554. doi: 10.1038/onc.2015.415. [DOI] [PubMed] [Google Scholar]
- 56.LeFevre P.G., Marshall J.K. The attachment of Phloretin and analogues to human erythrocytes in connection with inhibition of sugar transport. J. Biol. Chem. 1959;234:3022–3026. [PubMed] [Google Scholar]
- 57.Andrisse S., Koehler R.M., Chen J.E., Patel G.D., Vallurupalli V.R., Ratliff B.A., Warren D.E., Fisher J.S. Role of GLUT1 in regulation of reactive oxygen species. Redox Biol. 2014;2:764–771. doi: 10.1016/j.redox.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material