

Abstract
Since the proposal of Anfinsen’s thermodynamic hypothesis in 1963, our understanding of protein folding and dynamics has gained significant appreciation of its nuance and complexity. Intrinsically disordered proteins, chameleonic sequences, morpheeins, and metamorphic proteins have broadened the protein folding paradigm. Here, we discuss noncanonical protein folding patterns, with an emphasis on metamorphic proteins, and we review known metamorphic proteins that occur naturally and that have been engineered in the laboratory. Finally, we discuss research areas surrounding metamorphic proteins that are primed for future exploration, including evolution, drug discovery, and the quest for previously unrecognized metamorphs. As we enter an age where we are capable of complex bioinformatic searches and de novo protein design, we are primed to search for previously unrecognized metamorphic proteins and to design our own metamorphs to act as targeted, switchable drugs; biosensors; and more.
Introductory biochemistry courses teach that a given protein sequence typically folds into one lowest-energy three-dimensional structure. This paradigm is attributed to Anfinsen, who postulated that under native conditions, the three-dimensional structure of a protein is the one in which the entire system has the lowest Gibbs free energy. Contributions to the Gibbs free energy of protein folding include both entropic and enthalpic effects such as solvent expulsion, van der Waals forces, hydrophobic effects, and charge–charge interactions. This led Anfinsen to the hypothesis that a protein, in favorable conditions, will fold consistently into a native state structure that is effectively encoded in its amino acid sequence.8
A supposition commonly derived from Anfinsen’s assertion is that each globular protein has only one native structure. This view reflects the nature of the most commonly employed structure determination techniques and the constraints they impose. X-ray crystallography, for instance, requires protein in the form of a single, diffractible crystal. Virtually all solved structures therefore exhibit one global conformation, reinforcing the idea that the native state is defined by a single folded configuration. By extension, it has been believed that each protein’s single three-dimensional structure evolved to better serve a single biological function.
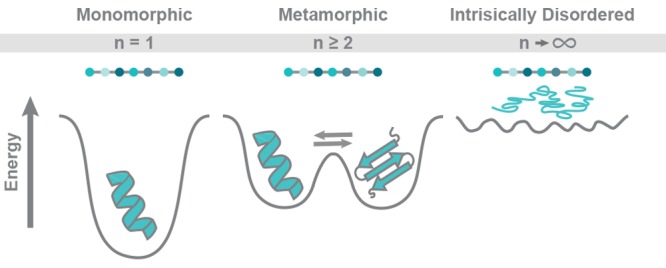
Advances in biochemical, biophysical, and computational techniques over the past 25 years have revealed several departures from the “one sequence, one structure, one function” canon, as detailed in Figure 1. In terms of function, it is now clear that many proteins “moonlight,” performing multiple functional roles that seem otherwise unrelated. Many moonlighting proteins have been identified,4,9 and the MoonProt database (moonlightingproteins.org) provides a curated catalog.10 Moreover, nuclear magnetic resonance spectroscopy (NMR)-based and computational studies have demonstrated that each protein sequence can have considerable structural plasticity, such that the “one sequence, one structure” dogma does not capture the complex nature of a protein’s structure. In fact, this flexibility is an intrinsic feature that contributes directly to the biological function of many proteins.11 The original protein folding paradigm has broadened considerably as a result, such that internal dynamics and conformational flexibility are necessary elements in a description of the native state. New categories that expand the protein folding universe include chameleonic sequences, intrinsically disordered proteins, morpheeins, and metamorphic proteins.
Figure 1.

History of protein folding. In 1973, Anfinsen proposed his Thermodynamic Hypothesis: in the correct conditions, a protein sequence will consistently adopt its native state fold.8 Shown here to represent this hypothesis is the paradigmatic protein folding funnel,7 diagramming the way in which protein sequences proceed to occupy their lowest energy folded states. In 1984, Kabsch and Sander searched the structures of 62 proteins and found six five-residue chameleonic sequences, or identical sequences which can fold as either helix or sheet in the context of different flanking structures.15 For example, shown here is part of mouse Disabled 1 and a peptide derived from mouse Disabled 2, which contain the same sequence (teal) that folds into a helix in one protein and a asheet in the other (PDB IDs 1P3R and 2LSW, respectively).16 In 1988, Piatigorsky et al. discovered the first instance of Moonlighting in delta-Crystallin, a lens protein.58 Shown for example is the well-known moonlighting protein aconitase, which functions as both a citric acid cycle enzyme and an mRNA binding protein. In 1996, Kriwacki et al. identified p21 as lacking a secondary or tertiary structure but remaining functional, making a strong case for the functionality of intrinsically disordered proteins.12 Jaffe introduced the term morpheeins in 2005 to describe proteins that dynamically interconvert between oligomeric states, with state changes being coupled to dissociation of the subunits and changes in subunit structure.3 The prototypical morpheein porphobilinogen synthase is shown as an example. In 2008, Murzin introduced the term metamorphic proteins, to describe a growing family of proteins that interconvert reversibly between two native folded states.1
In this review, we consider each of these classes and discuss how our understanding of protein folding has expanded in recent years. Because folding of prions and amyloidogenic proteins, for which the transition to a misfolded aggregate is typically irreversible, is well documented, we limit the scope of this review to systems that exhibit equilibrium folding rearrangements. Instead, our focus is on metamorphic proteins and their implications in evolution, drug discovery, and protein engineering.
Intrinsically Disordered Proteins
The advent of molecular cloning and recombinant protein expression systems enabled the study of thousands of newly discovered genes and gene products. As the database of solved structures swelled, structural biologists recognized that some fraction of polypeptide sequences encoded in genomic DNA lacked the density of apolar amino acids needed to form a hydrophobic core and consequently had no intrinsic propensity to fold into a stable tertiary structure. Thermodynamically, this corresponds to an energy landscape in which no specific conformation has a significant margin of stability relative to the unfolded state. Instead, the native state is characterized by rapid interconversion between states that include both extended random coil and more compact configurations. These proteins have come to be known as intrinsically disordered (or unstructured) proteins. In 1996, Kriwacki and co-workers described one of these proteins, p21, as lacking stable secondary or tertiary structure in the free solution state as shown by proteolytic mapping, circular dichroism spectroscopy, and NMR.12 However, they also showed by NMR that p21 can bind and inhibit a cyclin-dependent kinase, whereupon it adopts an ordered conformation. This demonstrates that independent thermodynamically driven folding is not a requirement for biological function.
Within a few years of the first experimental descriptions of intrinsically disordered proteins, algorithms for the prediction of intrinsically disordered domains enabled systematic assessments of disorder in entire proteomes.13 It was found that while some proteins are predicted to be completely unfolded, intrinsically disordered regions are more frequently predicted to be found in the context of larger multidomain proteins. In fact, large-scale bioinformatic analysis of animal and plant genomes predicted that most eukaryotic proteins consist of combinations of structured and unstructured regions. Rather than acting as inert linkers between folded domains, many unstructured regions participate in protein–protein interactions, post-translational modifications, or other functions that require conformational flexibility. In contrast to other categories of conformationally variable proteins discussed below, most intrinsically disordered protein sequences are unlikely to also exhibit spontaneous folding into one or more compact structured states in the absence of an interacting partner or other external driving force. Nevertheless, it is now clear that spontaneously folded domains and intrinsically disordered regions constitute the two fundamental classes of functional building blocks of proteins.14
Chameleonic Sequences
The protein data bank has been mined several times for amino acid sequences that exist as either a helix or sheet in different structures, as shown in the chameleon section of Figure 1. The first search, performed by Kabsch and Sander in 1983, yielded 25 such pentapeptides from 62 protein structures.15 The term chameleonic sequence was later introduced by Minor and Kim in 1996 upon engineering an 11-amino-acid sequence capable of folding as either an α-helix or a β-sheet depending on its position in the IgG-binding domain of Protein G (GB1).2 Most recently, a 2015 search by Li and co-workers identified naturally occurring chameleonic sequences up to 10 residues in length.16 In this study, 19 603 chameleonic sequences of 6–10 residues were discovered. Of those sequences, 20 were identified in pairs of proteins that share a common structure, and 12 were associated with specific biological functions, such as substrate binding or oligomerization. In a few cases, the chameleonic sequence was observed in different structures of the same protein. For example, a chameleonic sequence was found that folded differently in respiratory syncytial virus fusion protein’s prefusion and postfusion structures. While short polymorphic peptides do not identify or define metamorphic proteins, chameleonic sequences are likely to be more abundant in metamorphic proteins, due to the requirement for adopting two or more folds.
Morpheeins
Morpheeins represent another class of proteins with unique structural behavior. The morpheein model was first described by Jaffe in 20053 and is a current target of active exploration.17,18 Morpheeins occupy multiple distinct oligomeric states and transition between these states dynamically in solution. These transitions are coupled to dissociation into subunits, followed by a change in the structure of each subunit prior to reassociation.3 The prototype morpheein porphobilinogen synthase (PBGS), a key enzyme in respiration and photosynthesis, occupies two oligomeric states: a high-activity octamer and a low-activity hexamer.19 Interconversion between assemblies proceeds via dissociation to dimer, a change in conformation, and reassociation, as shown schematically in the morpheein section of Figure 1.3 A shift from hexamer to octamer is favored by increased concentrations of magnesium, an allosteric activator of PBGS.19 While the unique oligomeric states of PBGS have different abilities to carry out the same task, other morpheeins’ distinct assemblies perform different functions from one another.20 For example, VP40 is a morpheein found in ebolavirus, whose three different assemblies each perform a distinct function in the viral life cycle.20 In its dimer state, VP40 traffics to the cellular membrane, where electrostatic interactions trigger rearrangement into a hexamer. Hexameric VP40 then forms a multilayered, filamentous matrix structure that is important for budding. A third, octameric assembly regulates viral transcription in infected cells by binding to RNA.
The study of morpheeins provides unique opportunities for the development of targeted therapeutics.17,18 The equilibrium of different morpheein assemblies can be shifted by changes in temperature, pH, and ionic strength, as well as single point mutations.18,20 It follows that identification of small molecules that shift morpheein equilibria, likely by targeting allosteric sites, should be possible. Morpheeins’ targetable allosteric sites are readily identifiable due to their shape-shifting mechanism of interconversion between oligomeric states, making them opportune targets for drug discovery.17 Small molecules that shift the morpheein equilibrium to favor one oligomeric state could be designed to direct biologic outcomes, just as allosteric sites are increasingly popular targets for drug development.
Metamorphic Proteins
Metamorphic proteins spontaneously adopt two or more different native state structures.1 The term “metamorphic proteins” was coined in 2008 by Murzin to describe this phenomenon.1 Murzin defined a metamorphic protein as a single amino acid sequence that adopts multiple folded conformations under native conditions and interconverts reversibly between states.1 An example is XCL1, a small secreted protein that adopts two diverse native state conformations with unique secondary and tertiary structures and interconverts between the two structures at a rate of ∼1 s–1.21 This definition thus excludes prions and amyloidogenic proteins, for which the transition to a misfolded aggregate is typically irreversible. It can be challenging to separate metamorphic proteins from the many others that undergo significant conformational changes. In this review, we cover proteins that have been described in the literature to date as being metamorphic. Initially viewed as an anomaly, numerous analyses suggest that metamorphic proteins may be more common than first expected,1,22−26 and their unique biophysical properties have sparked growing interest.22−24,26,27 As new metamorphic proteins are discovered, it is important to understand the biological role of each native conformation and explore methods to manipulate their function in useful ways.
The known naturally occurring metamorphic proteins have been discovered serendipitously. Other metamorphs likely occur in nature but remain unappreciated due to the predisposition of commonly used structure solving techniques to exclude one or more native states of a metamorphic protein. X-ray crystallography is a powerful approach for determining protein structure and has been used to solve about 90% of structures in the PDB. Crystallography works best with stable, homogeneous proteins, or with proteins that can be stabilized via mutagenesis, removal of flexible regions, or antibody binding. To the contrary, proteins that interconvert between several native states do not readily form diffractible crystals or are forced into a single structure via the above methods. Thus, while crystallography can be useful in the study of each folded state of a metamorphic protein individually, it is unlikely to discover novel metamorphs. Methods such as NMR spectroscopy that can capture dynamic information are better suited to the discovery and analysis of metamorphic native structures. Present day techniques to study proteins via NMR now include many pulse schemes to parse out dynamic properties and ensembles. It is useful to note that for many known metamorphic proteins, one can gain preferential access to one metamorphic native state at a time by altering solution conditions such as ionic strength, pH, and temperature, facilitating structural characterization by crystallography and NMR. In all, it is important to use a variety of complementary techniques to truly gain a full understanding of metamorphic protein structures and functions.
Combining experimental work with computational techniques can help to corroborate experimental data, generate models and hypotheses, and guide future efforts. Molecular simulations, such as molecular dynamics, provide information on the motion of a protein in solution and bear insight regarding specific residues that may be involved in facilitating metamorphic transitions. Additionally, bioinformatic techniques can be developed to facilitate searches for metamorphic proteins.
Naturally Occurring Metamorphic Proteins
IscU
Iron–sulfur cluster protein U (IscU) is a scaffold protein for iron–sulfur cluster biosynthesis and delivery in E. coli.28 As with other metamorphic proteins, IscU exists as an equilibrium of two folded states. IscU’s two states slowly interconvert (kex ≈ 1 s–1) and are approximately equally populated under native conditions, although changes in pH and temperature can shift this equilibrium.28 One state is largely structured with four α-helices and three antiparallel β-strands (S state). Unlike other metamorphic proteins described here, the other native state is dynamically disordered (D state).28 IscU’s S and D structures have been shown to preferentially interact with different proteins involved in the assembly or transfer of iron–sulfur clusters, suggesting that access to both conformations is required for full biologic function.29 The D state preferentially binds to the cysteine desulfurase, which transfers sulfur to IscU. The S state preferentially interacts with chaperone proteins HscB and HscA, leading to eventual binding of IscU’s D state and release of the bound iron–sulfur cluster. The two structures of IscU perform different functions but work in concert to facilitate a single process, similarly to Mad2 and XCL1. To facilitate comparison, key features of the metamorphic proteins described in this section are detailed in Table 1.
Table 1. Comparison of Proteins with Shape-Shifting Behavior.
| Structure 1 | Structure 1 PDB ID | Structure 1 function | Structure 2 | Structure 2 PDB ID | Structure 2 Function/ Activity | |
|---|---|---|---|---|---|---|
| IscU | S (Structured) State: 4 α-helices, 3 β-strands | 24LX | Interaction with DnaJ-type co-chaperone (HscB) | D (Dis-ordered) State: Dynamically disordered | N/A | Cysteine desulfurase (IscS) binding |
| CLIC1 | α3+β4 N-terminal domain, soluble, monomeric | 1K0M | Unknown | All-α N-terminal domain, oligomeric, membrane-associated | 1RK4 | Chloride ion channel |
| RfaH | “Closed” state: all-α fold, interdomain salt bridge | 2OUG | Auto-inhibition restricts recruitment to selected targets | “Open” state: all-β fold, no salt bridge | 2LCL | Interacts with RNAP and the ribosome to inhibit termination and activate translation |
| Selecase | slc1: Monomeric, active form | 4QHF | Metallo-peptidase | slc4: Tetrameric, inactive form | 4QHH | Unknown |
| Mad2 | O-Mad2: Open conformation | 1DUJ | Under investigation; required for full Mad-2 function | C-Mad2: Closed conformation | 1S2H | Spindle Assembly Checkpoint (Cdc20 trapping) |
| Arc/ Switch Arc | Arc: β-strand homodimer | 1BDT | Binds DNA and represses transcription | Switch Arc: engineered variant of Arc with a helix replacing the β-strand | 1QTG | N/A |
| XCL1 | Chemokine structure | 1J9O | Chemokine receptor binding | β-sheet structure | 2N54 | Glycosamino-glycan binding |
CLIC1
CLIC1 (chloride intracellular channel protein 1) is a chloride ion channel protein with two native folded states.26 Like other metamorphic proteins, CLIC1 interconverts reversibly between these two structures; however, this transition occurs only in response to changes in its local environment.26 Structural interconversion is driven by formation or breakage of an intramolecular disulfide bond (Cys24–Cys59) under reducing or oxidizing conditions.26 CLIC1’s soluble, monomeric conformation has an N-terminal α3+β4 structure and is preferentially populated in reducing conditions.26 CLIC1’s membrane-associated conformation has an all-α N-terminal domain and is preferentially populated in oxidizing conditions.26 These two structures are shown in Figure 2. In oxidizing environments, the all-α structure enters the membrane and oligomerizes, forming a transmembrane chloride ion channel. Whereas, under reducing conditions, CLIC1 occupies its soluble structure and cannot enter the membrane. Understanding of CLIC1’s structure and dynamics provides a basis for the design of drugs that would regulate molecular transport across membranes by affecting structural interconversion.
Figure 2.
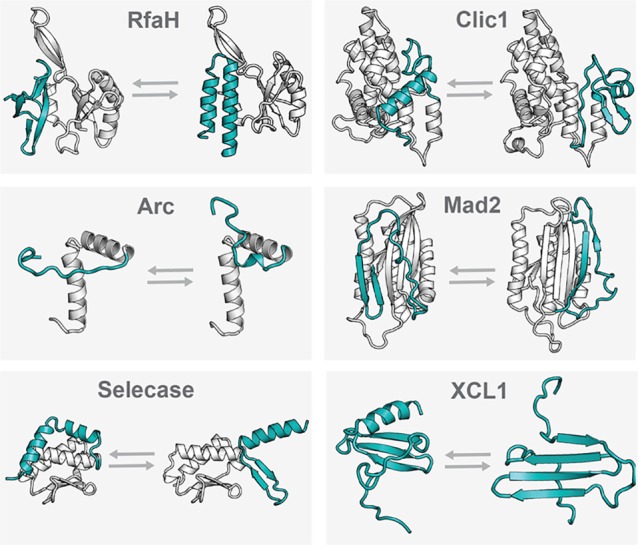
Comparison of the two folded states of some of the known metamorphic proteins. Regions that significantly rearrange folded structure were identified via differences in structural alignment performed in PyMol and are shown here in teal. Regions whose folded structures remain stable are shown in gray. For RfaH, linker regions are invisible in the crystal structure and are shown in gray dashed lines. For the RfaH open state (shown at top left), the RfaH C-terminus was crystallized independently of the N-terminus but is shown here with the 2OUG N-terminus for reference.31 While some of these metamorphic protein structures preferentially oligomerize in solution, they are all shown here as monomer subunits to enhance the clarity of structural comparison. IscU is not shown here due to the lack of a solved structure for the disordered (D) state. PDB IDs (left structure listed first): Rfah, 2LCL and 2OUG; CLIC1, 1RK4 and 1K0M; Arc, 1BDT and 1QTG; Mad2, 1DUJ and 1S2H; XCL1, 1J9O and 2JP1; Selecase, 4QHF and 4QHH. Abbreviations: CLIC1, chloride intracellular channel protein 1.
RfaH
The E. coli elongation factor RfaH is part of the NusG family of transcription factors, which is conserved across all three domains of life (archaea, bacteria, and eukarya).30 RfaH has two native states with different C-terminal structures that work together to regulate the coupling of transcription to translation.31 RfaH’s “closed” state has an all-α coiled-coil structure and an interdomain salt bridge, while the “open” state has an all-β structure and no salt bridge, as shown in Figure 2. In the autoinhibitory closed state, the α-helical hairpin inhibits RfaH from binding to RNA polymerase (RNAP) and to the ribosome. Interaction with ops DNA sequences in the transcription complex causes the helical hairpin to dissociate from RfaH’s N-terminal domain, exposing the RNAP binding domain and allowing the C-terminal domain to convert to occupy a β-barrel structure. This open, β-barrel state couples transcription to translation by simultaneously binding to RNAP and the ribosome. Thus, in wild type RfaH, interaction with DNA is required to allow the C-terminal domain to switch between structures. On the other hand, an engineered RfaH variant (E48S) exchanges between the open and closed conformations as discussed further below.31
It was first hypothesized that RfaH was metamorphic in 2007, upon determination of its crystal structure.32 Structures of RfaH’s ancestor, the general transcription factor NusG, had previously been solved, showing that NusG’s N-terminal structure closely resembles that of RfaH.33 However, NusG’s C-terminal structure is strikingly different from RfaH’s, despite maintenance of statistically significant sequence similarity.32 In their respective crystal structures, the NusG C-terminal domain occupies a β-sheet fold, whereas the RfaH C-terminal domain occupies its “closed” coiled-coil fold.32 Interestingly, homology modeling showed that the sequence of the RfaH C-terminal domain could be integrated into the NusG C-terminal structure without energy minimization and without producing unfavorable contacts.32 This led Artsimovitch and co-workers to hypothesize that the RfaH C-terminus was capable of occupying both a coiled-coil as well as the β-sheet fold seen in NusG, but the crystal structure had only captured one of its folded states.32 In 2012, this was found to be true: upon dissociation from its N-terminal domain, the C-terminal domain of RfaH can indeed refold into a β-sheet.31 This example offers promise to the approach of searching for more metamorphic proteins using evolutionary information (as discussed later) and suggests that while crystallography is unlikely to reveal metamorphic fold switching, it can still play an important role in the study of metamorphic proteins. Molecular dynamics simulations of RfaH interconversion highlight the utility of current computational approaches for understanding metamorphic fold switching.34
Metamorphic folding of the RfaH C-terminal domain presents an illustration of how the evolution of metamorphic folding can alter protein function and introduce new regulatory mechanisms.35 RfaH only transitions to its “open” state upon binding to ops DNA, thus restricting its activity to a narrow set of targets. In contrast, its ancestor NusG is always in an open conformation and does not show any DNA sequence specificity. Although both RfaH and NusG bind to RNAP, they have opposite functions: Rho-dependent termination is decreased by RfaH but increased by NusG. NusG is essential for cell survival, and if RfaH were always in its open conformation, it would bind more tightly to RNAP and outcompete NusG, leading to cell death. However, due to the metamorphosis-mediated regulation of RfaH binding to RNAP, cell survival is facilitated, allowing RfaH and NusG to coexist and execute their unique functions independently.
Selecase
Selecase is a metallopeptidase from Methanocaldococcus jannaschii that occupies three different C-terminal structures. These conformations are preferentially monomeric, dimeric, and tetrameric (called Slc-1, Slc-2, and Slc-4, respectively), and they exist in equilibrium.36 Slc-1 has a C-terminal α-helix, which swings outward away from its core in the Slc-2 structure, facilitating oligomerization by allowing domain swapping of the C-terminal α-helix. In the Slc-4 structure, the C-terminal helix switches to occupy a β-sheet folded conformation.36 The structures of Slc-1 and Slc-4 are compared in Figure 2. In contrast to the metamorphic proteins, whose distinct conformations perform different functions, the Selecase monomer is active while the higher oligomers are inactive.36 The use of metamorphosis as a mechanism for regulating activity levels is also seen with CLIC1. As the concentration of Selecase increases, the occupancy of the conformations that prefer to oligomerize increases.36 In other words, the occupancy of the active monomer structure is highest (70%) when Selecase concentrations are lowest (below 0.15 mg mL–1).36 Thus, unlike most enzymes, Selecase shows decreased activity at increased enzyme concentrations.36
Mad-2
Mitotic Assembly Deficient 2 (Mad2), an essential cell cycle checkpoint for spindle assembly, has two native folded states: a closed state (C-Mad2) and an open state (O-Mad2).37 These two structures have similar Gibbs free energies and exist in equilibrium under physiologic conditions, in about a 90:10 C-Mad2/ O-Mad2 ratio.38 During interconversion between conformations, Mad2’s C-terminal structure rearranges significantly, such that it is stabilized by an entirely different hydrogen bonding network.37 The core and N-terminal structures (about 70% of the Mad2 sequence) largely maintain their secondary structural characteristics and hydrophobic packing.39 Mad2’s folded states interconvert spontaneously, but the interconversion is slower than that of XCL1 and IscU (t1/2 ∼ 9 h).37
Mad2, along with other proteins in the spindle assembly checkpoint (SAC), prevents the transition from metaphase to anaphase by targeting Cdc20.40 C-Mad2 binds and traps Cdc20, while O-Mad2 does not. Still, it has been shown that the interaction of O-Mad2 and C-Mad2 is essential to sustain the SAC.40 The reason for this is not clearly established, but it is thought to be partly due to preferential recruitment of soluble O-Mad2 to kinetochores, where it dimerizes with C-Mad2 and is converted to its active form.40 Mad2’s use of its two structures in concert to perform different parts of the same task makes it similar to IscU and XCL1. Due to its involvement in a wide variety of physiologic processes, Mad2 presents an interesting target and tool for drug discovery efforts.
XCL1
XCL1, also known as Lymphotactin, is a member of the XC family of chemokines. Under physiologic conditions, XCL1 exists as an equilibrium of two approximately equally populated native state conformations, which interconvert spontaneously.41 XCL1’s global conformational shift between structures occurs without ligand binding or significant environmental changes.21 The two conformations populated by XCL1 are (1) the canonical chemokine structure, which exists in solution as a monomer and binds to XCL1’s chemokine receptor XCR1, and (2) a four-stranded β-sheet that forms a dimer and binds to glycosaminoglycans (GAGs).21 Full chemokine function in vivo requires chemokines to bind GAGs and to bind and activate G protein-coupled chemokine receptors, and most chemokines have a single native fold that can accomplish both of these tasks.42 Because each conformational species of XCL1 performs only one of these essential roles,21 its metamorphosis is likely required for full biologic activity. XCL1’s diverse folded conformations also play unique roles in infection. The GAG-binding conformation is a broad-spectrum inhibitor of HIV43 and has antimicrobial activity,44 whereas the receptor-binding conformation does not.
XCL1’s two native structures have entirely different tertiary and quaternary contacts that are stabilized by two mutually exclusive hydrogen bonding networks.21 It is thought that interconversion between XCL1’s two native states proceeds via global unfolding.45 The two species interconvert on a time scale of ∼1 s–121,45 with a Keq of ∼1 under physiologic conditions (37 °C, 150 mM NaCl).45 When comparing the chemokine structure and β-sheet structure, there is a very small free energy difference (ΔGstructure1–structure2 = 0.8 kcal/mol) between the two folded states.45 Each of XCL1’s native states are favored by certain environmental conditions, but these conditions are not required to stimulate interconversion. The chemokine structure is preferred at 10 °C in high salt solutions, whereas the β-sheet structure is favored at 40 °C in low salt solutions.21 Additionally, XCL1 is a valuable model system by which to study metamorphic proteins. Engineered XCL1 mutants have been designed that lock it into its chemokine structure, β-sheet structure, and unfolded state, allowing us to better study its two conformations, and aiding us as we work toward a better understanding of which primary sequence traits are required for metamorphosis.46
Engineered Metamorphic and Related Proteins
RfaH E48S
The E. coli transcription factor RfaH has two native folded states that do not spontaneously interconvert in solution but rather require changes in binding partners to undergo structural rearrangement.31 When interacting with RfaH’s N-terminal domain, the C-terminal domain is entirely helix. However, when the C-terminal domain is released from these interactions, it switches to occupy a β-barrel structure. These structures are shown in Figure 2. A single amino acid substitution (E48S) RfaH variant has been engineered to populate both the α-helical and the β-barrel C-terminal folded states in equimolar equilibrium under physiologic solution conditions.31 This variant undergoes a large-scale, reversible shift in the conformation of its C-terminal domain spontaneously in solution, while its N-terminal domain remains stable.31 The E48S substitution breaks an intramolecular salt bond, which is present in wild type RfaH, allowing the C-terminal domain the freedom to adopt both conformations.31 MD simulations performed for wild type RfaH and RfaH E48S showed that the E48S variant’s C-terminal domain always sampled structures with greater root mean squared distance (RMSD) than the wild type’s C-terminal domain.47 In these simulations, the full α to β conformational switch was not observed, likely because the shift occurs on a slower time scale than the 2.1 μs length of the simulation.47
Switch Arc
Arc is a repressor protein found in the Salmonella bacteriophage P22, which normally adopts a homodimer conformation that binds to DNA.48 In 1999, the Sauer group created a “switch” mutant of Arc, in which Leu12, a hydrophobic core residue, is interchanged with Asp11, a polar surface residue.48 This causes a dramatic shift in the fold of residues 8 to 14, in which a right-handed helix replaces a β-strand, accompanied by side chain repacking. The secondary structure of the sequence outside of this short region remains the same. Shortly thereafter in 2000, the Sauer group created another Arc mutant (N11L), which can interchange reversibly between the wild type arc fold and the switch arc fold freely in solution on the millisecond time scale.49 The equilibrium between the two structures depends on temperature, solvent conditions, and ligand binding.49 These findings illustrate the potential of a single point mutation to significantly alter protein folding, thus exemplifying how protein structures can shift greatly during evolution. Additionally, while the region of Switch Arc that switches conformations is relatively short, these studies provide a basis for future efforts to engineer larger metamorphic domains.
Protein G
In the 2000s, in order to better understand the evolution of protein fold and function, Bryan and co-workers set out to create two proteins with different structures and different functions but nearly identical sequences. Their model system was developed from the two binding domains of Streptococcus Protein G, GA and GB, which occupy a 3-α fold that binds to human serum albumin and a 4-β+α fold that binds IgG, respectively. Variants of GA and GB were generated that are 77% identical yet maintain their wild type folds and natural biologic functions.50 With these variants as a starting point, a systematic search identified a single amino acid substitution that could trigger a complete shift in both folded state and function, without significant population of the unfolded state.51 Specifically, a variant known as GA98 occupies the 3-α structure (90% populated, pH 7.2, 20 °C) and binds to human serum albumin. L45Y substitution yields a variant known as GB98, which occupies the 4-β+α structure (90% populated, pH 7.2, 20 °C) and binds to IgG. Interestingly, GA98 can also bind to IgG, suggesting that it samples the 4-β+α folded state, but this conformation was undetectable by NMR likely due to low occupancy. In other words, while this remains to be demonstrated, it is possible that GA98 is metamorphic, but its second native state structure is not highly populated. It was later discovered that there are multiple different single amino acid substitutions at diverse positions throughout the sequence of GA98 that can lead to a similar shift to the 4-β+α conformation.52 This work demonstrated that short mutational pathways can result in dramatic changes in protein structure without any complete losses of function or structure along the way. It also provided insights that will be valuable to the pursuits of engineering metamorphic proteins and determining features that promote metamorphosis.
Future Directions, Questions, and Challenges
Metamorphosis and Evolution
Multiple theories have been proposed to explain the role of protein metamorphosis in evolution. One possibility is that metamorphic proteins are evolutionary intermediates, caught in the process of evolving a new structure but not having yet evolved away from their previous structure. Complete fold switching during evolution has indeed been observed in certain proteins, dubbed evolutionary metamorphs, such as the Cro family of bacteriophage transcription factors.53 Ancestral Cro family members have an N-terminal helix-turn-helix motif.53 Some Cro family members have evolved an N-terminal α-helical fold; others, an N-terminal β-sheet fold.53 The possibility of very few mutations to drive a complete shift in conformation is also demonstrated by the protein G studies detailed above.51,52 In such a case of evolutionary metamorphosis, it could be possible for a single sequence to adopt both structures before splitting off.
On the other hand, it is possible that metamorphic folding is an evolved trait that confers enhanced fitness. Metamorphosis can allow proteins to accomplish multiple functions or allow for novel regulatory mechanisms. Many known morpheeins and metamorphs, such as XCL1,21 IscU,28 and Mad2,40 require access to both of their folded states for full biologic function. In other cases, metamorphosis functions as a regulatory mechanism. For example, Selecase is autoinhibitory at high concentrations due to its structure switching.36 CLIC1 only adopts its membrane channel conformation in oxidizing conditions,26 and RfaH only switches to occupy its RNAP- and ribosome-binding structure upon interacting with specific DNA sequences.31 Another way metamorphic character could enhance fitness is by increasing evolvability, the capability to produce heritable phenotypic modifications along evolutionary time.6 A component of evolvability is innovability, or the ability to acquire large changes in fold and/or function via relatively few mutations.54 Due to their flexible structures, low energy barriers between states, and shallower energy wells of their folded states, metamorphic proteins may be especially well prepared to further evolve new structures and functions as time goes on, making them especially innovable.
New-function mutations tend to destabilize the structures of proteins with a stable native conformation.55 However, destabilizing mutations can be deleterious because they can lead to protein unfolding, aggregation, degradation, and loss of function.55 Thus, after a destabilizing, new-function mutation, a stabilizing compensatory mutation is frequently necessary for the new-function mutation to be accommodated.55 For metamorphosis to evolve, mutations that allow for fold interconversion would have to be destabilizing enough for interconversion to be possible, but not so much so that they could not be accommodated. This phenomenon could potentially be facilitated by chaperones. By achieving this optimum, perhaps protein metamorphs are also able to maintain their previous function while simultaneously developing a new function. These hypotheses about the evolution of protein metamorphosis are illustrated in Figure 3. In all, the role of protein metamorphosis in evolution remains unclear. Further study of the emergence of metamorphic folding is needed to understand the frequency with which it contributes to neofunctionalization and to stimulate new directions for protein design and engineering.
Figure 3.
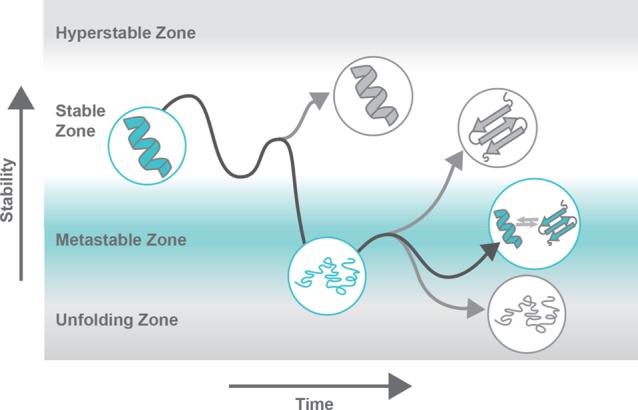
Metamorphic folding in protein evolution. Over time, mutations lead to stabilization or destabilization of protein structures. New-function mutations tend to be destabilizing and are thus frequently followed by compensatory stabilizing mutations to improve overall “protein fitness.” Protein metamorphosis requires a degree of stability that allows for adoption of stable conformations, while still permitting structural interconversion; here, this is labeled as the metastable zone. After a gain of function mutation, compensatory stabilizing mutations could allow stabilization to the degree that the protein is not unfolded or prone to aggregation but is not so stable that its previous function is lost. The dark gray line represents a potential evolutionary path of a protein that gains fitness with access to a second native state conformation. The lighter gray lines represent other possible evolutionary paths that would lead to evolution of nonmetamorphic folding. Inspired by ref (59). Copyright 2009 American Association for the Advancement of Science.
Prospects for Drug Discovery
The metamorphic proteins described in this review participate in a variety of essential biological processes, including chemotaxis,56 cell cycle regulation,37 ion transport,56 and regulation of transcription and translation.31 In some cases, metamorphic proteins are presumed to interconvert between two distinct structures because each structure performs an important function. In fact, it has been shown that the full biologic function of several metamorphic proteins requires access to both of their native states.21,31,40 Therefore, controlling the ability of metamorphic proteins to interconvert could be used as a biologic switch to temporally control critical cell processes often altered in disease. In other words, if one folded state is implicated in pathology, shifting the equilibrium to deplete the pathological isoform could be a powerful therapeutic strategy. Each folded state of a metamorphic protein can be expected to display a distinct set of sites that are potentially druggable. For example, a preliminary computational solvent mapping analysis of XCL1 predicted hotspots for ligand binding that were comprised of different sets of amino acids on its two native state structures. Thus, metamorphic proteins might represent at least two, nonequivalent drug targets within the same polypeptide sequence.
Engineered metamorphic proteins can be envisioned as molecular switches, self-assembling nanostructures, or self-modulating catalysts. Metamorphic molecular switches could be designed such that their metamorphic equilibrium is shifted to favor an active structure exclusively in certain environments, such as in the presence of certain metal ions, temperature, or pH conditions. For example, a cancer therapy could be designed such that its toxic, active conformation is only populated in an acidic tumor environment, reducing side effects and improving efficacy. One could also imagine morpheein-like3 drugs, whose topological rearrangement is only triggered in certain environments, allowing for controlled switching between oligomeric states. Such drugs could be used to build nanostructures under certain conditions, or conversely, to disassemble and release a trapped drug in only the desired locations. Additionally, drugs could be designed to modulate their own catalytic activity via concentration-dependent shifts in structure and oligomerization state, similarly to Selecase.36 Biosensors and other biologic tools could also be designed using these principles.
The Quest for Other Metamorphic Proteins
A search for new metamorphic proteins has the potential to yield productive and valuable results. So far, all known naturally occurring metamorphs have been discovered serendipitously. As discussed above, current structure determination techniques are unlikely to detect new metamorphic proteins, and many protein structures remain unsolved. Thus, it seems likely that additional metamorphic proteins remain undiscovered. Implementation of a PDB search for metamorphic proteins could lead us to find potential metamorphs. Some common characteristics of metamorphic proteins are already discernible and could aid in the determination of search criteria. For example, metamorphosis necessitates a sufficiently low energy barrier between the protein’s two native states, or a barrier that can be reduced under certain conditions, to allow for dynamic interconversion in solution. This is facilitated when the free energy minima of the folded states are less negative, which can be in part due to the presence of flexible regions, for example, the flexible linker in RfaH,1 the flexible tail of O-Mad2,37 and Selecase’s flexible C-terminal domain.25 In theory, chaperones could facilitate metamorphic folding and interconversion, and chaperone involvement could be a useful search criterion. Additionally, while it has been assumed that moonlighting proteins perform multiple functions using the same native state, it is quite possible that some moonlighting is accomplished via protein metamorphosis. Moonlighting behavior may thus provide another appropriate criterion in initial searches for new metamorphs.
A recent report suggests that discrete molecular dynamics (DMD) calculations enriched with coevolutionary information might be used to identify proteins with a propensity for metamorphic folding.57 The approach of Sfriso et al. requires the analysis of large multiple sequence alignments to identify pairs of coevolving sequence positions that are not in contact in the known structure.57 Their premise was that each non-native coevolving pair could report on the existence of an alternative conformation in which the two amino acids are brought into contact.57 Steered DMD simulations that incorporated the coevolutionary information were able to capture known alternative conformations in a majority of the 92 proteins evaluated in this manner.57 The authors concluded that coevolving sequence positions provide useful clues to the potential for access to alternative conformations.57
Since their existence is known, efforts to identify metamorphic proteins throughout the available genome are easily justified. Steady advances in computational power and molecular simulation methods will permit exhaustive search efforts. It will be exciting to observe the outcome of experimental analysis of new metamorphic protein candidates. We expect the results to raise new questions about protein evolution, propel innovative strategies for drug development, and stimulate the design and engineering of protein-based nanomaterials with switchable properties based on native state rearrangements.
Acknowledgments
We thank M. M. Babu for helpful discussions.
Glossary
Keywords
- Metamorphic Protein
a protein that, under given solution conditions, spontaneously adopts two or more different native folds and interconverts reversibly between them1
- Chameleonic Sequence
an amino acid sequence that exists as both helix and sheet conformations in different proteins2
- Morpheein
a protein that exists as a dynamic equilibrium of distinct oligomeric states, where a change from one oligomeric state to another requires dissociation into subunits and a conformational change in each subunit3
- Moonlighting Protein
a protein that performs multiple functional roles that seem otherwise unrelated4
- Thermodynamic Hypothesis
the hypothesis that, under native conditions, a protein sequence will consistently adopt its native three-dimensional fold, which is the structure that minimizes the Gibbs free energy of the whole system5
- Protein Folding
the physical process by which a polypeptide folds from a random coil into its native three-dimensional structure, which is typically biologically functional
- Evolvability
a system’s capacity to generate adaptive genetic diversity and acquire new functional attributes over time6
- Folding Funnel
the conformational free energy landscape a protein can sample, where the native state corresponds to the global minimum7
This work was supported in part by National Institutes of Health Grants AI058072 and AI103225 (to B.F.V.). A.F.D. is a member of the NIH supported (T32 GM080202) Medical Scientist Training Program at the Medical College of Wisconsin (MCW).
The authors declare the following competing financial interest(s): B.F.V. holds an ownership interest in Protein Foundry, LLC.
References
- Murzin A. G. (2008) Metamorphic Proteins. Science 320, 1725–1726. 10.1126/science.1158868. [DOI] [PubMed] [Google Scholar]
- Minor D. L.; Kim P. S. (1996) Context-dependent secondary structure formation of a desigend protein sequence. Nature 380, 730–734. 10.1038/380730a0. [DOI] [PubMed] [Google Scholar]
- Jaffe E. K. (2005) Morpheeins--a new structural paradigm for allosteric regulation. Trends Biochem. Sci. 30, 490–497. 10.1016/j.tibs.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Jeffery C. J. (1999) Moonlighting Proteins. Trends Biochem. Sci. 24, 8–11. 10.1016/S0968-0004(98)01335-8. [DOI] [PubMed] [Google Scholar]
- Anfinsen E. H. a. C. B. (1962) Side-chain Interactions Governing the Pairing of Half-cystine Residues in Ribonuclease. J. Biol. Chem. 237, 1839–1844. [PubMed] [Google Scholar]
- Kirschner M.; Gerhart J. (1998) Evolvability. Proc. Natl. Acad. Sci. U. S. A. 95, 8420–8427. 10.1073/pnas.95.15.8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leopold P. E.; Montal M.; Onuchic J. N. (1992) Protein folding funnels: A kinetic approach to the sequence-structure relationship. Proc. Natl. Acad. Sci. U. S. A. 89, 8721–8725. 10.1073/pnas.89.18.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anfinsen C. B. (1973) Principles that Govern the Folding of Protein Chains. Science 181, 223–230. 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- Jeffery C. J. (2014) An introduction to protein moonlighting. Biochem. Soc. Trans. 42, 1679–1683. 10.1042/BST20140226. [DOI] [PubMed] [Google Scholar]
- Mani M.; Chen C.; Amblee V.; Liu H.; Mathur T.; Zwicke G.; Zabad S.; Patel B.; Thakkar J.; Jeffery C. J. (2015) MoonProt: a database for proteins that are known to moonlight. Nucleic Acids Res. 43, D277–282. 10.1093/nar/gku954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henzler-Wildman K.; Kern D. (2007) Dynamic personalities of proteins. Nature 450, 964–972. 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- Kriwacki R. W.; Hengst L.; Tennant L.; Reed S. I.; Wright P. E. (1996) Structural studies of p2lWa1CiPl/Sdil in the free and Cdk2-bound state: Conformational disorder mediates binding diversity. Proc. Natl. Acad. Sci. U. S. A. 93, 11504–11509. 10.1073/pnas.93.21.11504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldfield C. J.; Ulrich E. L.; Cheng Y.; Dunker A. K.; Markley J. L. (2005) Addressing the intrinsic disorder bottleneck in structural proteomics. Proteins: Struct., Funct., Genet. 59, 444–453. 10.1002/prot.20446. [DOI] [PubMed] [Google Scholar]
- van der Lee R.; Buljan M.; Lang B.; Weatheritt R. J.; Daughdrill G. W.; Dunker A. K.; Fuxreiter M.; Gough J.; Gsponer J.; Jones D. T.; Kim P. M.; Kriwacki R. W.; Oldfield C. J.; Pappu R. V.; Tompa P.; Uversky V. N.; Wright P. E.; Babu M. M. (2014) Classification of intrinsically disordered regions and proteins. Chem. Rev. 114, 6589–6631. 10.1021/cr400525m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W.; Sander C. (1984) On the use of sequence homologies to predict protein structure: Identical pentapeptides can have completely different conformations. Proc. Natl. Acad. Sci. U. S. A. 81, 1075–1078. 10.1073/pnas.81.4.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; Kinch L. N.; Karplus P. A.; Grishin N. V. (2015) ChSeq: A database of chameleon sequences. Protein Sci. 24, 1075–1086. 10.1002/pro.2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe E. K. (2010) Morpheeins - a New Pathway for Allosteric Drug Discovery. Open Conf. Proc. J. 1, 1–6. 10.2174/2210289201001010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence S. H.; Ramirez U. D.; Tang L.; Fazliyez F.; Kundrat L.; Markham G. D.; Jaffe E. K. (2008) Shape shifting leads to small-molecule allosteric drug discovery. Chem. Biol. 15, 586–596. 10.1016/j.chembiol.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breinig S.; Kervinen J.; Stith L.; Wasson A. S.; Fairman R.; Wlodawer A.; Zdanov A.; Jaffe E. K. (2003) Control of tetrapyrrole biosynthesis by alternate quaternary forms of porphobilinogen synthase. Nat. Struct. Mol. Biol. 10, 757–763. 10.1038/nsb963. [DOI] [PubMed] [Google Scholar]
- Bornholdt Z. A.; Noda T.; Abelson D. M.; Halfmann P.; Wood M. R.; Kawaoka Y.; Saphire E. O. (2013) Structural rearrangement of ebola virus VP40 begets multiple functions in the virus life cycle. Cell 154, 763–774. 10.1016/j.cell.2013.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuinstra R. L.; Peterson F. C.; Kutlesa S.; Elgin E. S.; Kron M. A.; Volkman B. F. (2008) Interconversion between two unrelated protein folds in the lymphotactin native state. Proc. Natl. Acad. Sci. U. S. A. 105, 5057–5062. 10.1073/pnas.0709518105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodchild S. C.; Curmi P. M. G.; Brown L. J. (2011) Structural gymnastics of multifunctional metamorphic proteins. Biophys. Rev. 3, 143. 10.1007/s12551-011-0053-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lella M.; Mahalakshmi R. (2017) Metamorphic Proteins: Emergence of Dual Protein Folds from One Primary Sequence. Biochemistry 56, 2971–2984. 10.1021/acs.biochem.7b00375. [DOI] [PubMed] [Google Scholar]
- Bryan P. N.; Orban J. (2010) Proteins that switch folds. Curr. Opin. Struct. Biol. 20, 482–488. 10.1016/j.sbi.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knauer S. H.; Rosch P.; Artsimovitch I. (2012) Transformation: the next level of regulation. RNA Biol. 9, 1418–1423. 10.4161/rna.22724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littler D. R.; Harrop S. J.; Fairlie W. D.; Brown L. J.; Pankhurst G. J.; Pankhurst S.; DeMaere M. Z.; Campbell T. J.; Bauskin A. R.; Tonini R.; Mazzanti M.; Breit S. N.; Curmi P. M. (2004) The intracellular chloride ion channel protein CLIC1 undergoes a redox-controlled structural transition. J. Biol. Chem. 279, 9298–9305. 10.1074/jbc.M308444200. [DOI] [PubMed] [Google Scholar]
- Yadid I.; Kirshenbaum N.; Sharon M.; Dym O.; Tawfik D. S. (2010) Metamorphic proteins mediate evolutionary transitions of structure. Proc. Natl. Acad. Sci. U. S. A. 107, 7287–7292. 10.1073/pnas.0912616107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markley J. L.; Kim J. H.; Dai Z.; Bothe J. R.; Cai K.; Frederick R. O.; Tonelli M. (2013) Metamorphic protein IscU alternates conformations in the course of its role as the scaffold protein for iron-sulfur cluster biosynthesis and delivery. FEBS Lett. 587, 1172–1179. 10.1016/j.febslet.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothe J. R.; Tonelli M.; Ali I. K.; Dai Z.; Frederick R. O.; Westler W. M.; Markley J. L. (2015) The Complex Energy Landscape of the Protein IscU. Biophys. J. 109, 1019–1025. 10.1016/j.bpj.2015.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner F.; Grohmann D. (2011) Evolution of multisubunit RNA polymerases in the three domains of life. Nat. Rev. Microbiol. 9, 85–98. 10.1038/nrmicro2507. [DOI] [PubMed] [Google Scholar]
- Burmann B. M.; Knauer S. H.; Sevostyanova A.; Schweimer K.; Mooney R. A.; Landick R.; Artsimovitch I.; Rosch P. (2012) An alpha helix to beta barrel domain switch transforms the transcription factor RfaH into a translation factor. Cell 150, 291–303. 10.1016/j.cell.2012.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belogurov G. A.; Vassylyeva M. N.; Svetlov V.; Klyuyev S.; Grishin N. V.; Vassylyev D. G.; Artsimovitch I. (2007) Structural basis for converting a general transcription factor into an operon-specific virulence regulator. Mol. Cell 26, 117–129. 10.1016/j.molcel.2007.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner T.; Marinkoviç S.; Huber R.; Wahl M. C.; Kaiser J. T. (2002) Crystal structures of transcription factor NusG in light of its nucleic acid- and protein-binding activities. EMBO Journal 21, 4641–4653. 10.1093/emboj/cdf455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gc J. B.; Bhandari Y. R.; Gerstman B. S.; Chapagain P. P. (2014) Molecular dynamics investigations of the alpha-helix to beta-barrel conformational transformation in the RfaH transcription factor. J. Phys. Chem. B 118, 5101–5108. 10.1021/jp502193v. [DOI] [PubMed] [Google Scholar]
- Belogurov G. A; Mooney R. A.; Svetlov V.; Landick R.; Artsimovitch I. (2009) Functional specialization of transcription elongation factors. EMBO J. 28, 112–122. 10.1038/emboj.2008.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Pelegrin M.; Cintas-Pedrola A.; Herranz-Trillo F.; Bernado P.; Peinado J. R.; Arolas J. L.; Gomis-Ruth X; Cerda-Costa N. (2014) Multiple Stable Conformations Account for Reversible Concentration-Dependent Oligomerization and Autoinhibition of a Metamorphic Metallopeptidase. Angew. Chem. 126, 10800–10806. 10.1002/ange.201405727. [DOI] [PubMed] [Google Scholar]
- Luo X.; Tang Z.; Xia G.; Wassmann K.; Matsumoto T.; Rizo J.; Yu H. (2004) The Mad2 spindle checkpoint protein has two distinct natively folded states. Nat. Struct. Mol. Biol. 11, 338–345. 10.1038/nsmb748. [DOI] [PubMed] [Google Scholar]
- De Antoni A.; Pearson C. G.; Cimini D.; Canman J. C.; Sala V.; Nezi L.; Mapelli M.; Sironi L.; Faretta M.; Salmon E. D.; Musacchio A. (2005) The Mad1/Mad2 complex as a template for Mad2 activation in the spindle assembly checkpoint. Curr. Biol. 15, 214–225. 10.1016/j.cub.2005.01.038. [DOI] [PubMed] [Google Scholar]
- Luo X.; Yu H. (2008) Protein metamorphosis: the two-state behavior of Mad2. Structure 16, 1616–1625. 10.1016/j.str.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mapelli M.; Massimiliano L.; Santaguida S.; Musacchio A. (2007) The Mad2 conformational dimer: structure and implications for the spindle assembly checkpoint. Cell 131, 730–743. 10.1016/j.cell.2007.08.049. [DOI] [PubMed] [Google Scholar]
- Kuloğlu E. S.; McCaslin D. R.; Kitabwalla M.; Pauza C. D.; Markley J. L.; Volkman B. F. (2001) Monomeric Solution Structure of the Prototypical ‘C’ Chemokine Lymphotactin. Biochemistry 40, 12486–12496. 10.1021/bi011106p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudfoot A. E. I.; Handel T. M.; Johnson Z.; Lau E. K.; LiWang P.; Clark-Lewis I.; Borlat F.; Wells T. N. C.; Kosco-Vilbois M. H. (2003) Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl. Acad. Sci. U. S. A. 100, 1885–1890. 10.1073/pnas.0334864100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzzo C.; Fox J.; Lin Y.; Miao H.; Cimbro R.; Volkman B. F.; Fauci A. S.; Lusso P. (2013) The CD8-derived chemokine XCL1/lymphotactin is a conformation-dependent, broad-spectrum inhibitor of HIV-1. PLoS Pathog. 9, e1003852. 10.1371/journal.ppat.1003852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevins A. M.; Subramanian A.; Tapia J. L.; Delgado D. P.; Tyler R. C.; Jensen D. R.; Ouellette A. J.; Volkman B. F. (2016) A Requirement for Metamorphic Interconversion in the Antimicrobial Activity of Chemokine XCL1. Biochemistry 55, 3784–3793. 10.1021/acs.biochem.6b00353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler R. C.; Murray N. J.; Peterson F. C.; Volkman B. F. (2011) Native-state interconversion of a metamorphic protein requires global unfolding. Biochemistry 50, 7077–7079. 10.1021/bi200750k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox J. C.; Tyler R. C.; Guzzo C.; Tuinstra R. L.; Peterson F. C.; Lusso P.; Volkman B. F. (2015) Engineering Metamorphic Chemokine Lymphotactin/XCL1 into the GAG-Binding, HIV-Inhibitory Dimer Conformation. ACS Chem. Biol. 10, 2580–2588. 10.1021/acschembio.5b00542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xun S.; Jiang F.; Wu Y. D. (2016) Intrinsically disordered regions stabilize the helical form of the C-terminal domain of RfaH: A molecular dynamics study. Bioorg. Med. Chem. 24, 4970–4977. 10.1016/j.bmc.2016.08.012. [DOI] [PubMed] [Google Scholar]
- Cordes M. H. J.; Walsh N. P.; McKnight C. J.; Sauer R. T. (1999) Evolution of a Protein Fold in Vitro. Science 284, 325–327. 10.1126/science.284.5412.325. [DOI] [PubMed] [Google Scholar]
- Cordes M. H. J.; Burton R. E.; Walsh N. P.; McKnight C. J.; Sauer R. T. (2000) An evolutionary bridge to a new protein fold. Nat. Struct. Biol. 7, 1129–1132. 10.1038/81985. [DOI] [PubMed] [Google Scholar]
- He Y.; Chen Y.; Alexander P.; Bryan P. N.; Orban J. (2008) NMR structures of two designed proteins with high sequence identity but different fold and function. Proc. Natl. Acad. Sci. U. S. A. 105, 14412–14417. 10.1073/pnas.0805857105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander P. A.; He Y.; Chen Y.; Orban J.; Bryan P. N. (2009) A minimal sequence code for switching protein structure and function. Proc. Natl. Acad. Sci. U. S. A. 106, 21149–21154. 10.1073/pnas.0906408106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y.; Chen Y.; Alexander P. A.; Bryan P. N.; Orban J. (2012) Mutational tipping points for switching protein folds and functions. Structure 20, 283–291. 10.1016/j.str.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newlove T.; Konieczka J. H.; Cordes M. H. (2004) Secondary structure switching in Cro protein evolution. Structure 12, 569–581. 10.1016/j.str.2004.02.024. [DOI] [PubMed] [Google Scholar]
- Wagner A. (2011) The Origins of Evolutionary Innovations: A Theory of Transformative Change in Living Systems, OUP, Oxford. [Google Scholar]
- Tokuriki N.; Tawfik D. S. (2009) Stability effects of mutations and protein evolvability. Curr. Opin. Struct. Biol. 19, 596–604. 10.1016/j.sbi.2009.08.003. [DOI] [PubMed] [Google Scholar]
- Lei Y.; Takahama Y. (2012) XCL1 and XCR1 in the immune system. Microbes Infect. 14, 262–267. 10.1016/j.micinf.2011.10.003. [DOI] [PubMed] [Google Scholar]
- Sfriso P.; Duran-Frigola M.; Mosca R.; Emperador A.; Aloy P.; Orozco M. (2016) Residues Coevolution Guides the Systematic Identification of Alternative Functional Conformations in Proteins. Structure 24, 116–126. 10.1016/j.str.2015.10.025. [DOI] [PubMed] [Google Scholar]
- Piatigorsky J.; O'Brien W. E.; Norman B. L.; Kalumuck K.; Wistow G. J.; Borras T.; Nickerson J. M.; Wawrousek E. F. (1988) Gene sharing by 6-Crystallin and argininosuccinate Iyase. Proc. Natl. Acad. Sci. U. S. A. 85, 3479–3483. 10.1073/pnas.85.10.3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuriki N.; Tawfik D. S. (2009) Protein Dynamism and Evolvability. Science 324, 203–207. 10.1126/science.1169375. [DOI] [PubMed] [Google Scholar]