


Abstract
Kleefstra syndrome, a disease with intellectual disability, autism spectrum disorders and other developmental defects is caused in humans by haploinsufficiency of EHMT1. Although EHMT1 and its paralog EHMT2 were shown to be histone methyltransferases responsible for deposition of the di-methylated H3K9 (H3K9me2), the exact nature of epigenetic dysfunctions in Kleefstra syndrome remains unknown. Here, we found that the epigenome of Ehmt1+/− adult mouse brain displays a marked increase of H3K9me2/3 which correlates with impaired expression of protocadherins, master regulators of neuronal diversity. Increased H3K9me3 was present already at birth, indicating that aberrant methylation patterns are established during embryogenesis. Interestingly, we found that Ehmt2+/− mice do not present neither the marked increase of H3K9me2/3 nor the cognitive deficits found in Ehmt1+/− mice, indicating an evolutionary diversification of functions. Our finding of increased H3K9me3 in Ehmt1+/− mice is the first one supporting the notion that EHMT1 can quench the deposition of tri-methylation by other Histone methyltransferases, ultimately leading to impaired neurocognitive functioning. Our insights into the epigenetic pathophysiology of Kleefstra syndrome may offer guidance for future developments of therapeutic strategies for this disease.
INTRODUCTION
Disruption of epigenetic marks has been frequently implicated in the pathophysiology of cognitive disorders and intellectual disability (1,2). Kleefstra syndrome, caused by haploinsufficiency of EHMT1, is characterized by severe intellectual disability (ID), autism spectrum disorders (ASD), developmental delay, childhood hypotonia and craniofacial abnormalities (3,4). EHMT1 and its closely related paralog EHMT2 are histone methyltransferases primary responsible for deposition at euchromatic DNA of the di-methylated H3K9 (H3K9me2) (5–9). H3K9me2 and especially H3K9me3, which is produced by the addition a third methyl group to H3K9me2, are repressive marks that are non-permissive for transcription. Many of the human clinical features of Kleefstra syndrome are recapitulated in mice carrying constitutive haploinsufficiency of Ehmt1 (Ehmt1+/−), making it a highly valid mammalian model (10–12) for the human counterpart. Recently, first insights into the molecular pathophysiology of Kleefstra syndrome have been obtained. For instance, the cranial abnormalities were associated to a reduction of H3K9me2 and consequent UP-regulation of bone morphogenetic genes (12).
Furthermore, EHMT1 deficiency was associated with impaired synaptic scaling, a mechanism that maintains neuronal network activity homeostasis (13), delayed neuronal network development (14) and compromised neuronal identity (15). Given the many reports linking Ehmt1 or Ehmt2 complete knock-outs to loss of H3K9me2 and consequent up-regulation of gene expression (5,16–19), impaired H3K9me2/3 is suspected to be the main cause of the Kleefstra syndrome. However, no quantitative genome-wide studies have yet been reported for constitutive haploinsufficency of Ehmt1 and for this reason the epigenetic dysfunctions causing impaired brain development and mental retardation in Kleefstra patients are still largely unknown. Furthermore, EHMT1 and EHMT2 reside in common complexes (6), but it is unclear whether also haploinsufficiency of EHMT2 could result in a Kleefstra-like syndrome.
MATERIALS AND METHODS
Mutant mouse lines
The Ehmt1+/− heterozygous knockout mice were obtained from Dr Shinkai and Dr Tachibana (Kyoto University, Japan (5)). Ehmt1+/− heterozygous knockout mice were genotyped and characterized (protein level) in previous works (10–12). The Ehmt2+/− heterozygous knockout mice were obtained from Dr Feil (Institute of Molecular genetics at Montellier, France (20,21)). EHMT2 knockout efficiency and specificity was confirmed by RNA-sequencing (–44%, RNAseq, Supplementary Figure S8A) and western blotting (–78%, Supplementary Figure S8B).
Phenotyping
Animals
A cohort of 8 wt, 8 Ehmt1+/− and 8 Ehmt2+/−, mutant male mice bred at the Celphedia-ICS were used for behavioural phenotyping. They had mix genetic background (B6N*B6J 75/25) and were group housed (2–4 per cage) and allowed 2 weeks acclimation in the phenotyping area with controlled temperature (21–22°C) under a 12–12 h light–dark cycle (light on at 07 a.m.), with food and water available ad libitum. Behavioral testing started at 10-week-old and was carried out in agreement with the EC directive 2010/63/UE86/609/CEE and with the agreement of the local Ethics committee under the license number 2012–139.
Gross neurological examination
The general health and basic sensory motor functions were evaluated using a modified SHIRPA protocol (EMPRESS, eumorphia.org). This analysis provides an overview of physical appearance, body weight, body temperature, neurological reflexes and sensory abilities.
Rotarod test
This test measures the ability of an animal to maintain balance on a rotating rod (Bioseb, Chaville, France). Mice were given three testing trials during which the rotation speed accelerated from 4 to 40 rpm in 5 min. Trials were separated by 10–15 min interval. The average latency was used as index of motor coordination performance.
Grip test
This test measures the maximal muscle strength (g) using an isometric dynamometer connected to a grid (Bioseb). Each mouse was submitted to three consecutive trials immediately after the modified SHIRPA procedure. Once the animal was holding the grid with its all paws it was slowly moved backwards until it released it.
Hot plate test
The mice were placed into a glass cylinder on a hot plate adjusted to 52°C (Bioseb). The latency of the first reaction (licking, flinches, …) was recorded, with a maximum of 30 s.
Shock threshold test
The mouse is placed in the fear-conditioning chamber and allowed to habituate for 30 s. Foot-shock is then manually applied for 1 s, and behavioral responses noted. Shock levels began at 0.05 mA, and increased in 0.05 mA steps with 30 s interval between the shocks, until both flinch (any detectable response) and vocalization are induced. After this point, shocks were increased in 0.1 mA steps until a jump (the mouse flinches such that the two hind paws leave the ground) is induced. A 1 mA cut-off is employed in this test.
Circadian activity
Spontaneous locomotor activity and rears were measured using 24 individual boxes equipped with infra-red captors. The quantity of water and food consumed was measured during the test period using automated pellet feeder and lickometer (Imetronic, Pessac, France). Mice were tested for 35 h in order to measure habituation to the apparatus as well as nocturnal and diurnal activities. Results are expressed per 1 h periods.
The open field test
Mice were tested in automated open fields (Panlab, Barcelona, Spain), each virtually divided into central and peripheral regions. The open fields were placed in a room homogeneously illuminated at 70 lx. Each mouse was placed in the periphery of the open field and allowed to explore freely the apparatus for 30 min, with the experimenter out of the animal's sight. The distance traveled, the number of rears, and time spent in the central and peripheral regions were recorded over the test session. The number of entries and the percentage of time spent in center area are used as index of emotionality/anxiety.
Elevated plus maze
The apparatus used was completely automated and made of PVC (Imetronic, Pessac, France). It consisted of two open arms (30 × 5 cm) opposite one to the other and crossed by two enclosed arms (30 × 5 × 15 cm). The apparatus was equipped with infrared captors allowing the detection of the mouse in the enclosed arms and different areas of the open arms. The number of entries into and time spent in the open arms were used as an index of anxiety. Closed arm entries were used as measures of general motor activity. Ethological parameters such as stretching, attempts and head dips, were also automatically scored.
Social recognition
This task is used to evaluate the preference of a mouse for a congener as compared to an object placed in an opposite compartment. Reduced social behaviour is observed in psychiatric disorders and mental retardation. The apparatus is composed with a central starting box and two goal boxes. In each goal box, a grid cup is positioned and in an object or a congener is placed during testing. Testing was performed for two consecutive days. On the first day, fresh bedding was placed in the goal boxes. The mouse was placed in start box for 30 s then allowed to explore freely the apparatus for 10 min in order to attenuate their emotionality and to evaluate any potential preference between the two compartments. On the second day, mice were tested for social preference (social recognition). A C57Bl/6 congener from the same gender was placed in one goal box and an object (dice for example) placed in the opposite one. The mouse was then placed in the start box for 30 s then allowed to explore freely the apparatus for 10 min. The position of the congener and object boxes was counterbalanced to avoid any potential spatial preference. Mice were then tested for evaluation social memory. A third trial (10 min-duration) is performed immediately after evaluation of the preference for congener/object. The congener (familiar) is remained in the same goal box and a new congener is placed in the other goal box (where the object was previously placed). The mouse to be tested is then placed in the start box (central box) for 30 s then allowed to explore freely the apparatus for 10 min. The duration of exploration of each goal box (when the mouse is sniffing the grid delimiting the goal box) was measured. The percentage of time the mouse takes to explore the congener versus object or the novel versus familiar congener were used as index of social preference (recognition preference) and social memory. The number of entries and the time spent in each goal box, and vertical activity in each box were also measured. A social recognition index (SR) is defined as (time Congener/(time Object + time Congener)) × 100. A social memory index is defined as (time Novel Congener/(time Novel Congener + time Familiar Congener)) × 100.
Auditory startle reflex reactivity and Pre-Pulse inhibition
Acoustic startle reactivity and pre-pulse inhibition of startle were assessed in a single session using standard startle chambers (SR-Lab Startle Response System, San Diego Instruments, USA). Ten different trial type were used: acoustic startle pulse alone (110-db), eight different prepulse trials in which either 70, 75, 85 or 90-dB stimuli were presented alone or preceded the pulse, and finally one trial (NOSTIM) in which only the background noise (65 dB) was presented to measure the baseline movement in the Plexiglas cylinder. In the startle pulse or prepulse alone trials, the startle reactivity was analyzed and in the prepulse plus startle trials the amount of PPI was measured and expressed as percentage of the basal startle response.
Object recognition task
The object recognition task was performed in automated open fields (see above). The open-fields were placed in a room homogeneously illuminated at 70 lx at the level of each open field. The objects to be discriminated were a glass marble (2.5 cm diameter) and a plastic dice (2 cm). Animals were first habituated to the open-field for 30 min. The next day, they were submitted to a 10-min acquisition trial during which they were placed in the open-field in presence of an object A (marble or dice). The time the animal took to explore the object A (when the animal's snout was directed towards the object at a distance ≤ 1 cm) is manually recorded. A 10-min retention trial is performed 3 h later. During this trial, the object A and another object B are placed in the open-field, and the times tA and tB the animal takes to explore the two objects are recorded. A recognition index (RI) is defined as (tB/(tA + tB)) × 100.
Y-maze spontaneous alternation
The apparatus was a Y-maze made of Plexiglas and having 3 identical arms (40 × 9 × 16 cm) placed at 120° from each other. Each arm had walls with specific motifs allowing distinguish it from the others. Each mouse was placed at the end of one of the three arms, and allowed to explore freely the apparatus for 5min, with the experimenter out of the animal's sight. Alternations are operationally defined as successive entries into each of the three arms as on overlapping triplet sets (i.e. ABC, BCA …). The percentage of spontaneous alternation was calculated as index of working memory performance. Total arm entries and the latency to exit the starting arm were also scored as indexes of ambulatory activity and emotionality in the Y-maze, respectively.
The pavlovian fear conditioning
Polymodal operant chambers (Coulbourn Instruments, Allentown, PA, USA) were used. Each chamber (18.5 × 18 × 21.5 cm) consisted of aluminum side walls and Plexiglas rear and front (the door) walls. A loudspeaker and a bright light constituted the sources of the cues during conditioning and cue-testing. The general activity of animals was recorded through the infrared cell placed at the ceiling of the chambers and was directly recorded on a PC computer using the Graphic State software (Coulbourn). For conditioning, mice were allowed to acclimate for 4 min, then a light/tone (10 kHz) CS was presented for 20 s and co-terminated by a mild (1 s, 0.4 mA) footshock (US). Mice were returned to their home cages 2 min later. Testing was performed 24 h following conditioning session. Testing for the context was performed in the morning. The mouse was placed back into the same chamber that was used for the conditioning and allowed to explore for 6 min without presentation of the light/auditory CS. Testing for the cue was performed in the afternoon (∼5 h after the context testing). The contextual environment of the chambers was changed (wall color, odor and floor texture). The mouse was placed in the new chamber and allowed to habituate for 2 minutes then presented with light/auditory cues for 2 min. This sequence was repeated once again. At the end of testing, animals were returned to their home cages.
Statistical analysis
Data were analyzed using unpaired Student's t-test or repeated measures analysis of variance (ANOVA) with one between factors (genotype) and one within factor (time). A Student-Neumann–Keuls post hoc analysis was used to compare data between each two groups. Additional one group t-test or paired t-test were used to compare performance to the chance level or between two conditions in the same group (e.g. familiar vs novel congener). Qualitative parameters (e.g. clinical observations, susceptibility to seizures) were analyzed using χ2 test. The level of significance was set at P < 0.05.
RNA-seq and ChIP-seq libraries
Tissue samples were dissected and snap-frozen in liquid nitrogen. Next, samples were grinded in a liquid-nitrogen chilled mortar and the resulting powder was split into multiple aliquots: for RNA extraction and ChIP. Total RNA was Trizol extracted and RNA was treated with DNase (Qiagen, Venlo, The Netherlands) and purified on the RNeasy MinElute Cleanup Kit (Qiagen). 2000 ng of total RNA were treated with the Ribo-Zero rRNA Removal Kit (Human/Mouse/Rat; Illumina MRZH11124, Eindhoven, The Netherlands). Depleted RNA was precipitated 1hr at -80C in three volumes of ethanol plus 1ug of glycogen. RNA was then washed and resuspended in 36 ul of Rnase free water. RNA fragmentation buffer (NEBNext® Magnesium RNA Fragmentation Module, NEB, Leiden, The Netherlands) was added to the solution and the RNA was fragmented by incubation at 95°C for 3 min. Reverse transcription: cDNA first strand synthesis was performed with random hexamer primers. cDNA second strand synthesis was performed with dUNTPs, to ensure strand specificity. The RNA-seq library was synthesized with KAPA Hyper prep kit (Kapa Biosystems, Wilmington, MA, USA): a treatment with USER enzyme (NEB, M5505L) was added to digest the unspecific strand.
ChIP-seq samples were prepared following the standardized Blueprint-IHEC (international human epigenome consortium) protocols and antibodies (http://www.blueprint-epigenome.eu/UserFiles/File/Protocols/Histone_ChIP_May2013.pdf): H3K27ac (Diagenode pAb-196-050), H3K4me3 (Diagenode pAb-003-050), H3K4me1 (Diagenode pAb-037-050), H3K27me3 (Diagenode pAb-195-050), H3K9me3 (Diagenode pAb-193-050), H3K36me3 (Diagenode pAb-192-050) or H3K9me2 (Abcam ab1220).
Briefly, the tissue power of the ChIP aliquot was fixed with 1% formaldehyde (Sigma) in a rotating 1.5 ml Eppendorf at room temperature for 5 min. Fixed tissue preparations were sonicated using a Diagenode Bioruptor UCD-300 for 4 min (30 s on; 30 s off). Next, 67 μl of chromatin were incubated overnight at 4°C with rotation with 229 μl dilution buffer, 3 μl protease inhibitor cocktail and 0.5–1 μg of H3K27ac, H3K4me3, H3K4me1, H3K27me3, H3K9me3, H3K36me3 or H3K9me2 antibodies. Protein A/G magnetic beads (Dynabeads™ Invitrogen, 10008D, 10009D) were washed in dilution buffer with 0.15% SDS and 0.1% BSA, added to the chromatin/antibody mix and rotated for 60 min at 4°C. Beads were washed with 400 μl buffer for 5 min at 4°C with five rounds of washes. After washing chromatin was eluted using elution buffer for 20 min. Supernatant was collected, 8 μl 5M NaCl, 3μl proteinase K were added and samples were incubated for 4 h at 65°C. Finally, samples were purified using QIAGEN; Qiaquick MinElute PCR purification Kit and eluted in 20 μl EB. ChIP-seq libraries were synthesized with KAPA Hyper prep kit (Kapa Biosystems, KK8504) following the manufacturer's instructions. The libraries were pooled (4/lane) and sequenced on the Illumina HiSeq 2000. RNA-seq libraries were mapped with GSNAP (22) (version 2015-06-23, RefSeq annotations) and ChIP-seq libraries with BWA (23) (0.6.2), genome build mm9.
GO enrichments
GO annotations are updated to 25/6/2015. Enrichments are made with hypergeometric test, bonferroni corrected.
DNA bisulfate sequencing
DNA was isolated using the Cell Culture DNA Midi Kit from QIAGEN (Cat No 13343) or GenElute™ Mammalian Genomic DNA Miniprep Kit (Cat No. G1N350, Sigma). BS-seq was performed as described previously (Lister et al., 2008). FASTQ sequence files generate for BS-seq were aligned to Mus musculus NCBIM37 genome assembly (mm9) using rmapbs-pe (https://github.com/smithlabcode/rmap) allowing a maximum of 10 mismatches for efficient mapping of reads containing bisulfite converted unmethylated cytosines. 5mC level were directly determined using MethPipe (24). In short, both mates of the paired-end sequencing were mapped separately by RMAPBS. Reads mapping equally well on multiple positions on the genome were excluded from further analysis. Mates mapping within a maximum distance of 500 bp were merged and other reads were excluded from further analysis. If multiple mated reads mapped on exactly the same genomic coordinates (duplicates), all but one were discarded. Within a CpG context, symmetric cytosines on both forward and reverse strands were combined. Cytosine methylation level was called per individual C as #C/(#C + #T). For WGBS a CpG coverage threshold of ≥10 was used.
Quantitative RT-qPCR—MIQE guidelines essential information
Experimental design: Three wild type P30 hippocampi, 3 Ehmt1+/– hippocampi, H3K4me3 ChIP sample.
Sample: H3K4me3 ChIP sample, stored at –80°C.
qPCR target information: genomic amplicon, details below.
| Length | Amplicon length | Melting temp. | gc% | seq | ||
|---|---|---|---|---|---|---|
| Pcdhb5 | LEFT PRIMER | 20 | 86 | 60.69 | 50 | TGCTTCCTGGTCCTTTGTGT |
| RIGHT PRIMER | 20 | 86 | 60.93 | 50 | CCCATTCAAGGTCGGGTATT | |
| Pcdhb14 | LEFT PRIMER | 20 | 100 | 60.1 | 50 | GCAAGTGACCGCCATTATCT |
| RIGHT PRIMER | 19 | 100 | 60.24 | 57.89 | CACCAAAGAGCCCCTGTCT | |
| Pcdhb15 | LEFT PRIMER | 19 | 108 | 59.78 | 63.16 | GGCACCCCTCTCTTCTCAC |
| RIGHT PRIMER | 21 | 108 | 59.66 | 42.86 | CCCCAAATTCCTTCCATTTAG | |
| Pcdhb22 | LEFT PRIMER | 21 | 80 | 60.01 | 47.62 | GGTGCAGAAGCAAAGATGAAG |
| RIGHT PRIMER | 20 | 80 | 59.84 | 50 | ACCACTCCCAGCAAGAGAAA |
qPCR protocol: Reaction volume 25 ul composed of 10.9 ul nuclease free (NF) water, 0.5 ul primers, 1.1 ul ChIP sample in NF water, 12.5 ul IQ Sybr Supermix (Biorad #1708884). Cycles: 1×) 95°C, 3 min 40×) 95°C 10 s, 58°C 40 s melting curve) 58–95°C, increment 0.5°C, 5 s. Optical head serial number: 785BR8460. Base serial number: CT004082. CFX Manager version: 3.0.1224.1015.
qPCR validation and DATA Analysis: Biorad CFX Manager version 3.0.1224.1015. Justification of number and choice of reference genes: not applicable. Two technical replicates (wells) for each sample. For other information see Supplementary Figure S6B.
Peak calling
Peak calling was performed for H3K4me1/2, H3K27ac/me3, H3K36me3 and H3K9me3 as follows. The 25% of the mappable genome with the lowest nonzero coverage was estimated as the background level. Next, regions displaying a signal higher than two times the background for at least 300 kb were considered as peaks. Peaks called from multiple samples of the same histone mark were than merged. Merged peaks and normalized tags counts are available in GEO. The code used for peak calling has been deposited in github https://github.com/iaconogi/Histone-mark-analysis.
Data normalization and DEseq2
Significant changes in expression or histone marks were calculated as follows. Initially, the uniquely mapped tags were counted in a given annotation list. Annotations used were (i) refseq mm9 genes for RNA-seq (exon data only), (ii) refseq mm9 gene bodies for H3K36me3, (iii) refseq mm9 gene promoters (TSS ± 2 kb) for H3K9me2/3, H3K27me3, H3K4me1 and DNA methylation. (iv) Previously called peaks for H3K4me3, H3K27ac. Next, tags counts were normalized to account for library size (geometric mean) and P-values were calculated in DEseq2, which estimates the likelihood of a change by means of a negative binomial distribution.
Analysis of RNAseq and H3K4me3 in postnatal development
Genes displaying a significant (P < 0.01) change in expression (RNAseq) or H3K4me3 (promoter) in at least one developmental stage were considered deregulated in Ehmt1+/− mice as opposed to wild type mice. This resulted into 255 genes. Next, wild type samples of postnatal (P1, P7, P15, P30, RNAseq and H3K4me3) stages were analyzed in order to define the genes changing expression in the course of wild-type development. Criteria for significant changes: max (FCi) > 2, min (Pvali) <2.9e–07, i = 1:C(n,k), where FC represent the fold change, Pval the P-value, n = 4 as the number of developmental stages, k = 2 and i as the couples of developmental stages. In other terms, pair wise comparison of expression are performed for all the 12 possible couples of developmental stages (as for example P1/P30 or P7/P15). A gene is considered significantly changed if it displays in at least one of these comparison a change of expression (RNAseq or H3K4me3) which is at least 2-fold and P < 2.9e–07 (equivalent to a Z-score of 5). Threshold for P-value is very stringent because postnatal transcriptome undergoes extensive changes and we aim to select the main developmental markers. Next, all genes were collected in four different expression patterns: UP, DN, STABLE, OTHERS. Given X as the expression value, genes in which XP15 > XP1, XP30 > XP1, XP7 < XP15 are classified as up-regulated. Opposite for DN-regulated. STABLE genes do not change expression, as they do not satisfy the criteria for significant changes. OTHERS change expression, as they satisfy the criteria, but with transient variations which are neither UP nor DN.
Sliding window analysis of H3K9me2, H3K9me3 and H3K27me3 ChIP-seq
The genome wide analysis of repressive marks H3K9me2, H3K9me3 and H3K27me3 were based on a sliding window approach. As a first step, genome is divided in overlapping bins of 5 kb starting every 1.5 kb. In this way, each bin overlaps of 3.5 kb with his neighbors. For the mouse mm9 genome, the total amount of bins is 1 817 079. Subsequently, ChIP-seq tags are counted for every bin and the resulting tag counts is given as input to DEseq2, an algorithm based on the negative binomial distribution which identifies the bins presenting a significant change in the number of tags among two different conditions (for example, wild type and Ehmt1+/− hippocampus). Raw results of DEseq2 consist of a list of all the bins with significant changes. We next transformed the lists of bins in lists of regions. To this end, whenever two overlapping bins are called as significant by DEseq2 we merged them. This procedure is iteratively repeated until all the overlapping bins are merged. In this way, when a change of H3K9me3 affects a broad region, we recreate the area by merging all the individually deregulated bins. Likewise, a circumscribed change of methylation within a narrow region is detected with a single bin. Therefore, this approach is very flexible, as it allows to isolate at the same time both broad and local changes.
Ehmt1/Ehmt2 comparison of D-scores
Genes with D-scoreehmt1 > 6, D-scoreehmt2 < 6 are considered as up-regulated specifically in Ehmt1+/−. Otherwise (D-scoreehmt1 > 6, D-scoreehmt2 > 6) they are considered as commonly up-regulated. Same reasoning goes for down-regulated genes and Ehmt2+/− specific deregulations. The genes of the six regions of the Venn diagrams are ranked in the Supplementary Table S1 (GEO).
GEO (gene expression omnibus data)
Data deposited in GEO, ID GSE89010. Elaborated data uploaded in GEO includes: D-scores for P30 Ehmt1 and Ehmt2 epigenomes, developmental Ehmt1 clusters, P30 Ehmt1 deregulated genes clusters, Ehmt1 vs Ehmt2 gene sets, Raw reads counts for RNAseq samples, Peak calls and raw reads counts for H3K4me1, H3K4me3, H3K36me3, H3K27ac, H3K9me3, H3K27me3.
Clustering
Clustering of ChIP-seq regions and developmental RNAseq/H3K4me3 data was achieved by means of a three-step, semi-supervised algorithm consisting of ‘trajectory clustering’ (1st step, to find the optimal number of clusters), ‘k-means, sq-euclidean distance’ (second step, to create fine-graded clusters) and an optional ‘parametric clustering’ (third and last step, to merge fine-graded clusters in final clusters). Given a input matrix m*n of expression values which needs to be clustered, ‘trajectory clustering’ simplifies the values of the input matrix such that each original value is reduced to only five possible values delimited by the 10th, 30th, 70th and 90th percentiles. These five values can be generally interpreted as ‘not changed’, ‘up(down)-regulated’, ‘strongly up(down)-regulated’ or, equally, ‘medium’, ‘above(below)-average’, ‘ strongly above(below)-average’. Subsequently, each element is associated with a trajectory (a unique tuple of these five values) and the trajectories collecting enough elements are recognized as the real/main trajectories. The minimal number of elements that a real trajectory must collect is set with the parameter min_el. Here, we generally used very low min_el in order to retain as many trajectories as possible, to minimize loss of information. More precisely, min_el was set to 0.1%, meaning that a trajectory is considered as real if it collects at least 0.1% of the total elements to be clustered. The final number of real trajectories is equivalent to the optimal number of clusters. Once determined the optimal amount of clusters, the m*n matrix (with its original values) is then clustered via standard k-means with squared Euclidean distance. Given that a low min_el was used, the number of clusters generated via k-means was generally very high, also ranging from 40 to 70 in most cases. We refer to these clusters as fine-graded clustering. When fine-graded clusters were too many, a parametric model is generated with the aim to further collapse the centroids and obtain the final clusters. A parametric model establishes a rigid set of values (parameters) which allows to pool the centroids sharing similar features. Centroids which do not fit any of the parametric model are directly retained as individual, final clusters. At this final step, clusters which collect less than 1% of the total elements are considered outliers and discarded. The parametric models used in the manuscript are shown below.
3RD STEP: PARAMETRIC CLUSTERING OF BRAIN AREAS H3K9ME3 CENTROIDS (FC, frontal cortex, OB olfactory bulb, H hippocampus, CE cerebellum)


3RD STEP: PARAMETRIC CLUSTERING OF BRAIN Ehmt1+/− and Ehmt2+/− CENTROIDS (H3K9me3 and H3K27me3)

RESULTS
Increased H3K9 di- and tri-methylation in the adult hippocampus of Ehmt1+/− mice
To gain insight into the epigenetic changes associated with haploinsufficiency of Ehmt1 or Ehmt2 in mice, a comprehensive epigenetic dataset (seven histone marks, DNA methylation and RNAseq) encompassing 200 biological samples of multiple brain areas and developmental stages was generated according to the BLUEPRINT/IHEC standard (25) (Figure 1A). All ChIP-seq profiles show an excellent enrichment of the signal over the background (Figure 1B).
Figure 1.

Overview of the epigenetic dataset. (A) Samples table indicating the number of mice used in each experiment (wild type/mutant). (B) Examples of tracks. Y-axis: coverage is shown for ChIP-seq tracks. Methylation (0–1) is shown for DNAme track. Magnified plot for a region with increased H3K9me3 is shown (mean ± SEM). (C) Overview of genome-wide deregulations for histone marks. H3K4me3, H3K27ac, H3K36me3 and H3K4me1 peaks were called and DEseq2 was used to detect significant deregulations (P< 0.01) in Ehmt1+/− animals as compared to wild type littermates. For the repressive marks, generally distributed in extended stretches, a genome-wide sliding window analysis and DEseq2 were used to detect significant deregulations (P< 0.001, see methods).
We initially focused on the hippocampus, a crucial brain structure for a multitude of cognitive functions. A genome wide assessment of the epigenome of Ehmt1+/− (5) and wild type adult (P30) hippocampus revealed that heterozygous mice display extensive increase in H3K9me2 and H3K9me3, with more restricted changes of the other histone marks (Figure 1C). Specifically, instead of the expected decrease in H3K9 methylation, we found significantly (P<0.001, DEseq2 (26)) increased levels of H3K9me3 in Ehmt1+/− hippocampus in a total of 99.2 Mb (6009 regions, 73% intergenic, 15% promoters, 12% gene bodies) whereas only few regions (3.15 Mb, 587 regions) displayed reduced H3K9me3 (Figure 2A and B). This result was validated by the use of unbiased, non-parametric statistical tests and t-sne (27) analysis, which resulted in a clear segregation between heterozygous and wild type, as well as high concordance between biological replicates (Figure 2B and C). In the same regions displaying increased H3K9me3, H3K9me2 is increased too (Figure 2D).
Figure 2.

Epigenetic profiling of Ehmt1+/− unveils extensive up-regulation of H3K9me3. (A) Examples of regions showing a significant increase of intergenic H3K9me3 or unaltered H3K9me3 over Hoxa genes. Average H3K9me3 coverage ± SEM, P-values (DEseq2, see methods) and threshold for up/down-regulations (P< 0.001 equal to Z-score = 3). ( B) Predominant H3K9me3 up-regulation in Ehmt1+/− as show by the dimensionally reduced (t-sne) plots of deregulated regions where dot size is proportional to the average length (17 kb up, 5kb down). The clear segregation into two clouds indicates reproducibility of deregulations. (C) Clustering of statistical likelihood in H3K9me3 up-regulated regions segregates Ehmt1+/− and wild type samples (Kruskal–Wallis test, euclidean distance, hierarchical clustering with Ward's linkage). (D) Distribution of repressive epigenetic marks in the 6009 regions with increased H3K9me3. While H3K9me3 and H3K9me2 are strongly shifted towards positive values, indicating up-regulation, H3K27me3 and DNA methylation are centered on zero, indicating lack of correlation with H3K9me2/3. ( E) AU041133, the gene with the strongest epigenetic repression in Ehmt1+/−. Promoter (H3K4me1/3/K27ac/me3/K9me3), gene body (H3K36me3/K9me2), CpG island (DNAme). DEseq2 P-values *P< 0.05, **P< 0.025, ***P< 0.01 (see Materials and Methods) Y-axis, normalized tags, mean ± SEM.
One explanation that needed to be excluded is that the increased H3K9me3 levels could have been artificially generated by the normalization. The use of alternative statistical methods that are commonly used for library-normalization showed that increased H3K9me3 is consistently detected with significant P-values independently from the normalization approach (Supplementary Figure S1A).
Moreover, genes displaying increased H3K9me3 at their promoter also display reduced expression (RNA-seq), decreased active marking (H3K4me3, H3K4me1, H3K27ac, H3K36me3) and increased repressive H3K9me2 and DNA methylation (GWBS) (Figure 2E). These multiple, concordant changes in epigenetic makeup and RNA expression further support the notion that increase of H3K9me3 is not an artifact. Increased H3K9me3 mostly localizes over long stretches (from 25 kb up to 190 kb, Supplementary Figure S1B) silencing hundreds of genes belonging to large clustered families such as olfactory receptors (731/1102, P < 1e–1000), taste receptors (23/65, P < 2.3e–08), pheromone receptors (62/99, P < 1.5e–46) among others.
Increased H3K9 tri-methylation is conserved in multiple brain regions and developmental stages
We next sough to determine whether the observed increase of H3K9me3 found in hippocampus is also present in other brain regions. Analysis of wild type and Ehmt1+/− H3K9me3 samples from frontal cortex, olfactory bulb and cerebellum (20 samples, littermates) showed that increased H3K9me3 was consistently found in all investigated brain regions, whereas consistent down-regulation of H3K9me3 could not be detected (Supplementary Figure S2).
We next surmised that the observed increase of H3K9me2/3 in Ehmt1+/− could be the consequence of events occurring earlier in brain development. Therefore, we asked whether and when H3K9me3 is increased in earlier stages of postnatal hippocampal development (P1-P30 developmental stages, 20 samples, littermates). Interestingly, H3K9me3 appeared markedly higher in Ehmt1+/− hippocampi in every developmental stage tested, starting already from postnatal day 1 (Supplementary Figure S3A and B), indicating that aberrant H3K9me3 is deposited before birth at embryonic stage.
Given the remarkable extent of H3K9me3 up-regulation in multiple brain areas and developmental stages (conserved up-regulation in 51 Mb Supplementary Figure S1B and C) and the minimal (0.02 Mb) down-regulation, we assessed whether the expression of any of the known H3K9 (de)methyltransferases (namely Suv39h1/2, SetDB1/2, Prdm2, Kdm4A-D) or (de)acetyltransferases are deregulated in Ehmt1+/− brain. Intriguingly, none of these regulators appeared deregulated in any brain area or developmental stage of our dataset (Supplementary Figure S3C).
Ehmt2+/− mice present a mild increase of H3K9me3
Despite the large functional overlaps reported between the paralogous genes Ehmt1 and Ehmt2 (5,7–9,15,18), to date no GWAS study has yet found any ID patient with mutations in Ehmt2. We therefore wanted to investigate whether also Ehmt2+/− (20,21) mice show increased H3K9 methylation levels like Ehmt1+/− mice. Furthermore, given that both Ehmt1 and Ehmt2 were linked to in vivo and in vitro to tri-methylation of H3K27 (19,28,29), we determined the regions displaying significant (P < 0.01) deregulations in H3K9me3 or H3K27me3 in Ehmt1+/− or Ehmt2+/− hippocampi (22 samples, littermates). As a result, we found that in 15% of the regions H3K9me3 is up-regulated to nearly similar extent in Ehmt1+/− and Ehmt2+/− (C1) whereas in the remaining regions up-regulation in Ehmt2+/− is much weaker (44% of regions) or totally absent (39%) (Figure 3). This indicates that increase of H3K9me3 occurs also in Ehmt2+/− mice, but not with the same strength and extension as detected in Ehmt1+/− mice and hence only in a subset of regions. Alterations in the level of H3K27me3 are highly marginal in both mutants, indicating that this mark shows minimal to absent deregulation (Figure 3). Overall, these results indicate that heterozygous deletion of Ehmt2 causes an increased H3K9me3 which underpins the findings with Ehmt1+/−. However, the increase is weaker compared to Ehmt1+/-and less extensive, indicating that haploinsufficiency of Ehmt2 does not alter the epigenetic makeup as strongly as Ehmt1.
Figure 3.

Ehmt1+/− and Ehmt2+/− comparative analysis of repressive histone marks. (A) Clustering of the regions with significant (P< 0.001) H3K9me3 or H3K27me3 deregulations in Ehmt1+/− or Ehmt2+/− samples. Left: Pie chart size representing the size of each cluster in terms of base pairs covered, genome-wide. Right: Centroids mean, 25th and 75th percentiles for clusters C1–4. For each cluster the total amount of regions and their total length (size in base pairs) is indicated. Only in 15% of regions (C1) Ehmt2+/− displays an increase of H3K9me3 comparable to Ehmt1+/−. Changes in H3K27me3 are negligible (C4, 2%). (B) Example of H3K9me3 tracks. Average +/- SEM of a region formed by elements of C1 and C2 and encompassing several olfactory receptors (from Olfr697 to Olfr711, not shown).
Ehmt1+/− mice display impaired expression of protocadherins
We next set out to unravel the molecular dysfunctions of Ehmt1+/− brain by analyzing the epigenome (seven histone marks, RNA-seq and DNA methylation) of adult (P30) hippocampus at actively expressed genes. Integrated analysis of histone marks, RNA-seq and DNA methylation (see Supplementary Figure S4A) revealed 53 up-regulated and 160 down-regulated genes in Ehmt1+/− compared to wild type. In the 53 up-regulated genes a very mild decrease of the repressive mark H3K9me2 at promoters correlates with a marked increase of the four activating marks (especially H3K4me3) and RNA expression levels (Supplementary Figure S4B). Vice versa, down-regulated genes generally display a marked increase of H3K9me2/3 and DNA methylation at their promoters concomitantly with a decrease of active marks and RNA expression (Figure 4A, Supplementary Figure S4B). GO (gene ontology) analysis of the down-regulated genes revealed exclusively a significantly enrichment in clustered Protocadherins (20/59, P < 8.5e–33) whereas no enrichments were found for up-regulated genes. Interestingly, we found that only members of clustered Protocadherins (Pcdhs) genes (alpha, beta and gamma families) are down-regulated, whereas non-clustered Pcdhs genes are unaffected. Clustered Pcdhs are a major family (59 members) belonging to cadherin superfamily and expressed primarily in the vertebrate nervous system. Similarly to olfactory receptors, these Pcdhs are clustered in the genome in order to allow combinatorial and monoallelic expression which is fundamental for the proper development of dendritic arbors (30,31). The abundance of clustered Pcdhs among epigenetically repressed genes in Ehmt1+/− represents an interesting finding and prompted for a more detailed investigation. Analysis of all clustered Pcdhs promoters (59) revealed that the activating marks H3K4me3, H3K27ac, H3K36me3 are globally decreased in Ehmt1+/− (84%, 90%, 90% of promoters, respectively) whereas H3K9me2/3 is globally increased (>90%, Figure 4B and C). Furthermore, besides hippocampus, also frontal cortex, cerebellum and olfactory bulb show decreased H3K4me3 at Pcdhs promoters (Supplementary Figure S5A) indicating that multiple regions in Ehmt1+/− brain suffer epigenetic silencing of Pcdhs. Notably, the top down-regulated Pcdhs are not randomly scattered but instead sequential ones (Figure 4H, Supplementary Figure S6). In fact, down-regulation follows a graded pattern with peaks of repression in specific groups, for instance Pcdhb13–16 in the beta cluster (Figure 4H). Ehmt1 and H3K9 methylation were already known to be important for stochastic expression of olfactory receptors (18), and here we extend its importance also to the clustered Pcdhs.
Figure 4.

Epigenetic profiling of Ehmt1+/− developmental and adult hippocampus. (A) Increase of H3K9 methylation and repression of expression. Boxplot, mean ± SEM and 99% confidence interval for the genes of down-regulated cluster 1. Other deregulated genes are shown in Supplementary Figure S4B. (B) Epigenetic repression of protocadherins in Ehmt1+/− hippocampus. Smoothed bin count, percentages represent number of promoters <0 (down) or >0 (up). (C) Pcdhs beta subfamily is the most affected (9/21, 42% are repressed), example of Pcdhb15 (mean ± SEM, *P< 0.05, **P< 0.025, ***P< 0.01). (D) Analysis of postnatal development: 255 were found deregulated (P< 0.01). The genes were divided in 5 clusters according to the profile (see methods). The biggest cluster, C1, is shown, mean+/-SEM, 99% confidence interval (additional clusters in Supplementary Figure S5C-F). For C1, Kruskal-Wallis test rejects the hypothesis of different means, both in RNA-seq and H3k4me3, indicating stability of repression from P1 to P30. (E) Deregulated Ehmt1+/− epigenomes are largely stable during development, implying that deregulations are established during embryogenesis. Stacked centroids of the five clusters (C1–C5) transformed in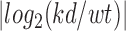 each weighted for its size. (F–G) Examples of Ehmt1+/− deregulated genes in development. Y-axis, number of tags, normalized to P1, mean ± SEM. DEseq2 P-values (*P< 0.05, **P< 0.025, ***P< 0.01). (H) Overview of Pcdhs beta cluster, from Pcdhb1 to Pcdhb22, red-colored proportionally to the average RNAseq and H3K4me3 down-regulation. Down-regulation has a clear spatial distribution, with Pcdhb1 and Pcdhb14/15 being at the center of strongest repressed loci.
each weighted for its size. (F–G) Examples of Ehmt1+/− deregulated genes in development. Y-axis, number of tags, normalized to P1, mean ± SEM. DEseq2 P-values (*P< 0.05, **P< 0.025, ***P< 0.01). (H) Overview of Pcdhs beta cluster, from Pcdhb1 to Pcdhb22, red-colored proportionally to the average RNAseq and H3K4me3 down-regulation. Down-regulation has a clear spatial distribution, with Pcdhb1 and Pcdhb14/15 being at the center of strongest repressed loci.
Deregulations of gene expression in Ehmt1+/− hippocampus are detected already at postnatal day 1
Epigenetic analysis revealed that a relatively reduced (213) set of specific genes is deregulated in adult (P30) Ehmt1+/– hippocampus. However, EHMT1 protein was shown to be 10x more expressed at P4 as compared to P30, suggesting a prominent role for EHMT1 in the early postnatal development (11). Therefore, we set to quantitatively define the interplay of EHMT1 with postnatal development by selecting the genes displaying a significant (P < 0.01) change in expression (RNAseq) or H3k4me3 (promoter) in at least one developmental stage (P1, P7, P15 or P30), which yielded 255 genes. Interestingly, most the genes deregulated at P30 appear deregulated already at P1 (Figure 4D and E), indicating that deregulation is established during embryonic stage, same as found for the H3K9me3 increase. Several Pcdhs (19/71) are already repressed at P1, as for instance Pcdhb14 (Figure 4F). This finding is highly relevant because it means that Pcdhs expression is impaired during the synaptogenesis and sprouting of dendrites, which can account for the reduced dendritic arborizaton found in Ehmt1+/− brain (15). Finally, of the 5549 genes found to be regulated during wild-type hippocampus development (Supplementary Figure S5B), only 109/5549 (<2%, such as Zfp951 in Figure 4G) are deregulated in Ehmt1+/−, indicating that the influence of Ehmt1 in regulation of gene expression during postnatal developing is limited and specific. This most likely occurs because the majority (98.6%) of postnatally regulated genes does not colocalize with the long stretches of H3K9me3 which are deregulated in Ehmt1+/−.
Ehmt2+/− displays mild deregulations of gene expression which are occluded by Ehmt1+/−
It is hitherto unknown whether haploinsufficiency of Ehmt2 would result in a Kleefstra-like phenotype as with Emht1+/− (10,11,12). To further test the similarities and dissimilarities between Ehmt1 and Ehmt2 epigenomes, we compared Ehmt1+/− and Ehmt2+/− mutant epigenomes (seven histone marks, RNA-seq and DNA methylation) in P30 hippocampus with a focus on deregulations of gene expression (see Materials and Methods).
Comparative epigenetic analysis revealed that, in both mutants, down-regulated genes outnumber up-regulated genes (>3× Ehmt1+/−, >2× Ehmt2+/−), in agreement with the observed increase in the repressive mark H3K9 methylation. However, Ehmt1+/− displays a larger ensemble of deregulated genes compared to Ehmt2+/− (227 versus 102).
In the up-regulated genes (Figure 5A), Ehmt1+/− and Ehmt2+/− display a reduced intersection (10 genes, example Tfip2, Figure 5B) and larger sets of specific genes (44 and 22 respectively). Interesting examples are Tdo2, an enzyme implicated in serotonin metabolism, which is epigenetically up-regulated only in Ehmt1+/− (Figure 5B, details in Supplementary Figure S4A) and Glo1, a metabolic detoxification enzyme, which is epigenetically up-regulated only in Ehmt2+/− (Figure 5B).
Figure 5.

Ehmt1+/− and Ehmt2+/− comparative analysis of deregulated genes. (A) Venn diagrams comparing Ehmt1+/− and Ehmt2+/− deregulated genes. Diagrams are based on D-scores which take into account expression and epigenetic makeup (see Materials and Methods). (B) Most representative examples (as ranked in Supplementary Table S1, GEO). Only deregulated marks are shown and D-scores of each gene is indicated for each mutant model (E1 = Ehmt1+/−, E2 = Ehmt1+/-).
Vice versa, in down-regulated genes we found a larger inclusion of the Ehmt2+/– ensemble into the Ehmt1+/– ensemble, as 28 genes (60%) are down-regulated also in Ehmt1+/– (Figure 5A). Commonly repressed genes include a large amount of zinc finger proteins (>35%, >10-fold enrichment, P < 1e–7, example Zfp951). In contrast, extensive repression of Pcdhs is specific to Ehmt1+/– (22 genes, >12-fold enrichment). These data indicate that haploinsufficiency of Ehmt2 causes milder deregulations of gene expression compared to Ehmt2+/−, which is in agreement with the milder H3K9me3 increase. Furthermore, several of Ehmt2 deregulations are occluded by Ehmt1+/−, supporting the latter paralog as the primary one in brain development and functioning.
Ehmt2+/− mice do not present the severe cognitive deficits presented by Ehmt1+/− mice
Phenotyping of Ehmt1+/− have previously highlighted severe developmental delay, hypoactivity and mental retardation (10–12). However, phenotyping of Ehmt2+/– mice have not been reported so far. Given that Ehmt1 and Ehmt2 work in heterodimeric complex and often display overlapping, non-redundant functions (5,6), we compared the phenotypes of Ehmt1+/– and Ehmt2+/-mice using a broad pallet of tests to evaluate general health, cognitive and behavioral features. Comprehensive phenotyping of Ehmt1+/− and Ehmt2+/− mice (>20 behavioral and cognitive tests, Figure 6A) revealed that Ehmt1+/− but not Ehmt2+/- are hypoactive (circadian activity Figure 6B, exploration of openfield Figure 6C, social behavior Figure 6D, habituation in fear conditioning Figure 6F). Different tests found alteration of learning and memory only in Ehmt1+/-mice, including a significant reduction of object recognition index (Figure 6E) and a significantly lower suppression of activity than wild type mice during re-exposure to the same context in the fear conditioning/context test (Figure 6F–G). Impaired sensory motor gating (Supplementary Figure S7D) was found in both mutants, for the 75 dB prepulse for Ehmt2+/− mice and through the different prepulses for Ehmt1+/− mice (significant for 90 dB). In social behavior, both Ehmt1+/− and Ehmt2+/− mice showed normal social preference but impairment of social memory (Supplementary Figure S7E,F). No other significant changes in mutants as opposed to wild-types were found. These data indicate that Ehmt1+/– but not Ehmt2+/− mice show impaired cognitive abilities and hypoactive behavior.
Figure 6.

Phenotyping (A) overview. (B) Circadian activity, actimetric cages. Locomotion is measured by the number of consecutive beam breaks (separated by 7.5 cm). There are two beams in the cage (front and back). When the mouse go front, (break the front beam) then back (break the back beam), the system records 1 unit. Ehmt1+/− mice display decreased locomotor activity and rears during the habitation phase. (C) Openfield, Ehtm1+/− display reduced locomotory activity and rears. (D) The number of visits and number of rears in the social recognition test. (E) Duration of object exploration during acquisition, and object recognition performance during retention. (F–G) Duration of immobility during habituation to the conditioning cage and during context in the fear conditioning, and contextual freezing performance (*P< 0.05, **P< 0.01, ANOVA, Student–Newman–Keuls, §P< 0.05, one group t-test, see Materials and Methods).
DISCUSSION
We describe here the detailed epigenetic and phenotypic characterization of the Kleefstra syndrome Ehmt1+/-mouse models and of its paralog Ehmt2+/−. Unexpectedly, we determined that constitutive haploinsufficiency of EHMT1 and, to a lower extent, EHMT2 results in increased H3K9 trimethylation in targeted genetic loci. An extensive number of biochemical studies have firmly established EHMT1 and -2 as methyltrasferases (28,32), and that complete loss of either Ehmt1 or Ehmt2 leads to a reduction of H3K9me2 (5,7–9,16–19) and also H3K9me3 (18). While we did find changes in genes previously shown to be targets of EHMT1/2 (i.e. olfactory receptors) (18,33), the overriding change we observed in our epigenomic profiling of Ehmt1+/− brain was paradoxically increased and not decreased H3K9me3. Importantly, the biological material we have analyzed is different from that in previous studies in at least two ways: firstly, our mutation is constitutively present throughout the development, unlike other models inducing EHMT1 deficiency at postnatal stages only (15). Secondly, we studied haploinsufficency, which in humans causes Kleefstra syndrome, and not complete loss of Ehmt1 as in homozygous knockouts (5,15,16,19).
Interestingly, a previous study showed that homozygous Ehmt2−/− embryos, but not heterozygous Ehmt2+/– embryos present severe growth retardation, early lethality and drastically reduced H3K9me2 (western blot Figure 3 (16)). Likewise, Ehmt1−/− but not Ehmt1+/− embryos were shown to present severe growth retardation and early lethality to the same extent of Ehmt2−/− embryos (5). These evidences suggest that the embryonic loss of H3K9me2 is not present in heterozygous mutants, in agreement with the lack of a global reduction of H3K9me2 in our heterozygous mice.
We envisage that compensatory effects took place during development of Ehmt1 and Ehmt2 heterozygous embryos. Specifically, while Ehmt1/Ehmt2 are the main H3K9me1/2 HMTs, they were reported to be inefficient in depositing H3K9me3 (7,34–36). Yet, we unveiled a strong increase especially of H3K9me3 suggesting that other HMTs, possibly Suv39H1/2 or SetDB1/2 are involved. Interestingly, it was previously hypothesized that EHMT1/2 via their ankyrin repeats bind to H3K9me1/2 quenching the deposition of tri-methylation by other HMTs (7). Our novel data is compatible with this hypothesis. Given (37) that H3K9me0→1 is methylated faster than H3K9me1→2, one possible scenario is that reduced levels of EHMT1 are enough to establish H3K9me1 but not to prevent the methylation to H3K9me3 by other HMTs. Another plausible hypothesis is that the excessive H3K9 methylation detected at postnatal stages occurred at earlier development stages and is subsequently maintained. Specifically, haploinsufficiency of Ehmt1 could cause aberrant activity of other HMTs such as Suv39H1/2 or SetDB1/2 in the dividing radial glia and/or neuronal progenitors, which are abundant in embryonic stage (38). Next, the replication machinery would maintain the aberrant H3K9 methylation up to the post-mitotic cells of the postnatal brain (39). This scenario is consistent with our findings in Ehmt1+/− hippocampus of increased H3K9me3 already at P1 and stable postnatal epigenetic deregulations.
Our work provides major advancements in the understanding of Ehmt1, Ehmt2 and their interplay in cognitive disorders. Ehmt1 and Ehmt2 have been reported to work as a heterodimeric complex (5,6). In line, deficiency of either Ehmt1 or Ehmt2 has often been associated to similar phenotypic/epigenetic outcomes (5,7–9,15,18). Conversely, we find that haploinsufficiency of Ehmt2 has a considerably lower epigenetic and behavioral impact than Ehmt1, indicating the latter as more crucial in brain. Our result is compatible with a previous report showing that EHMT1, but not EHMT2, is a limiting factor controlling the amount of EHMT1/EHMT2 holoenzyme complex. Collectively, these results may explain why patients with Ehmt2 genetic defects have not yet been reported in ID/ASD population.
Interestingly, we also found several genes specifically deregulated only in Ehmt1+/− or only in Ehmt2+/−. This is in agreement with other recent findings suggesting that Ehmt1 and Ehmt2, although being able to form heterodimers, may have functions independent of each other. Specifically, (i) findings of functional differences between Ehtm1 and Ehmt2, as for instance diverse binding affinity towards histone substrates (32), (ii) different tissue-specific expression profiles, as shown at the protein level in the auditory brainstem and adipose tissue (40,41) and at the RNA level in human tissue atlas (42) and (iii) markedly different phenotypes of Ehmt1 and Ehmt2 mutants with impaired ankyrin domains (17).
Interestingly, commonly repressed genes in both Ehmt1+/− and Ehmt2+/− hippocampi include zinc finger proteins, which were previously shown to be binding factors recruiting EHMT1 and EHMT2 to the histone substrates (7,43–45).
Our epigenetic analysis disclosed the potential molecular mechanisms impaired in the Kleefstra syndrome. Interestingly Tdo2, an enzyme indirectly involved in serotonin metabolism (46), is in our data up-regulated only in Ehmt1+/− (2.2-fold H3K4me3, 1.7 RNA-seq plus changes in H3K4me1, H3K27ac, H3K36me3, DNA methylation), suggesting that serotonin deregulation could be one underlying cause of the depressive/mood disorders we observed in patients with Kleefstra syndrome (47). Also the Protocadherins (Pcdhs), cell-adhesion molecules that confer unique identities to individual neurons (30,31), appeared extensively repressed specifically in Ehmt1+/− brain.
Recently, Pcdhs were shown to be stochastically expressed in single cells via differential DNA methylation at individual promoters (48). Specifically, in a Dnmt3b knockout model, impaired DNA methylation was associated with up-regulation of multiple alternative Pcdhs isoforms in single neurons, with no detectable change in global (bulk) brain levels. Our finding does not seem to be compatible this scenario because we did not find significant changes in DNA methylation of Pcdhs promoters. Furthermore, unlike Toyoda and coworkers (48), we detect a global change (reduction) of Pcdhs bulk expression. In addition to DNA methylation, Pcdhs expression is also regulated by long range interactions between enhancers and promoters mediated by CTCF-cohesin (49,50). To date, several different enhancers have been found for Pcdhs alpha, beta and gamma (48–51) and a model of a ‘transcriptional hub’ recruiting promoters to enhancers has been proposed (49). Disruption of interaction mechanisms by knocking out CTCF resulted in a global reduction in Pcdhs bulk expression similar (albeit stronger) to ours (50). Furthermore, a targeted deletion of DNase I hypersensitive (HS) site HS5–1, an enhancer for the alpha cluster, caused a graded down-regulation of Pcdhs (51) which resembles the graded patterns found in Ehmt1+/-samples. Graded down-regulation may be due to different binding affinities of the enhancers to the individual Pcdhs promoters, as shown for the alpha cluster (49). Interestingly, the zinc finger protein Wiz, previously shown to form a repressive Wiz/EHM1/EHMT2 tri-complex (45), was recently found to localize at CTCF biding-sites and to act as a transcriptional activator of Pcdhs. This could explain how a decrease of EHMT1 can cause a down-regulation of Pcdhs. In conclusion, we believe that our data is compatible with an altered chromatin looping caused by aberrant H3K9 methylation and impaired accessibility of enhancers. Still, the nature of our data (bulk) prevents us to draw exact conclusions about the deregulation of Pcdhs in individual cells.
The main phenotype associated with Pcdhs malfunctioning is a reduced dendritic arborization together with reduced mEPCSs (miniature excitatory postsynaptic potentials) (30,31,50,52). Interestingly, the same phenotype was found in Ehmt1+/− hippocampal neurons (11). Whether in Ehmt1+/− brain the repression of Pcdhs is the effect and not the cause of reduced dendritic arborization and synaptic changes is unlikely considering that Pcdhs appeared repressed already at P1 stage, which is before the formation of dendrites and synapses. At present, there is only one case (53) of a syndrome in which gene expression analysis in a mouse model has revealed a down-regulation of Pcdhs, the Cornelia de Lange syndrome (developmental abnormalities, intellectual disability, autistic-like behaviors) (54). It is intriguing to speculate that both Kleefstra and Cornelia de Lange syndromes could actually belong to a novel, broader class of ID syndromes sharing malfunctioning of Pcdhs at the molecular level.
DATA AVAILABILITY
Data deposited in GEO under accession number GSE89010 (https://www.ncbi.nlm.nih.gov/geo/). The code used for peak calling has been deposited in github https://github.com/iaconogi/Histone-mark-analysis.
Supplementary Material
ACKNOWLEDGEMENTS
Acknowledgements to Dr Yochi Shinkai and Dr Makoto Tachibana (Kyoto University, Japan) for providing the Ehmt1+/– mice and to Dr Feil (Institute of Molecular genetics at Montellier, France) for providing the Ehmt2+/– mice.
Author contributions (CRediT taxonomy): Conceptualization, G.I. and H.G.S.; Methodology, G.I. and H.G.S.; Software, G.I.; Validation, G.I. and M.B.; Formal analysis, G.I.; Investigation, G.I.; Resources, G.I, M.B., A.D, N.N.K.; Data curation, G.I.; Writing-original draft, G.I., H.G.S.; Visualization, G.I.; Supervision, H.G.S.; Project administration, H.G.S., H.B. and Y.H.; Funding acquisition, H.G.S., H.B. and Y.H.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
GENCODYS, an EU FP7 large-scale integrating project [241995 to H.B., Y.H., H.G.S.]. Funding for open access charge: European Union 7th framework program [Health-241995].
Conflict of interest statement. None declared.
REFERENCES
- 1. Lagali P.S., Corcoran C.P., Picketts D.J.. Hippocampus development and function: role of epigenetic factors and implications for cognitive disease. Clin. Genet. 2010; 78:321–333. [DOI] [PubMed] [Google Scholar]
- 2. Hsieh J., Eisch A.J.. Epigenetics, hippocampal neurogenesis, and neuropsychiatric disorders: unraveling the genome to understand the mind. Neurobiol. Dis. 2010; 39:73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kleefstra T., Brunner H.G., Amiel J., Oudakker A.R., Nillesen W.M., Magee A., Geneviève D., Cormier-Daire V., van Esch H., Fryns J.-P. et al. . Loss-of-function mutations in euchromatin histone methyl transferase 1 (ehmt1) cause the 9q34 subtelomeric deletion syndrome. Am. J. Hum. Genet. 2006; 79:370–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kleefstra T., van Zelst-Stams W.A., Nillesen W.M., Cormier-Daire V., Houge G., Foulds N., van Dooren M., Willemsen M.H., Pfundt R., Turner A., Wilson M. et al. . Further clinical and molecular delineation of the 9q subtelomeric deletion syndrome supports a major contribution of ehmt1 haploinsufficiency to the core phenotype. J. Med. Genet. 2009; 46:598–606. [DOI] [PubMed] [Google Scholar]
- 5. Tachibana M., Ueda J., Fukuda M., Takeda N., Ohta T., Iwanari H., Sakihama T., Kodama T., Hamakubo T., Shinkai Y.. Histone methyltransferases g9a and glp form heteromeric complexes and are both crucial for methylation of euchromatin at h3-k9. Genes Dev. 2005; 19:815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tachibana M., Matsumura Y., Fukuda M., Kimura H., Shinkai Y.. G9a/glp complexes independently mediate h3k9 and dna methylation to silence transcription. EMBO J. 2008; 27:2681–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Collins R., Cheng X.. A case study in cross-talk: the histone lysine methyltransferases g9a and glp. Nucleic Acids Res. 2010; 38:3503–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Benevento M., van de Molengraft M., van Westen R., van Bokhoven H., Kasri N.N.. The role of chromatin repressive marks in cognition and disease: A focus on the repressive complex glp/g9a. Neurobiol. Learn. Mem. 2015; 124:88–96. [DOI] [PubMed] [Google Scholar]
- 9. Shinkai Y., Tachibana M.. H3k9 methyltransferase g9a and the related molecule glp. Genes Dev. 2011; 25:781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Balemans M.C.M., Huibers M.M.H., Eikelenboom N.W.D., Kuipers A.J., van Summeren R.C.J., Pijpers M.M.C.A., Tachibana M., Shinkai Y., van Bokhoven H., Van der Zee C.E.E.M.. Reduced exploration, increased anxiety, and altered social behavior: Autistic-like features of euchromatin histone methyltransferase 1 heterozygous knockout mice. Behav. Brain Res. 2010; 208:47–55. [DOI] [PubMed] [Google Scholar]
- 11. Balemans M.C.M., Kasri N.N., Kopanitsa M.V., Afinowi N.O., Ramakers G., Peters T.A., Beynon A.J., Janssen S.M., van Summeren R.C.J., Eeftens J.M., Eikelenboom N., Van der Zee C.E.E.M. et al. . Hippocampal dysfunction in the euchromatin histone methyltransferase 1 heterozygous knockout mouse model for kleefstra syndrome. Hum. Mol. Genet. 2013; 22:852–866. [DOI] [PubMed] [Google Scholar]
- 12. Balemans M.C.M., Ansar M., Oudakker A.R., van Caam A.P.M., Bakker B., Vitters E.L., van der Kraan P.M., de Bruijn D.R.H., Janssen S.M., Kuipers A.J., Huibers M.M.H. et al. . Reduced euchromatin histone methyltransferase 1 causes developmental delay, hypotonia, and cranial abnormalities associated with increased bone gene expression in kleefstra syndrome mice. Dev. Biol. 2014; 386:395–407. [DOI] [PubMed] [Google Scholar]
- 13. Benevento M., Iacono G., Selten M., Ba W., Oudakker A., Frega M., Keller J., Mancini R., Lewerissa E., Kleefstra T. et al. . Histone methylation by the kleefstra syndrome protein ehmt1 mediates homeostatic synaptic scaling. Neuron. 2016; 91:341–355. [DOI] [PubMed] [Google Scholar]
- 14. Bart Martens M., Frega M., Classen J., Epping L., Bijvank E., Benevento M., van Bokhoven H., Tiesinga P., Schubert D., Nadif Kasri N.. Euchromatin histone methyltransferase 1 regulates cortical neuronal network development. Sci. Rep. 2016; 6:35756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schaefer A., Sampath S.C., Intrator A., Min A., Gertler T.S., Surmeier D.J., Tarakhovsky A., Greengard P.. Control of cognition and adaptive behavior by the glp/g9a epigenetic suppressor complex. Neuron. 2009; 64:678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tachibana M., Sugimoto K., Nozaki M., Ueda J., Ohta T., Ohki M., Fukuda M., Takeda N., Niida H., Kato H. et al. . G9a histone methyltransferase plays a dominant role in euchromatic histone h3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 2002; 16:1779–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu N., Zhang Z., Wu H., Jiang Y., Meng L., Xiong J., Zhao Z., Zhou X., Li J., Li H. et al. . Recognition of h3k9 methylation by glp is required for efficient establishment of h3k9 methylation, rapid target gene repression, and mouse viability. Genes Dev. 2015; 29:379–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lyons D.B., Magklara A., Goh T., Sampath S.C., Schaefer A., Schotta G., Lomvardas S.. Heterochromatin-mediated gene silencing facilitates the diversification of olfactory neurons. Cell Rep. 2014; 9:884–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mozzetta C., Pontis J., Fritsch L., Robin P., Portoso M., Proux C., Margueron R., Ait-Si-Ali S.. The histone h3 lysine 9 methyltransferases g9a and glp regulate polycomb repressive complex 2-mediated gene silencing. Mol. Cell. 2014; 53:277–289. [DOI] [PubMed] [Google Scholar]
- 20. Auclair G., Borgel J., Sanz L.A., Vallet J., Guibert S., Dumas M., Cavelier P., Girardot M., Forné T., Feil R. et al. . Ehmt2 directs dna methylation for efficient gene silencing in mouse embryos. Genome Res. 2016; 26:192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wagschal A., Sutherland H.G., Woodfine K., Henckel A., Chebli K., Schulz R., Oakey R.J., Bickmore W.A., Feil R.. G9a histone methyltransferase contributes to imprinting in the mouse placenta. Mol. Cell. Biol. 2008; 28:1104–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu T.D., Nacu S.. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010; 26:873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li H., Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Song Q., Decato B., Hong E.E., Zhou M., Fang F., Qu J., Garvin T., Kessler M., Zhou J., Smith A.D.. A reference methylome database and analysis pipeline to facilitate integrative and comparative epigenomics. PLoS ONE. 2013; 8:e81148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hsieh J., Eisch A.J.. Neurobiol. Dis. 2010; 39:73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. van der Maaten L., Hinton G.. Visualizing high-dimensional data using t-sne. Journal of Machine Learning Research. 2008; 9:2579–2605. [Google Scholar]
- 28. Tachibana M., Sugimoto K., Fukushima T., Shinkai Y.. Set domain-containing protein, g9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone h3. J. Biol. Chem. 2001; 276:25309–25317. [DOI] [PubMed] [Google Scholar]
- 29. Wu H., Chen X., Xiong J., Li Y., Li H., Ding X., Liu S., Chen S., Gao S., Zhu B.. Histone methyltransferase g9a contributes to h3k27 methylation in vivo. Cell Res. 2011; 21:365–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Keeler A.B., Molumby M.J., Weiner J.A.. Protocadherins branch out: Multiple roles in dendrite development. Cell Adhesion Migration. 2015; 9:214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hirano K., Kaneko R., Izawa T., Kawaguchi M., Kitsukawa T., Yagi T.. Single-neuron diversity generated by protocadherin-β cluster in mouse central and peripheral nervous systems. Front. Mol. Neurosci. 2012; 5:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Collins R.E., Northrop J.P., Horton J.R., Lee D.Y., Zhang X., Stallcup M.R., Cheng X.. The ankyrin repeats of g9a and glp histone methyltransferases are mono- and dimethyllysine binding modules. Nat. Struct. Mol. Biol. 2008; 15:245–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Magklara A., Yen A., Colquitt B.M., Clowney E.J., Allen W., Markenscoff-Papadimitriou E., Evans Z.A., Kheradpour P., Mountoufaris G., Carey C. et al. . An epigenetic signature for monoallelic olfactory receptor expression. Cell. 2011; 145:555–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Collins R.E., Tachibana M., Tamaru H., Smith K.M., Jia D., Zhang X., Selker E.U., Shinkai Y., Cheng X.. In vitro and in vivo analyses of a phe/tyr switch controlling product specificity of histone lysine methyltransferases. J. Biol. Chem. 2005; 280:5563–5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Trojer P., Zhang J., Yonezawa M., Schmidt A., Zheng H., Jenuwein T., Reinberg D.. Dynamic histone h1 isotype 4 methylation and demethylation by histone lysine methyltransferase g9a/kmt1c and the jumonji domain-containing jmjd2/kdm4 proteins. J. Biol. Chem. 2009; 284:8395–8405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Patnaik D., Chin H.G., Estève P.-O., Benner J., Jacobsen S.E., Pradhan S.. Substrate specificity and kinetic mechanism of mammalian g9a histone h3 methyltransferase. J. Biol. Chem. 2004; 279:53248–53258. [DOI] [PubMed] [Google Scholar]
- 37. Zee B.M., Levin R.S., Xu B., LeRoy G., Wingreen N.S., Garcia B.A.. In vivo residue-specific histone methylation dynamics. J. Biol. Chem. 2010; 285:3341–3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Iacono G., Benevento M., Dubos A., Herault Y., van Bokhoven H., Nadif Kasri N., Stunnenberg H.G.. Integrated transcriptional analysis unveils the dynamics of cellular differentiation in the developing mouse hippocampus. Sci. Rep. 2017; 7:18073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xu M., Wang W., Chen S., Zhu B.. A model for mitotic inheritance of histone lysine methylation. EMBO Rep. 2011; 13:60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ebbers L., Runge K., Nothwang H.G.. Differential patterns of histone methylase ehmt2 and its catalyzed histone modifications h3k9me1 and h3k9me2 during maturation of central auditory system. Cell Tissue Res. 2016; 365:247–264. [DOI] [PubMed] [Google Scholar]
- 41. Ohno H., Shinoda K., Ohyama K., Sharp L.Z., Kajimura S.. Ehmt1 controls brown adipose cell fate and thermogenesis through the prdm16 complex. Nature. 2013; 504:163–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. GTEx Consortium The genotype-tissue expression (gtex) project. Nat. Genet. 2013; 45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nishida M., Kato M., Kato Y., Sasai N., Ueda J., Tachibana M., Shinkai Y., Yamaguchi M.. Identification of znf200 as a novel binding partner of histone h3 methyltransferase g9a. Genes Cells. 2007; 12:877–888. [DOI] [PubMed] [Google Scholar]
- 44. Vassen L., Fiolka K., Möröy T.. Gfi1b alters histone methylation at target gene promoters and sites of gamma-satellite containing heterochromatin. EMBO J. 2006; 25:2409–2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ueda J., Tachibana M., Ikura T., Shinkai Y.. Zinc finger protein wiz links g9a/glp histone methyltransferases to the co-repressor molecule ctbp. J. Biol. Chem. 2006; 281:20120–20128. [DOI] [PubMed] [Google Scholar]
- 46. Nabi R., Serajee F.J., Chugani D.C., Zhong H., Huq A.H.M.M.. Association of tryptophan 2,3 dioxygenase gene polymorphism with autism. Am. J. Med. Genet. B, Neuropsychiatric Genet. 2004; 125:63–68. [DOI] [PubMed] [Google Scholar]
- 47. Vermeulen K., de Boer A., Janzing J.G.E., Koolen D.A., Ockeloen C.W., Willemsen M.H., Verhoef F.M., van Deurzen P.A.M., van Dongen L., van Bokhoven H. et al. . Adaptive and maladaptive functioning in kleefstra syndrome compared to other rare genetic disorders with intellectual disabilities. Am. J. Med. Genet. A. 2017; doi:10.1002/ajmg.a.38280. [DOI] [PubMed] [Google Scholar]
- 48. Toyoda S., Kawaguchi M., Kobayashi T., Tarusawa E., Toyama T., Okano M., Oda M., Nakauchi H., Yoshimura Y., Sanbo M. et al. . Developmental epigenetic modification regulates stochastic expression of clustered protocadherin genes, generating single neuron diversity. Neuron. 2014; 82:94–108. [DOI] [PubMed] [Google Scholar]
- 49. Guo Y., Monahan K., Wu H., Gertz J., Varley K.E., Li W., Myers R.M., Maniatis T., Wu Q.. Ctcf/cohesin-mediated dna looping is required for protocadherin α promoter choice. PNAS. 2012; 109:21081–21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hirayama T., Tarusawa E., Yoshimura Y., Galjart N., Yagi T.. Ctcf is required for neural development and stochastic expression of clustered pcdh genes in neurons. Cell Rep. 2012; 2:345–357. [DOI] [PubMed] [Google Scholar]
- 51. Yokota S., Hirayama T., Hirano K., Kaneko R., Toyoda S., Kawamura Y., Hirabayashi M., Hirabayashi T., Yagi T.. Identification of the cluster control region for the protocadherin-beta genes located beyond the protocadherin-gamma cluster. J. Biol. Chem. 2011; 286:31885–31895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Weiner J.A., Jontes J.D.. Protocadherins, not prototypical: a complex tale of their interactions, expression, and functions. Front. Mol. Neurosci. 2013; 6:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kawauchi S., Calof A.L., Santos R., Lopez-Burks M.E., Young C.M., Hoang M.P., Chua A., Lao T., Lechner M.S., Daniel J.A. et al. . Multiple organ system defects and transcriptional dysregulation in the nipbl(+/-) mouse, a model of cornelia de lange syndrome. PLos Genet. 2009; 5:e1000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Parisi L., Di Filippo T., Roccella M.. Behavioral phenotype and autism spectrum disorders in cornelia de lange syndrome. Mental Illness. 2015; 7:5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data deposited in GEO under accession number GSE89010 (https://www.ncbi.nlm.nih.gov/geo/). The code used for peak calling has been deposited in github https://github.com/iaconogi/Histone-mark-analysis.