


Abstract
Protein engineering is used to generate novel protein folds and assemblages, to impart new properties and functions onto existing proteins, and to enhance our understanding of principles that govern protein structure. While such approaches can be employed to reprogram protein–protein interactions, modifying protein–DNA interactions is more difficult. This may be related to the structural features of protein–DNA interfaces, which display more charged groups, directional hydrogen bonds, ordered solvent molecules and counterions than comparable protein interfaces. Nevertheless, progress has been made in the redesign of protein–DNA specificity, much of it driven by the development of engineered enzymes for genome modification. Here, we summarize the creation of novel DNA specificities for zinc finger proteins, meganucleases, TAL effectors, recombinases and restriction endonucleases. The ease of re-engineering each system is related both to the modularity of the protein and the extent to which the proteins have evolved to be capable of readily modifying their recognition specificities in response to natural selection. The development of engineered DNA binding proteins that display an ideal combination of activity, specificity, deliverability, and outcomes is not a fully solved problem, however each of the current platforms offers unique advantages, offset by behaviors and properties requiring further study and development.
INTRODUCTION
Understanding the molecular mechanisms that dictate the affinity and specificity of protein–DNA recognition is an area of investigation that remains critical for many fields of research, including protein engineering. This is particularly important for the purpose of creating novel target specificities for enzymes that act upon DNA targets, including those that are used for targeted genome modification (such as recombinases and endonucleases). The earliest illustrations of protein–DNA recognition mechanisms, provided in part via crystallographic structures of protein–DNA complexes (1–4), emphasized the formation of directional hydrogen bonds between protein side chains and complementary acceptor and donor atoms presented by nucleotide base pairs in the major groove of a DNA duplex. These observations, along with the obvious steric complementarity between protein α-helices and the DNA major groove, led to an appreciation of the importance of such contacts for sequence-specific DNA recognition (5). A broad collection of research literature surrounding protein–DNA binding and recognition refers to the exploitation of these features as ‘direct’ readout of DNA target sequences.
Investigators have also recognized the importance of entropic changes (largely driven by the ordering of protein backbone and side chains during binding, as well as the release of ordered water molecules from the protein–DNA interface) and DNA bending as additional factors that further dictate DNA binding affinity and specificity. In particular, the ability of DNA sequences to adopt or prefer unique structural shapes and features (ranging from subtle alteration of duplex dimensions, to more significant short- and/or long-range deformations) provides additional strategies for sequence-specific readout by DNA binding proteins. These collective features have often been referred to in the literature as ‘indirect readout’ and/or as ‘shape-based’ recognition (see (6,7) for more comprehensive reviews).
Continuing studies have further enhanced our understanding of the complex balance of contacts and forces that lead to protein–DNA recognition. Examination of highly diverse DNA binding protein systems have demonstrated how recognition of the shape and structural features of a potential DNA target can augment the specificity imparted by contacts to the chemically distinct sequence of individual nucleotide base pairs. This includes recognition of altered minor groove dimensions (and corresponding changes in the surrounding surface electrostatic potential) in response to DNA bending (8,9); recognition of altered DNA conformations as a result of base modifications (such as cytosine methylation) and other epigenetic modifications (10); recognition of the structural effects of non-canonical base pairs in the target (11), and the contribution of flanking DNA sequences on target shape and conformation (12).
A relatively recent review article (13) focused on how transcription factors limit their interactions with potential targets in various cell types and tissues, and described how DNA recognition involves the presence and exploitation of many layers of unique structural features beyond DNA sequence (including shape, flexibility, accessibility and cooperativity between multiple DNA binding proteins). Overall, simple codes or correlation between protein and DNA sequences that might be predictive of protein–DNA recognition are largely absent (14), except for rare examples of extremely modular DNA-binding proteins (such as TAL effectors) (15,16).
Whereas considerable progress has been made in engineering novel protein folds (17) and protein–protein recognition (18), engineering of protein-nucleic acid recognition remains difficult, and engineering and redesign of the recognition specificity of a DNA-binding protein is currently a challenging area of research and development (19). This disparity is attributable in part to the differing composition of these two types of molecular interfaces, with protein–DNA interactions involving large numbers of directional hydrogen bonds, electrostatic contacts, ordered solvent molecules and bound counterions. As well, the changes in DNA backbone conformation and its base pair geometries that are induced by protein binding are challenging to computationally sample and predict. Many projects that involve the retargeting of protein–DNA specificity thereby require the development and use of selection and screening strategies, usually in concert with structure-based analyses and guidance.
However, considerable progress has nonetheless been reported over the past several years on the combined use of structural modeling, structure-based computational engineering, and structurally informed selections and screens to alter the DNA recognition properties of a wide variety of DNA binding proteins and enzymes (20–23). Many of these advances have been driven by activities related to targeted genome engineering and targeted gene modification, which require the creation and use of sequence-specific endonucleases, recombinases and integrases.
Here, we summarize recent approaches and successes in the creation of lab-generated DNA binding proteins with altered target specificity. The highlights of engineering studies results for each protein system also summarized in Table 1. These systems range from highly modular TAL effectors, to somewhat modular, but more challenging zinc finger proteins, to several types of distinctly non-modular DNA binding proteins and enzymes (recombinases, homing endonucleases, and restriction endonucleases). Each type of protein displays unique features of ‘evolvability’ (i.e. molecular structures and interactions that allow natural selection and the passage of time to efficiently modify DNA recognition) that clearly influence their corresponding ‘engineerability’ (leading to similar alterations of recognition specificity, executed in a laboratory).
Table 1. Summary of many of the significant attempts to engineer the DNA recognition properties of the protein systems discussed in this review. While not intended to be entirely comprehensive and complete, this table is intended to assist reviewers in following the details of the main text.
| Platform and year(s) | Targets and development | Engineering approach | References |
|---|---|---|---|
| Zinc Fingers | |||
| 1992–1993 | Novel DNA triplets | Structure-based modeling | (49–51) |
| 1994–1995 | Novel DNA triplets | Phage Display | (52–55) |
| 1999 | Novel sites with GNN triplets | (56) | |
| 2000 | Novel 9 basepair targets | Bacterial two-hybrid selections | (61) |
| 2001 | Novel triplets with ANN and CNN triplets | Phage Display | (57,58) |
| 2001 | Novel 9 basepair targets | Phage Display; hybrid 3 finger library panning | (60) |
| 2001 | Novel 12 to 18 basepair targets | Assembly of two-finger ZFP subunits | (64) |
| 2002 - 2003 | Drosophia yellow gene target | Modular assembly | (31,32) |
| 2005 | Human IL2Rg gene target | Zinc finger selections and assembly | (22) |
| 2008 | Novel 9 basepair targets | Bacterial two-hybrid selections | (62) |
| 2011 | Novel 9 basepair targets | Informatics-driven, Context-dependent ZFP assembly | (63) |
| Meganucleases | |||
| 2002 | Single basepair target variants (I-CreI) | Bacterial gene elimination assay/screen | (97) |
| 2002 | Activity-based selection (I-SceI) | Bacterial gene elimination assay/screen | (98) |
| 2002 | Hybrid nuclease generation (I-CreI/I-DmoI –> H-DreI | Structure-based computational redesign | (110) |
| 2003 | Single basepair target variants (PI-SceI) | Bacterial two-hybrid selections | (96) |
| 2006 | Single basepair target variant (I-MsoI | Structure-based computational redesign | (99) |
| 2006 | Multiple base pair target variants (I-CreI) | Bacterial ene elmination assay/screen | (100) |
| 2006 | Multiple basepair target variants (I-CreI) | Eukaryotic gene recombination assay/screen | (101–103) |
| 2009 | Individual and multiple base pair target variants (I-AniI) | Structure-based computational redesign and bacterial selections | (89) |
| 2009–2010 | Monomerization of homodimeric meganuclease (I-CreI) | Structure-based modeling and activity-based selections | (113,114) |
| 2009 | Activity-based selections (I-AniI) | Yeast surface display | (130) |
| 2010 | Maize liguless gene target (I-CreI) | Structure-based modeling and activity-based selections | (113) |
| 2010 | Multiple basepair target variants (I-MsoI) | Structure-based computational redesign | (105) |
| 2014 | Human Brutons Tyrosine Kinase (Btk) get target (I-AniI) | Structure-based computational redesign and Yeast Surface Display | (107) |
| 2007–2013 | Various eukaryotic gene targets (I-CreI) | Structure-based modeling; bacterial selections; eukaryotic selections | (117–129) |
| 2014–2015 | Human TCRa and CCR5 gene targets (I-OnuI) | Yeast surface display meganuclease selections and MegaTAL | (115,116) |
| 2014–2015 | Human CFTR gene target (I-OnuI) | In vitro compartmentalization | (133,134) |
| 2017 | Various eukaryotic gene targets (I-OnuI) | Yeast surface display and bacterial selections | (23) |
| TAL effectors | |||
| 2009 | TAL effector code determination and first designer TALs | Tandem repeat assembly | (16,136) |
| 2010–2011 | TAL Nuclease Creation and Initial refinement | Tandem repeat assembly and FokI fusion | (171–173) |
| 2012 | Improved design using additional RVDs; G-specific RVDs | Tandem repeat assembly using new specificity determinants and data | (141–143) |
| 2012–2013 | Increasing mismatch tolerance of C-terminal repeats | Tand repeat assembly and DNA substrate sequence variation | (151,152) |
| 2013–2014 | Altered specificity at base 0 | Modification of cryptic repeat sequences; RVD at position1, and context | (132,149,150) |
| 2014 | Aberrant repeats that allow frameshift binding | Incorporation of natural repeat variants with small insertions or deletions | (191) |
| 2014–2015 | Expanded repertoire of RVDs for fine-tuned targeting | Characterization of specificities and affinities of 400 RVDs | (185,186) |
| 2016–2017 | Modularion of TAL effector binding strength and TALEN efficiency | Varying the backbone (non-RVD) sequecnes of the repeats | (188,189) |
| 2017 | Optimized length for maximum specificity | Varying the number of repeats | (153) |
| Site specific recombinases | |||
| 1999 | Circumvent need for accessory factors (Tn3 resolvase) | Error prone PCR, galK-based colored colony selection | (203) |
| 2009 | Increased efficiency/selectivity (PhiC31 Integrase) | Error prone PCR, lacZ selection & GFP expression | (204) |
| 1988 | Enhanced and altered activity (gin) | Chemical mutagenesis and bacterial selection | (205) |
| 2000 | Circumvent need for accessory factors (lambda integrase) | GFP based fluorescence | (206) |
| 2015 | Targeting CCR5 and AAVS1 safe harbor locus (Bin and Tn21 recombinases) | Site specific sequence randomization and error prone PCR, antibotic selection | (207) |
| 2003 | Altered loxP sequence (Cre) | site specific sequence randomization, GFP expression and FACS | (218,219) |
| 2001–2011 | Altered loxP sequence,HIV LTR sequences (Cre) | Substrate linked protein evolution | (207,221,222, 224,226) |
| 2013 | HIV LTR (improved activity) (Cre) | Molecular modeling and dynamics | (227) |
| 2017 | HIV LTR (improved activity) (Cre) | Observations based on the crystal structure | (228) |
| 2008 | Mutants that promote heterotetramers (Cre) | Structure-based selection of interfactial residues to be randomized | (232) |
| 2015 | Mutants that promote heterotetramers (Cre) | Protein design via molecular modeling | (233) |
| 2013 | Weakened protein–protein interactions to enhance specificity (Cre) | Random mutagenesis and bacterial selection | (234) |
| 1988 | Enhanced activity (Flp) | Substrate linked protein evolution | (235) |
| 2004 | Mutants that promote heterotetramers (Flp) | Error prone PCR, blue/white selection | (236) |
| 2003–2006 | Altered FRT sequence, interleukin 10 target (Flp) | Error prone PCR and randomization of specific sites, LacZ and RFP reporters | (237,238) |
| 2016 | Enhanced activity (R and TD recombinases) | Sequence truncation, random mutagenesis | (240) |
| 1995 | Relaxed specificity (lambda integrase) | Analysis of chimeric integrases | (245) |
| 2015 | Human genome target (lambda integrase) | Beta-lactamase inhibitor based screen | (246) |
| 2001 | Human chromosome 8 target (PhiC31) | Blue/white selection | (248) |
| 2003–2011 | Hybrid reslovase/ZFN targets (serine recombinases) | Truncated resolvases with zinc finger fusion (varied linkers) | (249–251) |
| 2011–2014 | Mutants to promote heterodimers (resolvases) | Rational design and directed evolution | (255,256) |
| 2011–2014 | Altered specificity of catalytic domains (serine recombinases) | Random mutagenesis of selected residues and directed evolution | (257,258,261) |
| Restriction endonucleases | |||
| 1987–1999 | 1st Attempts to alter specificity (EcoRI, EcoRV, BamHI) | Structure-based modeling | (270–273) |
| 2002–2006 | Additional attempts to alter specificity (BstYI, NotI) | Directed evolution and selection | (274,275) |
| 2003 | Alteration of specificity of bifunctional RM enzyme (Eco57I) | Directed evolution and selection for altered methylation specificity | (278) |
| 2009 | Alteration of specificity of type IIG enzyme (MmeI) | Informatics covariation analysis and structure-based modeling | (21) |
ZINC FINGER AND ZINC FINGER NUCLEASE ENGINEERING
Overview
The C2H2 zinc finger is one of the most common DNA binding motifs in multicellular organisms (24). Individual fingers contain about 30 amino acids and these units typically occur as tandem repeats of two or more fingers (Figure 1A) (25). When the structure of this motif bound to DNA was first solved, its modularity immediately suggested that a ‘mix and match’ strategy could be used to bind essentially any desired sequence in a complex genome (Figure 1B) (26). Fusing the nonspecific DNA cleavage domain of the Type II restriction enzyme, FokI, to a zinc finger protein (ZFP) allows the resulting zinc finger nuclease (ZFN) to cleave DNA at a sequence determined by the ZFP (Figure 1C) (27,28). A previous demonstration that mammalian genomes, engineered to contain a target site for the homing endonuclease I-SceI, could be manipulated at positions near a nuclease-induced double-strand break (29,30) raised the possibility that ZFNs could also be used to manipulate the genomes of complex organisms at will. Work from multiple laboratories on both ZFP engineering and additional aspects of ZFN function and specificity eventually led to the targeting of endogenous loci in Drosophila melanogaster (31,32) and in human cells (22). Successful targeting of endogenous loci in other organisms soon followed including a wide variety of model organisms including Arabidopsis (33,34), C. elegans (35), zebrafish (36–38), mice (39), rabbits (40), and rats (41,42). Important crop species such as corn (43) and soybean (44) and economically important animals such as pigs (45) and cattle (46) have also been targeted with ZFNs. The California-based biotechnology company Sangamo Therapeutics is also testing zinc finger nucleases in human clinical trials as potential treatments for HIV/AIDS (47), Hemophilia B (https://clinicaltrials.gov/ct2/show/NCT02695160), and lysosomal storage disorders (https://clinicaltrials.gov/ct2/show/NCT02702115 and https://clinicaltrials.gov/ct2/show/NCT03041324).
Figure 1.
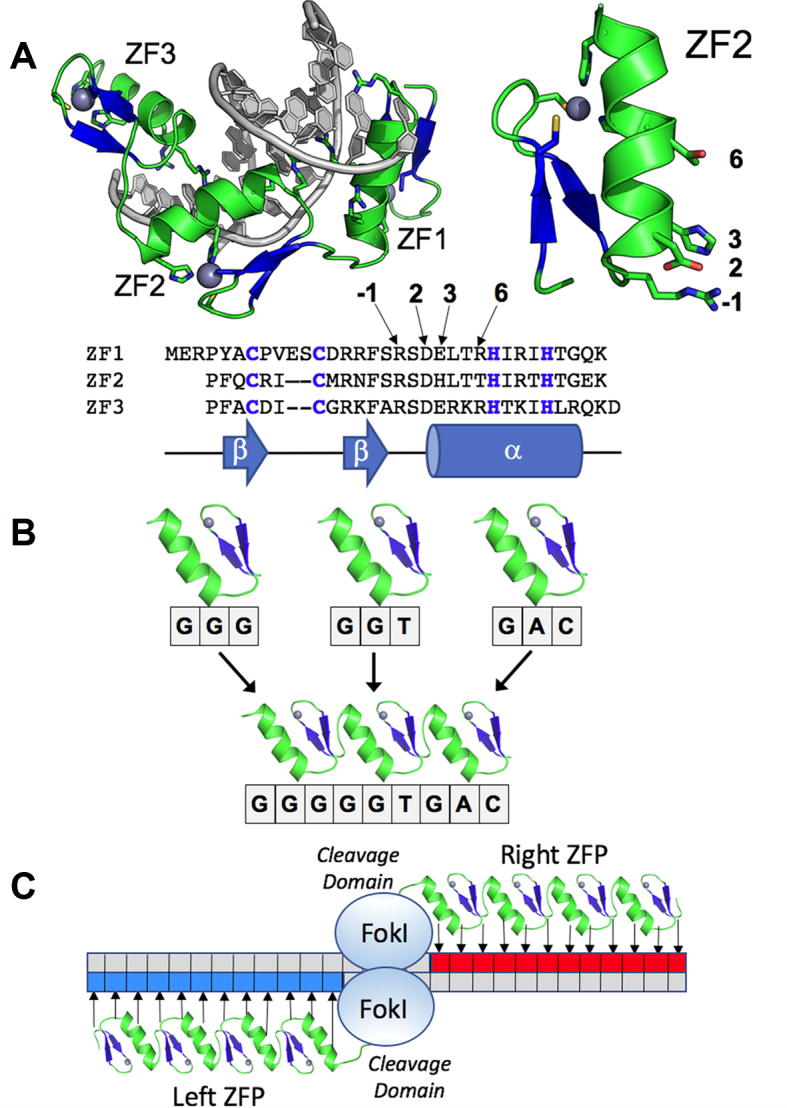
Structure and mode of action of zinc fingers and zinc finger nucleases. Panel A:Structure of the Zif268-DNA complex showing the three zinc fingers of Zif268 bound in the major groove of the DNA. Fingers are spaced at 3-bp intervals. The DNA is grey; the zinc ions are dark teal spheres. The structure and primary DNA contacting residues of zinc finger #2 (ZF2) are indicated to the right. A sequence alignment of the 3 fingers of Zif268 is shown below. The zinc binding Cys2-His2 motif is indicated with blue bold font; the canonical DNA-contacting residues are indicated by arrows. Panel B: Modular assembly of a three-finger protein from individual fingers. To generate a zinc finger protein (ZFP) with specificity for the sequence GGGGGTGAC, three fingers are identified that each bind a component triplet. These fingers are then linked. Panel C:Sketch of a pair of zinc finger nuclease (ZFN) subunits bound to two halves of a DNA target. Each ZFN contains the cleavage domain of FokI linked to an array of three to six zinc fingers (four are shown here) that have been designed to specifically recognize sequences (blue and red boxes) that flank the cleavage site. A small number of bases separate the ZFN targets. The FokI nuclease domains transiently dimerize across those central bases and cleave each DNA strand to generate a double strand break with 5′ overhangs averaging 4 bases in length.
Re-targeting zinc finger proteins
The structure of Zif268 bound to DNA (Figure 1A) provided the first detailed view of how Cys2His2 zinc fingers interact with DNA (26). Each finger is comprised of a simple ββα fold, wherein the α helix fits into the major groove of the DNA target. Adjacent fingers are spaced at 3 bp intervals along the DNA, and the residues at four key positions of the α-helix make base-specific contacts to each finger's portion of the DNA target site. In principle, altering these four amino acid residues in each finger should allow targeting of any desired sequence. But in practice, altering three additional residues interspersed between the four key residues usually gives the best results and allows protein–DNA contacts that do not match the ‘canonical’ pattern observed in Zif268 (48). An additional complexity of engineering zinc finger proteins is that individual zinc fingers do not behave in a completely modular fashion, and strategies to deal with this ‘context dependence’ are critical to achieving optimal results (25).
Initial attempts to retarget zinc finger proteins relied upon a small panel of alternative amino acid residues at key DNA base contacting positions, based upon a bioinformatic search of naturally occurring ZFPs. This yielded some ZFP variants with altered specificity (49–51), but it was not clear that this approach could be extended to recognize all possible DNA sequences. Around the same time, multiple groups began testing a more powerful approach to engineer zinc fingers that involved using phage display to simultaneously test up to 109 variant zinc finger sequences for binding to a desired sequence (25). These initial efforts involved randomizing the four key residues of a single finger of Zif268 and yielded zinc finger variants specific for some, but not all of the targeted sites (52–55). But even library sizes of 109 variants are not large enough to properly sample all possible variants in multiple zinc fingers simultaneously so other strategies had to be employed to target a zinc finger protein to a completely novel site.
Choo and Klug were the first to target a completely novel sequence of interest by combining individual zinc fingers that had been selected separately (52), but the resulting protein had somewhat modest affinity and was only used to target expression of a reporter gene on a plasmid with multiple copies of the targeted binding site (25). The Barbas lab achieved greater success by combining fingers from separate selections to target sites of the form GNNGNNGNN (56), but a pair of such sites separated by 6 bp was required for the first generation of ZNFs; such pairs occur only about once per 4096 bp. They attempted to extend this approach to additional types of sites (57,58), but this resulted in less promising results (59). Another strategy involved separate selections corresponding to the N-terminal or C-terminal half of a three-finger protein (one and a half fingers per selection), followed by combining the results of two such selections to create a completely novel three finger protein (60). A hybrid approach, that took advantage of a newly developed bacteria selection system (61), was more tractable (62), but was still too labor-intensive for widespread adoption. A refined version of this system used a computational approach to determine viable module-module pairings (63). But these methods were primarily geared towards generation of 3-finger ZFPs with 9 bp binding sites.
Other groups pursued an approach that involved mixing and matching pre-selected two-finger units (64); this approach yields ZFPs with between four and six zinc fingers that have been used to successfully target a wide variety of endogenous genes (65,66). Based mainly on these improvements in retargeting ZFPs, the precision of targeting desired regions of the genome has gradually increased from one in 4096 bp to coding sequences of genes of interest (22,62,67) to the ability to generally target individual point mutations or short regulatory regions (68,69). However, even the most successful version of this strategy still relies on assembling and testing multiple pairs of ZFNs to generate optimal activity and results (65).
Cleaving DNA
While the application of zinc finger nucleases (ZFNs) for genome engineering and gene editing is not the focus of this review, examination of ZFNs have provided the most detailed analyses of the specificity of engineered ZFPs, relying upon genome-wide DNA cleavage assays as a reporter of zinc finger DNA recognition specificity. Because ZFPs and corresponding ZFNs were among the very first engineered protein systems of this type, they stand out as some of the most rigorously characterized of all designed DNA binding proteins.
Creation of ZFNs involves fusing ZFPs possessing desired DNA binding properties to a DNA cleavage domain (typically the non-specific cleavage domain of FokI (27)). Although it was not understood initially, FokI must dimerize in order to cleave DNA (70,71) and thus a pair of ZFNs that bind their target sequence with the appropriate orientation and spacing are required to cleave DNA. It was not originally known if the prokaryotic cleavage domain from FokI would function on DNA in eukaryotic cells, but experiments in Xenopus oocytes using an extrachromosomal substrate (72) and experiments in human cells using an integrated reporter construct (73) both demonstrated that engineered ZFNs could indeed target reporter constructs in higher eukaryotes.
Two initial questions were exactly how to connect the FokI cleavage domain to the engineered ZFP and how much of a gap was required between the binding sites for the left and right ZFNs. Initial studies showed that a short linker and half-sites spaced by 6 bp worked well in Xenopus oocytes (72). Additional work explored some variant linker sequences and demonstrated that gaps of 5–7 bp can be targeted (74,75). A FokI cleavage domain variant with increased catalytic activity has also been generated (76). However, the majority of the FokI engineering work has focused on reducing off-target DNA cleavage. One approach has been to engineer obligate heterodimer variants of FokI (77–79). The basic concept of obligate heterodimer ZFNs is to build two variants of the FokI cleavage domain that can cut DNA when paired with each other, but can’t cut DNA when paired with a second copy of themselves. This approach has shown a substantial reduction of off-target cleavage for a ZFN pair that targets the human CCR5 gene (67). Another approach to engineer FokI domains to reduce off-target cleavage is to create a ZFN that can only nick DNA rather than cleave both strands (80,81). This is desirable in the presence of a homologous donor DNA construct because nicked DNA can still potentiate homology directed repair of DNA without leading to DNA double-strand breaks. However, published zinc finger nickase constructs tend to exhibit lower levels of the desired homology directed repair activity than comparable zinc finger nucleases.
Specificity
Genome engineering with artificial nucleases is premised on the fact that DNA cleavage is focused mainly at the desired target site. High levels of off-target DNA cleavage could be toxic to the cells and even low levels of off-target cleavage at certain genomic loci could be problematic for applications such as human therapeutics. Some early ZFN work monitored off-target cleavage by measuring various effects of overall DNA cleavage in cells. This included staining cells for foci of DNA repair proteins that are indicative of a DNA double-strand break (67) and monitoring the amount of phosphorylated H2AX histone in a cell by flow cytometry since H2AX is phosphorylated in response to DNA damage (78). However, these methods become less effective for comparing different nucleases as the specificity of ZFNs improved to the point where ZFN-induced breaks are not detectable over the background level of DNA breaks in the cells of interest (67). Thus, many groups started developing assays to monitor double-strand breaks (DSBs) at specific off-target loci.
Initial attempts to identify individual ZFN off-target loci used either a bioinformatics approach to search the relevant genome for sites homologous to the intended target or a combination of bioinformatics and a biochemical DNA specificity assay (67). However, in practice this method still missed numerous off-target sites that were identified by more sophisticated methods developed later. The first method capable of directly monitoring ZFN cleavage of a complex mixture of potential target sites used a large library of potential ZFN cleavage sites. This library of potential sites was digested in a cell-free system and the results were obtained by high throughput DNA sequencing (82).
Later, a method was developed that could identify sites of ZFN cleavage genome-wide in human cells. This method relies on the observation that DSBs can capture exogenous DNA with a mechanism that doesn’t rely on sequence homology (83). Briefly, integrase deficient lentivirus (IDLV) and ZFNs are co-transfected into human cells, IDLV is captured at sites of DSBs, and then Linear Amplification Mediated PCR (LAM-PCR) followed by high-throughput sequencing is used to identify genomic loci where IDLV integration has occurred. This analysis was applied to variants of ZFN pairs targeted to the human CCR5 and IL2Rg genes. For CCR5-targeted ZFN variants with heterodimer FokI variants, off-target sites were identified, but even in aggregate these off-target sites were less than the cleavage at the intended target. This type of assay probably won’t be able to detect weak off-target sites and it is formally possible that a ‘long tail’ of weak off-target sites could still cause a substantial amount of off-target DSBs in each cell. But sequencing the genome of a ZFN treated C. elegans did not identify any ZFN induced indels other than the intended target (84) and exome sequencing of ZFN-treated induced pluripotent stem (iPS) cells did not identify any ZFN induced changes other than the intended change (69).
HOMING ENDONUCLEASE (MEGANUCLEASE) ENGINEERING
Overview
Homing endonucleases (now usually termed ‘meganucleases’) are primarily associated with mobile self-splicing elements (introns and inteins) that display genetic mobility and evolutionary persistence within all known forms of microbial life. Like zinc finger nucleases, meganucleases have been studied since the mid-1990s as for use in genome editing applications (29,30,85). As the drivers of DNA invasion events, meganucleases face strong evolutionary pressure to continuously alter their DNA recognition specificity. By doing so, they increase their ability to invade new DNA sequences and targets, while also persisting in their current host genes. This (along with various mechanisms to control the timing and level of their expression) provides the endonuclease with sufficient specificity to avoid overt toxicity to their host organism, while still accommodating individual polymorphisms within their targets that naturally occur over the course of host genetic drift (86).
At least five distinct structural families of meganucleases have been visualized and extensively characterized. Of these proteins, those from the ‘LAGLIDADG’ family (Figure 2) have been extensively developed and engineered for genome engineering. LAGLIDADG endonucleases correspond both to homodimeric and single-chain monomeric proteins (for the latter, the N- and C-terminal domains display considerable structural similarity). In each case, residues corresponding to the interface between the protein domains, as well as metal-binding active site residues, form a 10-residue sequence motif represented by the consensus ‘LAGLIDADG’ nomenclature.
Figure 2.

Structure and reprogramming of a meganuclease. Panel A:Structure and original target site of the I-OnuI meganuclease. The protein is comprised of a single protein chain of 290 residues and is bound to a 22 base pair DNA target site. The N- and C-terminal domains of the endonuclease, which possess the same overall protein fold related by a pseudo two-fold symmetry axis, recognize and interact with the 5′ and 3′ half-sites of the DNA target site, respectively. The interface between the target 5′ half-site and the protein N-terminal domain is indicated by the oval. Panel B:Schematic of immediate contacts between the DNA 5′ half-site and the meganuclease N-terminal domain (corresponding to oval in panel a above). Bases and protein residues in blue boxes correspond to elements shown in panel C. Panel C:Region corresponding to the contacts between two consecutive base pairs in DNA target site (indicated by the blue box in panel B) and the six most near-neighboring protein side chains (also indicated in panel B with blue boxes). In a typical selection experiment, a cluster of at least six such residues are simultaneously randomized and incorporated into a combinatorial protein library for subsequent screening against a DNA substrate containing the desired base pairs at the corresponding nucleotide positions. Panel D:DNA-bound structure of a fully reprogrammed variant of the I-OnuI enzyme, harboring selected point mutations at 50 residues in the protein–DNA interface (corresponding to ∼17% of the total protein sequence; indicated with red spheres spanning the side chains of each altered residue). The engineered protein, which recognizes a DNA sequence that differs from the original target at over half of its base pair positions (12 out of 22; the altered basepairs are indicated by lower case letters) displays an rmsd across all backbone atoms of only 0.6 Å. A structural superposition of the wild-type enzyme and its fully redesigned variant is shown to the right (engineered enzyme is colored blue).
DNA recognition by this meganuclease family (Figure 2) is noteworthy with respect to the length of their target sites (usually 22 base pairs) and the size and complexity of their DNA-contacting surface (involving upwards of 50 amino acids). The mechanism of DNA recognition by these meganucleases, like many other DNA-binding proteins, involves a mixture of (i) contacts between protein side chains and nucleotide bases (largely concentrated within the major groove of the target site), (ii) significant DNA bending that results in a distortion of both major and minor groove dimensions, as well as alteration of molecular surface electrostatic distribution near the center of the site (87,88) and (iii) a variety of additional contacts within and near the minor groove, particularly at the bent target site center (87,88). The latter two features impose considerable specificity across the central 4 base pairs of the target site, in the absence of direct contacts to the protein. Specificity of recognition is strongly enforced during catalysis (i.e. at the DNA cleavage transition state) as well as through reduction of binding affinity (88). In some cases, the contribution of binding affinity versus cleavage activity towards overall nuclease specificity is strongly segregated between the two protein domains and corresponding DNA half-sites (89).
Overall, meganucleases display non-uniform recognition at individual positions across the DNA target site (ranging from nearly exclusive recognition at some nucleotide positions, to considerable promiscuity at nearby or adjacent positions) (86,89). The mechanism by which overall recognition specificity is enforced includes both considerable amounts of indirect ‘shape readout’ near the center of the DNA target, to a stronger reliance on direct readout of DNA base chemistries across the more distal ends of the DNA target sequence (88). Recent studies have demonstrated that even relatively moderate divergence of meganuclease sequences allows them to establish significantly altered DNA specificities, thereby enabling recognition and cleavage of new genomic targets (88). The ability of these proteins to efficiently generate new DNA target specificities, with relatively minor resculpting of their structures, is probably a consequence of their function as the catalysts of gene invasion, mobility and genetic persistence.
Some of the initial demonstrations that the action of a site-specific nuclease at a unique target within a mammalian genome could increase targeted gene modification events involved the use of I-SceI LAGLIDADG endonuclease (29,30,90). In those studies, the natural target site of that enzyme was first introduced into a desired chromosomal allele, prior to the subsequent expression and action of the meganuclease. Additional experiments using integrated I-SceI target site and introduction of wild-type enzyme have demonstrated correction of an exon disruption in the Artemis gene in mouse hematopoietic stem cells (91) and in vivo targeted recombination in mouse liver (92).
Subsequent to those experiments, it became clear that use of meganucleases for targeted genome modification would require substantial alteration of their recognition specificity. The first crystallographic structures of meganucleases (I-PpoI and I-CreI in 1998 (93,94) followed by I-MsoI, I-AniI and I-SceI in 2003 (87,95)) allowed identification of the amino acids in each system that were found within contact distance of base pairs in their DNA targets. With such information now available, it is possible to extensively and routinely retarget a meganuclease for the modification of unique genomic targets. The history of experiments that have led to this capability are summarized below.
Alteration of meganuclease target specificity at individual base pairs
Initial studies focused on the systematic alteration of individual residues within a meganuclease DNA-binding surface that might cause a change in specificity at a corresponding single base pair, coupled to in vitro or cellular assays of cleavage activity (96,97). These early investigations generally used either DNA binding reporter systems (such as bacterial two-hybrid screens (96)) or methods that coupled site-specific DNA cleavage to the elimination of a reporter gene (97,98). The results of these experiments indicated that at a limited number of individual DNA target positions and corresponding endonuclease contact residues, point mutants of the meganuclease could be identified that displayed a strong shift in specificity without a significant decrease in recognition fidelity at that position. Such positions in the protein–DNA interface typically corresponded to the most distal (outer-most) positions in the target site, where individual amino acid side chains (extending from protein loops at the periphery of the folded protein) often contact single nucleotide base pairs.
At the same time, a purely computational approach to accomplish the same purpose was also reported, using the Rosetta computational protein design algorithm. Similar to the results above, the altered enzyme cleaved its corresponding DNA target site (containing a single altered base pair) several orders of magnitude more effectively than did the wild-type enzyme, along with wild-type ability to discriminate between the two targets (99).
Combined alteration of specificity at multiple, adjacent base pairs
By 2006 it was clear that mutation of individual DNA-contacting residues (while otherwise maintaining an unchanged protein sequence) at certain positions in the DNA target site might sometimes result in desired changes in specificity at unique base pairs in the DNA target sequence (100). However, it was not obvious whether such changes in endonuclease sequence and function might be readily combined. To address these questions, a selection method to screen meganuclease libraries for altered DNA cleavage specificity was developed, in which endonuclease activity was coupled to reconstitution of a reporter gene via DSB-induced homologous recombination (101–103). This approach was used to systematically screen semi-randomized libraries of the I-CreI meganuclease. In this way, investigators were able to identify protein variants containing multiple alterations in its amino acid sequence, that could allow recognition of a DNA target site containing multiple adjacent base pair substitutions in a genomic target site (102,103). These experiments indicated that individual protein mutations that reduce activity or specificity on their own might function well in more extensively altered protein variants; conversely, some mutations of DNA-contacting residues that functioned well on their own were incompatible with protein mutations at nearby positions (reviewed in (104)). A similar effort to alter specificity across multiple consecutive base pairs, using a structure-based computational approach, further illustrated the importance of the context-dependent of protein–DNA interactions (105).
A series of studies to further improve the ability of structure-based computational redesign approaches was subsequently reported from 2009 through 2014. In those studies, the overall contribution of contacts to each base pair position on binding and/or DNA cleavage was determined (89), followed by improvements in the computational prediction of side-chain base interactions and conformations in the protein–DNA interface (106). This work led to the eventual redesign of the I-AniI meganuclease and its use in genome modification experiments both in mammalian cells (107) and in mosquitos (108). Details of strategies for the combined use of computations (using the Rosetta program suite) combined with selection experiments for nuclease activity, is documented in (19).
Hybrid meganucleases
A series of studies also demonstrated that while the DNA contacting elements and surfaces of meganucleases are distinctly non-modular, their N- and C-terminal domains can be structurally separated and recombined to form ‘hybrid’ meganuclease scaffolds that recognize chimeric DNA target sites (109–112). In addition, a homodimeric meganuclease (I-CreI) was turned into a functionally equivalent ‘single chain’ monomeric protein via introduction of a peptide linker between the two subunits of the dimeric enzyme (113,114). These experiments further enabled the development of novel meganuclease recognition, both by creating new starting protein scaffolds and specificities, and by reducing the process of engineering new specificity into two separate experimental tasks, for which the output could be fused into a final nuclease construct.
Complete retargeting of meganuclease specificity and application to genome editing
Multiple groups (in both industry and in academia) have exploited the results and observations summarized above to create extensively retargeted meganucleases for genome engineering and targeted gene modification. In all cases, the use of direct structure-based redesign and structure-based selection methods have each found a significant role in the engineering process, but the need for selection experiments as a fundamental requirement for high activity and requisite specificity has not been eliminated. In certain cases, the incorporation of such engineered meganuclease constructs into chimeric ‘MegaTAL’ architectures (comprised of N-terminal TAL effector domains tethered to C-terminal engineered nucleases; (115)) has facilitated the use of such enzymes for highly demanding applications in primary human cells as part of various therapeutic approaches (116).
Two separate biotechnology companies (Cellectis Inc. and Precision Biosciences Inc.) have described the generation of fully redesigned meganucleases, based on single chain versions of the I-CreI enzyme, and their subsequent use for targeted gene editing applications. Engineering and selection steps were separately focused on the N- and C-terminal protein domains (each targeting a half-site within the final genomic target) and then combined into a single polypeptide which is further refined for best in vivo performance. These two approaches largely converged on mutations of the same DNA-contacting protein side chains for various alterations of DNA recognition specificity.
The variants of single-chain I-CreI endonuclease created by these group include engineered meganucleases used for correction of the human XPC gene for the treatment of Xeroderma Pigmentosum (117–119), generation of cell lines harboring precisely generated genetic insertions and alterations (120,121), creation of genetically modified maize containing heritable disruptions of the ligueleless-1 and MS26 loci (113,122), modification of defined genomic regions in Arabidopsis (123), insertion and stacking of multiple trait genes in cotton (124), generation of Rag1 gene knockouts in human cell lines (125,126) and in transgenic rodents (127), disruption of integrated viral genomic targets in human cell lines (128), targeted exon deletions in the human DMD gene associated with Duchenne Muscular Dystrophy (129). Crystallographic structures of two of these fully reengineered variants (against the human Rag1 and XPC targets) have been solved and described (119,126).
Yet another biotechnology company (Pregenen, Inc.), in concert with several academic research labs, developed a high-throughput flow cytometric approach to screen semi-randomized endonuclease libraries for altered binding and cleavage specificity (130). Using this strategy, gene targeting nucleases have been created that cleave unrelated targets in human, viral or insect host genes. The resulting meganucleases have again been shown to be highly active in transfected primary human cells and transgenic insects, and display specificity profiles that rival or exceed the parental meganuclease. These enzymes drive the disruption of fertility-related genes as part of a gene drive strategy for the control of insect disease vectors (131), disrupt the gene encoding T-cell receptor α-chain gene (as part of a broader strategy to create engineered T-cells that can be used as anticancer immunotherapeutic reagents) or disrupt the gene encoding the human CCR5 gene that acts as a co-receptor for HIV (116). The details of the methods used by these latter investigators have been described in detail previously (109,132).
A complementary strategy for the purpose of retargeting of meganuclease specificity utilizes a technique known as in vitro compartmentalization (‘IVC’) (133,134). In this approach, the meganuclease is redesigned via activity selections within compartmentalized aqueous droplets. The method was illustrated by engineering several different meganucleases to cleave multiple human genomic sites, as well as variants that discriminates between single nucleotide polymorphic (SNP) variants.
Structural and functional outcomes of meganuclease engineering
Crystallographic and biophysical analyses of five different extensively retargeted variants of a single meganuclease, that have been shown to function efficiently in ex vivo and in vivo applications, has been more recently reported (23). The redesigned proteins harbor mutations at up to 53 residues (18% of their amino acid sequence), primarily distributed across the DNA binding surface, making them among the most significantly reengineered ligand-binding proteins to date (Figure 2D). Other than maintaining their original specificities across the central four base pairs of each target site (a constraint that is related to bending of the DNA), the base pair identities are changed liberally throughout the remainder of the DNA target, and many base pairs are present at least once at each position.
The reorganization and structural changes in these proteins that facilitate recognition of alternate DNA targets can be described as the sum of: (i) small protein backbone motions involving DNA-contacting β-sheets (that contribute the largest share of contacts to nucleotide bases throughout the major groove); (ii) much larger reorganization of flanking protein loops at both ends of the β-sheets, and (iii) extensive role-swapping throughout the entirety of the protein–DNA interface. ‘Role-swapping’ refers to protein residues that, after mutation, have switched from interacting with DNA to instead interacting solely with surrounding protein side chains, or vice-versa. Changes in overall DNA recognition specificity are facilitated by the ability of residues in or near the protein–DNA interface to readily exchange both form and function in this manner.
The fidelity of recognition is not precisely correlated with the fraction or total number of residues in the protein–DNA interface that are actually involved in DNA contacts, including directional hydrogen bonds. The plasticity of the DNA-recognition surface of this protein, which allows substantial retargeting of recognition specificity without requiring significant alteration of the surrounding protein architecture, reflects the ability of the corresponding genetic elements to maintain mobility and persistence in the face of genetic drift within potential host target sites. This demonstrates the extent to which a single meganuclease protein can be substantially reprogrammed for recognition of multiple unique genomic target sites, without the need for significant alteration of the surrounding protein scaffold.
TRANSCRIPTION ACTIVATOR-LIKE (TAL) EFFECTOR ENGINEERING
Overview
Similar to meganucleases, transcription activator-like (TAL) effectors evolved under a unique set of selective forces that shaped their unique DNA recognition properties. Despite the ‘-like’ in their name, TAL effectors are indeed transcription activators. Made by plant pathogenic bacteria in the genera Xanthomonas and Ralstonia, they enter host cells via the bacterial type III secretion system, translocate to the nucleus by virtue of C-terminal nuclear localization signals, bind to individual sequences in the host genome determined by a DNA recognition domain distinct for each effector, and directly upregulate downstream genes by virtue of a C-terminal acidic activation domain. TAL effectors have been selected that activate host genes whose expression facilitates bacterial multiplication and spread. Such genes are referred to as disease susceptibility or ‘S’ genes. The presence of an effector binding element (EBE) in the promoter of a so-called plant ‘executor resistance (R) gene’, however, will result in TAL effector-triggered host immunity, and exert negative selection pressure on the corresponding TAL effector. At the same time, sequence variation in the EBE of a major S gene can render a plant effectively resistant by preventing binding and activation by the TAL effector, resulting in loss of susceptibility and selection for TAL effectors that can accommodate the sequence variation. Several examples of both such host adaptations have been characterized in diverse plant species (135). Thus, TAL effectors can be presumed as a group to have been selected for the ability to rapidly evolve new specificities in order to probe the host genome for beneficial targets, and individually to have been subject to contrasting selective pressures - for stringent specificity to discriminate between potential EBE sequences in S vs. R genes, and for lax specificity to accommodate minor sequence polymorphism at S gene EBEs across different host genotypes.
The result of this selection (and/or the result of selection on ancestral proteins in other functional contexts not yet discovered) is a DNA recognition domain that functions via a modular mechanism, which allows evolution of new specificities by recombination-based shuffling and by point mutations within modules, and variation in the specificity profiles and affinity contributions of individual modules that confers plasticity in targeting stringency (16,136–138).
The TAL effector DNA binding domain forms a superhelical, monomeric protein chain that wraps around B form DNA in a right-handed manner, tracking the major groove without inducing any bend or other substantial structural distortion (Figure 3A and B). Its repeated modules form contiguous, two-helix bundles (Figure 3C and D), each of which comprises a highly conserved sequence of typically 33–35 amino acids and interacting with a single base, contiguously, on one strand of the DNA. The contribution of each module to specificity and affinity is determined predominantly and predictably by two residues within the repeat that vary, at positions 12 and 13, together referred to as the ‘repeat variable di-residue’ (RVD). The RVD resides in the loop connecting the two helices of a module together. Residue 12 interacts with the backbone at position 8 to stabilize and position the loop, and the side chain at position 13, which has been called the base-specifying residue (BSR), projects into the major groove to interact with the base at that position. RVDs HD, NG, NI and NN are the most common, and they are the most commonly used in engineering. HD specifies cytosine through van der Waals interaction and hydrogen bonding between the aspartic acid side chain and the base, resulting in high specificity and high affinity. NG specifies thymine via nonpolar van der Waals interaction between the backbone α carbon of the glycine residue and the methyl group of the thymine, again high specificity, but lower affinity. NI specifies adenine through nonpolar van der Waals contacts of the isoleucine side chain to the purine ring, which appear to desolvate at least one polar atom in that ring; though specific, this likely makes the interaction decidedly low-affinity. NN has dual specificity, making high affinity contact with guanine or adenine by hydrogen bonding between the BSR and N7 of either opposing pyrimidine.
Figure 3.

Structure of the TAL effector–DNA association and the basis of specificity. Panels A and B:The structure of PthXo1 binding region (comprised of 22 TAL effector repeats distributed along a single monomeric protein chain) bound to its DNA target site is shown from the side of the DNA duplex and looking down the axis of the DNA. The effector contains 22.5 repeat modules, each colored separately. In the side view, the N-terminal end of the protein is leftmost. The structure also contains two cryptic N-terminal repeats that engage the DNA backbone via a series of basic residues, and that contact a strongly conserved thymine at the 5′ position of the binding site. Panel C illustrates the contacts made by the HD RVD (residues 12 and 13) in repeat number 14. The histidine at position 12 in the repeat forms a hydrogen bond to the backbone carbonyl oxygen of residue 8 in the first a-helix, while the aspartate at position 13 forms a hydrogen bond to the extracyclic amino nitrogen of the cytosine base. Panel D shows repeats 14, 15 and 16 interacting with the DNA, illustrating that consecutive RVDs (HD, NG and NN, respectively in these repeats) contact consecutive bases (in this case cytosine, thymine, and guanine) on the same DNA strand. Figure adapted with permission from Figure 1 in Doyle et al. (2013) Trends in Cell Biology23 (8):390–398.
Several other, less common RVDs are found in native TAL effectors (15,16,139,140). Of particular note for engineering are NK, NH, N* and NS. NK and NH provide better specificity for guanine than the dual-specificity NN, though NK, and to a lesser extent NH, weaken overall interaction relative to NN (141–143). N*, in which the asterisk designates a missing amino acid at position 13 resulting in a slightly retracted interhelical loop (137), is found most often associated with thymine or cytosine in nature (16) and was shown to be a suitable alternative for HD when cytosine at the target might be methylated (144). In native TAL effector–target alignments, NS in native TAL effector–target alignments can be found in association with any of the four bases (16), and can be considered a ‘wildcard’ RVD for engineering.
The array of modules that determines DNA-binding specificity constitutes a domain called the central repeat region (CRR). Immediately N-terminal to the CRR, four additional two-helix bundles are present that do not match the repeat consensus sequence (137,138,145,146). Through lysine and arginine contacts with the DNA backbone, these ‘cryptic repeats’ can bind DNA independently, and are thought to nucleate interaction with the DNA for sequence-specific binding mediated by the CRR (146–148). The cryptic repeat closest to the CRR plays an important role in the general requirement of Xanthomonas TAL effectors for a thymine at position ‘0’ of the EBE, immediately 5′ of the first RVD-specified base: a tryptophan residue (W232) in the cryptic repeat coordinates with the thymine (137,149). Ralstonia TAL effectors (called ‘RipTALs’), which present an arginine instead of tryptophan at that position, require a guanine base opposite (140,149). The structural and biochemical basis for these specificities is poorly understood though. Influence of the CRR composition, particularly the RVD of the first repeat, and of the experimental context have been observed (149,150). Efforts to engineer altered specificities for base 0 have, nonetheless, met with some success (132).
The cryptic repeats may exert an effect on another property of TAL effector DNA recognition, increasing mismatch tolerance closer to the C-terminal end of the CRR (151,152). TAL effectors acquire their targets via a rotationally decoupled, linear search mechanism in which the cryptic repeats provide the major contribution to non-specific association with the DNA (147,148). Given this anchoring role of the cryptic repeats, it seems likely that the interaction transitions to specific binding via BSR-nucleotide contacts initiating from the end closest to the cryptic repeats, and that once the specific binding state is initiated by a certain number of these contacts, subsequent RVD-nucleotide pairings diminish in their influence on overall binding energy (153). Notably, proteins closely related to TAL effectors but found in the fungal endosymbiotic bacterium Burkholderia rhizoxinica lack two of the cryptic repeats, but make additional, non-specific contacts with the DNA backbone via non-RVD residues, often arginines and lysines, throughout the CRR (154). This observation suggests that it may be possible to engineer patterns of mismatch tolerance by modifying, moving, or replacing the cryptic repeats, or by modifying backbone residues of the CRR.
Assembly of custom TAL effector DNA recognition domains
The modularity of TAL effectors makes them easy to engineer for specificities of choice: coding sequences for the necessary modules are simply assembled in the correct order into a genetic backbone construct, which may include translational fusions to any of a variety of other protein domains. Many cloning kits and protocols are publicly available for such assembly. The earliest and among the most widely used of these take the Golden Gate approach (155), in which type IIS restriction enzymes (which cut at a distance from their recognition sites) are used to release cloned, module-spanning fragments each containing an RVD and all staggered such that cleavage results in sequentially matching 5′ overhangs; the overhangs drive ordered and oriented assembly of the fragments into a module array in a single tube ligation (156–159). PCR-based and other ligation-independent methods have also been developed (e.g., 160), as have sequential ligation but scalable assembly strategies for high throughput (161,162). Sakuma and Yamamoto (163) provide a comprehensive review.
Several web-based tools for design and off-target prediction have also been made available (see 164,165 for reviews). The earliest of these scores potential binding sites using a position weight matrix based on observed association frequencies (166). Others add a parameter to reflect the increasing mismatch tolerance of RVDs close to the C-terminal end of the array (167). The best performing tool, SIFTED, derives from extensive protein binding microarray data for 20 custom TALE effector proteins, and factors in observed, minor effects of neighbors, position in the array, and length of the array on individual RVD specificities (168). However, the TAL effectors assayed to develop SIFTED contained only the four most common RVDs, so the utility of the tool is limited to such proteins.
TAL effector-based DNA targeting applications
The first artificial TAL effectors were generated as part of the study that determined experimentally the RVD-nucleotide relationship ‘code’ that governs TAL effector DNA recognition (15). Apart from the customized CRR, these retained their native features as transcription activators and were assayed using a reporter gene assay in Nicotiana benthamiana leaves. Since then, such ‘ArtTALs’ (141), alternatively called ‘dTALEs’ (designer TAL effectors) (159), have been used extensively for functional validation of targets and candidate targets of native TAL effectors in the context of plant disease: if a dTALE activating a gene from a promoter binding site distinct from the EBE of the native TAL effector phenocopies that TAL effector, one can conclude that the gene is the relevant target of the native TAL effector (e.g.169,170).
Another early and widespread application of customized TAL effector DNA recognition domains is in TAL effector nucleases (TALENs) for genome editing. Originally developed by replacing the C-terminal TAL effector activation domain with a monomer of the catalytic domain of the type IIS restriction enzyme FokI, TALENs cleave in pairs, targeted to sequences on opposing DNA strands across a spacer (171–173). FokI fusions to full length TAL effectors, N- or C-terminal, and C-terminal fusions to TAL effectors missing the first 152 aa and all but the first 63 aa of the C-terminus (after the CRR) were also shown to function (172,173). The latter, compact configuration, referred to as the ‘Miller architecture’ has been widely adopted. Indeed, TAL effectors, the Miller architecture in particular, have proven amenable to a variety of fusions, including alternative activation domains, repressor domains, affinity and fluorescent tags, epigenetic modifiers, and others (149,174). They have also been tested as fusions to the restriction enzyme PvuII, the catalytic domain of the meganuclease TevI, and to the CRISPR-associated nuclease Cas9 toward developing monomeric TALENs for genome editing (175–177). As already mentioned, fusions of TAL effector DNA recognition domains to meganucleases have shown great promise as highly specific tools for therapeutic genome editing (115).
Future prospects: engineering targeting stringency
Most TAL effector assembly platforms use the Xanthomonas TAL effector backbone and repeat consensus sequence with the four most common RVDs, and in some cases one or more of the other RVDs discussed above. While the simplicity of assembling with such platforms TAL effectors that bind sequences of choice led to their widespread adoption and transformative impact in basic research, agriculture, and medicine (e.g. 178–184), several discoveries suggest important additional research directions and engineering approaches that could further enhance the utility of TAL effectors by taking advantage of underexploited properties to fine-tune binding specificity. This fine-tuning has the potential to be not only qualitative, i.e., to match a given target sequence, but quantitative, to modulate the stringency of that match, even non-uniformly across the target site.
First, two studies profiled the specificity and functionality of all 400 possible RVDs (185,186). The results revealed a large number of functional RVDs representing a striking diversity of specificity profiles that could be used in engineering. Protein-binding microarray analysis of dTALEs incorporating RVDs of interest, of the sort used to develop the SIFTED software for design and target prediction, would be an important further step toward precisely defining the behavior of these RVDs in different contexts.
Second, polymorphism in the backbone repeat sequence relative to Xanthomonas TAL effectors has been observed in RipTALEs (140) as well as the Burkholderia TAL-like proteins (BTLs) mentioned earlier (174), and in TAL effector-like sequences fished out of metagenomic data from marine samples (187). Engineering based on these polymorphisms achieved variation in strength of dTALE-DNA interactions (188). Separately, modification of backbone residues at positions 4 and 32 resulted in higher efficiency TALENs, ostensibly due to greater mobility along the superhelical axis that allowed better positioning of BSRs to interact with corresponding nucleotides throughout the array and limit the spatial distribution of the fused FokI domains for higher efficiency dimerization (189). Systematic characterization of the influence of backbone variation on the individual behaviors of different RVDs and on the overall dynamics of the TAL effector-DNA interaction could further expand the capacity to engineer specificity quantitatively. Incorporating variation in repeat backbone sequences could also guard against recombination of TAL effector constructs, which can be problematic in the context of viral systems for delivery (190).
Third, the earlier noted discovery of the relationship between structural variation outside the CRR and specificity for the base at position 0, as well as the degree and pattern of mismatch within the CRR, suggests that base 0 specificity and mismatch tolerance could be robustly engineered. Structure-function studies to better understand those relationships are needed however.
Fourth, so-called ‘aberrant repeats’ observed in some TAL effectors of the species Xanthomonas oryzae afford the opportunity, to our knowledge unique among characterized DNA binding proteins, to accommodate single base indels in a target (or set of targets). These aberrant repeats have short insertions or deletions of amino acids in the region that connects one repeat to the next. Though the repeats are functional, the particular indels observed render them capable of disengaging if a single base is missing at that position in a way that allows in-register binding of the remainder of the CRR to the DNA (191). Understanding the mechanistic basis for that capacity to disengage would inform use of such aberrant repeats in combination with other engineering-based modifications.
Finally, a recently characterized feature of TAL effectors that could be exploited in engineering is the influence of their length (number of RVDs) on overall specificity. With increasing length, TAL effectors exhibit exponentially decreasing gain in affinity for target DNA, yet less rapid deterioration of gain in affinity for non-target DNA (153). Using experimental data and simulations, plotting specificity as the affinity for target DNA relative to the affinity for non-target DNA across varying lengths results in a Gaussian curve with a peak centered between 14 and 22 RVDs depending on the overall RVD composition. Thus, length variation could be used to modulate specificity quantitatively. Further experimentation to better understand how RVD composition determines the optimum length for maximum specificity would benefit this goal.
Though not broadly distributed in nature, TAL effectors have been shaped by a unique set of selection pressures and provide a versatile platform for engineering protein–DNA interactions with precision and flexibility. As described above, to fully realize their potential, further characterization of the mechanistic basis for their unique properties and the influences of sequence variations on those properties is important. Not to be overlooked however, is the importance of continuing to identify and characterize related proteins, not only to understand the evolutionary origin of TAL effectors but to gain further insight into useful structural variation.
RECOMBINASE ENGINEERING
Overview
Members of the Int recombinase/topoisomerase family, also known as site specific recombinases (SSRs), have been used to edit DNA both in vitro and in vivo for decades, and some have been commercially developed to simplify cloning tasks (i.e. the Invitrogen Flp-In™ and Gateway® systems, based on Flp and λ-integrase, respectively). These proteins recognize, cleave and ligate double-stranded DNA during a multi-step reaction wherein the intermediate is covalently bound to the enzyme (Figure 4A and B). The details of this reaction differ significantly when comparing and contrasting tyrosine recombinases (such as Cre, Flp and λ-integrase, Figure 4C),versus serine recombinases (such as φC31 integrase and Gin, Figure 4D) (reviewed in (192)). Unlike tyrosine recombinases, where the determinants of DNA specificity generally lie within the same domains required for catalysis, serine recombinases are significantly modular in their form and function; their DNA binding domains can be replaced without affecting catalytic function (reviewed in (193)]). In recent years, a growing number of naturally-occurring recombinases with differing sequence specificities have been identified. Some of these will likely also prove useful in genome engineering applications (reviewed in (194)).
Figure 4.

Site specific recombinase modes of action. Panel A: SSRs are capable of catalyzing excision, insertion, or inversion reactions. Panel B: Tyrosine recombinases such as Cre, Flp and λ integrase proceed through a Holliday junction intermediate. The catalytic domains have pseudo four-fold symmetry and engage palindromic sequences (arrows). Panel C: The topology of the λ integrase catalytic domains is similar to that of simple tyrosine recombinases like Cre. However, λ integrase also contains N-terminal DNA-binding domains (top set with arrows) that are critical for site-specific DNA recognition. Panel D:The catalytic domains (center) of serine integrases break both DNA strands before rotating relative to one another and religation of the DNA. DNA binding domains (top and bottom) of the wild-type enzymes can be replaced with zinc fingers to customize specificity.
In contrast to nuclease-based DNA editing schemes, Cre, Flp and some other SSRs do not require cellular machinery or ancillary proteins for efficient recombination (195–198), and it has been shown with λ integrase and various serine recombinases, that even SSRs that require additional proteins and host factors can often be engineered to function in a cofactor-independent manner (199–203). Thus, many SSRs are well suited for use in heterologous organisms. Perhaps most importantly, SSRs generally act with single-nucleotide resolution, and they maintain their hold on the cut DNA ends throughout the reaction cycle. For this reason, they may prove safer in clinical applications than nuclease-based editing schemes which often result in unpredictable indels at the edited locus. Recombinase catalysis can result in insertion, deletion or inversion of large DNA fragments (>100 kb). SSR technology has, for instance, been used to exchange large segments of the mouse genome with the equivalent human region (204). With two different SSRs, cassette exchange reactions and translocations can also be efficiently catalyzed (205,206). The primary limitation of recombinase-based genome editing arises from the requirement that enzyme-specific sequences be present in the DNA to be edited. To overcome this, a variety of recombinases, some with dramatically altered target specificities, have been engineered using genetic selection schemes and structure-based modeling. These enzymes (also called integrases, resolvases and invertases depending on their primary natural function) are broadly separated into two classes; those with DNA intermediates covalently bound to an active site tyrosine and those where the intermediate is bound to serine. Both classes have been the subject of engineering.
Tyrosine recombinase engineering
P1 bacteriophage Cre recombinase, so named because it catalyzes recombination, is the prototypical member of the tyrosine recombinase family, and it is arguably the enzyme that has been the subject of the most engineering. For this reason, it will be discussed in somewhat more detail than the others. Like most other SSRs discussed here, wild-type Cre forms a homotetrameric complex with its DNA substrate (Figure 4C) (reviewed in (207)). The preferred substrate for wild-type Cre, called loxP, contains palindromic sequences 13 nucleotides long separated by a central, non-palindromic 8 bp ‘spacer’ region which imparts directionality to the recombination site. Each 13 bp ‘half-site’ is engaged by one copy of the recombinase. For recombination to occur, two loxP sites (hence four half-sites and four Cre monomers) must come together. Cleavage and ligation occurs after the first base of the spacer region on the top strand and after the seventh base on the bottom strand. Most of the DNA-protein interactions involve the palindromic segments. Having 13 bp palindromic regions and an 8 bp spacer is not universal. A number of tyrosine recombinases have 7 bp spacers, and the length of the palindromic regions varies from 12 to 18 bp (reviewed in (194)). Some tyrosine recombinases exhibit a degree of flexibility. For instance Flp, but not Cre, can act on substrates with spacers that are one nucleotide longer or shorter (208).
Cre and Flp are both composed of two globular domains that are connected by an extended linker segment. These domains form a C-shaped clamp that completely encircles their respective DNA targets. In both proteins, the globular domains make extensive interactions, mostly in the major groove of the DNA, though the C-terminal domain makes additional minor groove interactions. The specific sites of DNA interaction are widely distributed across the protein sequence and there are at least a dozen sequence-specific DNA contacts made by each recombinase monomer (reviewed in (194)). Many more residues make non-specific interactions with the DNA backbone, and there are water-mediated interactions with some of the DNA bases. In the case of Cre, it has been shown that disruption of these indirect interactions also alters sequence-specificity (192,209,210). loxP has been subject to extensive mutation and analysis. Many mutations in this DNA sequence, particularly those in the linker region, do not abrogate Cre's activity (211,212). This sequence promiscuity could be the reason that chromosomal abnormalities have sometimes been attributed to Cre-based genome editing ((213) and references therein).
Both site-directed and random mutagenesis approaches have been successfully used to alter Cre's specificity such that sites other than loxP are targeted for recombination. Most, but not all of the successful studies involved screening large pools of mutant recombinases. Notably, Santoro and Schultz succeeded in changing Cre's specificity by generating a library of mutated enzymes where the amino acid sequence diversity was limited to just a handful of amino acids involved in sequence-specific binding to DNA (214). A reporter plasmid encoding fluorescent proteins flanked by altered loxP sites allowed fluorescence activated cell sorting of recombined cells and discovery of two Cre variants that were able to recombine loxM7, a sequence that differs at three positions in the loxP half-site. The loxM7 sequence is not acted on efficiently by the wild-type enzyme (215). Crystal structures of the mutated Cre with loxM7 revealed a complex network of interactions; water molecules and molecular flexibility played important roles in the DNA recognition (209). Santoro and Schulz also showed that both positive and negative selection were important; without the latter, the most likely result was relaxed specificity rather than altered specificity. Rüfer and Sauer reached a similar conclusion when they used a selection scheme wherein Kanamycin resistance was triggered by recombination of an altered site, loxK2, which has 13 changes relative to loxP (210). In the later study mutations throughout the Cre gene were combined through DNA shuffling (216). The results highlight the importance of Glu 262, a critical residue which was independently identified in subsequent work (194).
Buchholz and Stewart used a different approach to identifying mutations that alter Cre specificity. They used error-prone PCR and DNA shuffling to generate random mutations throughout Cre, and they developed a method known as substrate-linked protein evolution (SLiPE) to separate active variants from inactive ones. In this scheme, the mutant enzymes are encoded by the same plasmid (pEVO) as the substrate sequence. pEVO recombination after induction with arabinose results in the loss of an Nde1 restriction site. Successful recombinants remain circular upon Nde1 treatment, while those that have not excised the restriction site are linearized. PCR primers that only yield products from the circularized plasmids are then used to recover the mutated recombination-competent Cre sequences. This process can be repeated, and still more mutations can be introduced into the pool of sequences that yield recombined products (217). This approach was initially used to develop a recombinase named Fre22, which has minimal activity against loxP. Fre22 contains 15 mutations relative to Cre and targets a sequence named loxH, with 13 changes relative to loxP (6 are within the symmetric, palindromic regions).
The SLiPE approach was later used to generate Tre and Brec1, engineered recombinases that target a 34 bp sequence within the LTRs that flank the genes of HIV after genome integration. Tre, which recognizes a sequence that is 50% identical to that of loxP has 19 mutations relative to Cre. Brec1, which recognizes a sequence that is only 32% identical, has 45 mutations. Sequencing the pools of successful variants has provided significant insight into the regions of Cre most important for substrate specificity (Figure 5). (20,218–222). Tre recombinase was further engineered using a structure-guided approach. Molecular dynamics simulations suggested that changing the lysines at positions 43 and 86 to glutamates should improve specificity for the HIV-derived target DNA, and this was confirmed experimentally, highlighting the power of combining selection-based and structural approaches (223). In addition, the crystal structure of Tre in complex with its LTR-derived target allowed Meinke et al. to correctly predict that reverting Val 30 back to its original amino acid, Met, would make Tre more active (224). Using a mouse model system both Tre and Brec1 have been shown to efficiently excise HIV provirus from human cells. No deleterious effects or chromosomal abnormalities were observed, even when the Brec1 recombinase was constitutively expressed for a period of 18 months (20,225). In contrast to the earlier, evolved recombinases, Tre and Brec1 were designed to target sequences that are highly asymmetric. Tre's target half-sites differ at 8 out of 13 positions, and are thus no longer palindromic. The sequence targeted by Brec1 differs in 6 out of 13 half-site positions. The crystal structure of Tre in complex with its LTR-derived target has clarified the structural basis for much of this dual specificity (224).
Figure 5.

Regions of Cre recombinase particularly important for recognition. The DNA is shown in grey, and key regions of the protein involved in DNA recognition as described in the main text are highlighted with labels and side chain atom spheres.
Targeting asymmetrical sequences with Cre has been a longstanding goal of the recombinase engineering field. One approach, as described above, evolves a single recombinase that acts on two different half-sites. A second approach is to engineer heterodimeric recombinase complexes. The XerC/XerD recombinase from Escherichia coli, is a natural example of such a heterodimeric complex (reviewed in (226)). It was shown in 2006 that mixtures of two Cre-like molecules, each with a different half-site specificity, can recombine asymmetrical sites that neither molecule alone acts on efficiently (227). To facilitate catalysis on asymmetrical substrates, obligate Cre heterodimers have been engineered. Gelato et al. accomplished this in three steps: (i) They created a library wherein all 20 amino acids were present at three key positions within the Cre protein–protein interface. The positions chosen (299, 304 and 334) are all hydrophobic in Cre, and they form a contiguous cluster. (ii) They designed a screen wherein bacterial survival is contingent upon recombination. This revealed an alternative, functional interface wherein the three key residues were mutated. (iii) They identified obligate heterodimers via visual analysis of the modeled protein–protein interface. The resulting, engineered interfaces are still hydrophobic, but the relative sizes of the interfacial sidechains are changed such that heterodimers are favored (228).
Zhang et al. redesigned a different region of the Cre interface (229). They used the Rosetta program suite to model changes at 10 positions, and made a series of mutations based on the computational results that selectively form heterodimers. The mutated residues were in Cre's N-terminal helix and the interacting amino acids across the interface. No selection scheme was used in the Zhang study; the mutated proteins were purified and assayed one at a time. Eroshenko et al. also altered the N-terminal helix of Cre, but for a different reason. They showed that recombinase accuracy can be increased by decreasing cooperative binding (230). An antibiotic selection scheme allowed them to identify mutants of R32, which makes a salt bridge across the protein–protein interface in the wild-type, as important for increased accuracy.
Flp, a recombinase encoded by the 2μ plasmid of Saccharomyces cerevisiae, has also been the subject of significant protein engineering efforts. Its thermostability has been improved using random mutagenesis and DNA shuffling (231), and as with Cre, engineering of the inter-protomer interface has allowed heterodimers to be favored over homodimers, facilitating the recognition of asymmetric DNA targets (232). The specificity of Flp has been altered through a bacterial selection scheme wherein lacZ or RFP reporters were flanked by mutated versions of FRT, the 34 bp Flp target (233). In this study, both PCR-based mutagenesis and randomization of specific codons provided the necessary genetic variability to recognize target sequences with single-base changes. Using a similar approach, specificity for two sequences close to the human interleukin 10 (IL-10) gene have also been engineered (234). As is the case with the HIV sequences targeted by Cre, these IL 10 targets are both asymmetrical and significantly different from the natural Flp target. Mutated Flp recombinases have also been shown to act in the context of mammalian genomes and to efficiently catalyze integration reactions. Furthermore, when paired with another recombinase the Flp variant targeting IL-10 was able to perform a recombinase mediated cassette exchange (RMCE) (235). Two other Flp-like recombinases have also been the subject of protein engineering efforts. The activity of the yeast R and TD recombinases has been enhanced, largely though truncating the sequences so they match Flp and other, shorter homologs and also through random mutagenesis (236).
Engineering of λ integrase and serine recombinases
The architectures of the tyrosine recombinase λ integrase and the serine integrases discussed below are distinct from each other and from the Cre-like enzymes. As in Cre, the λ integrase catalytic domains form tetramers and have Holliday junction intermediates (237). In contrast, serine integrases have intermediates with double-stranded breaks and are thought to undergo large swiveling motions during catalysis (238). More importantly from the standpoint of protein engineering, the integrases also contain separate DNA-binding domains that are largely responsible for sequence specificity. In λ integrase the catalytic domain is near the C-terminal end; in the serine integrases, the catalytic domain is at or near the N-terminal end (239,240). In both cases, the wild-type integrases typically join plasmid-encoded attP sites with bacterial attB sites creating distinct attL and attR sites that flank the integrated plasmid DNA. In contrast to those of the Cre-type reactions, the pre- and post- recombination sites after integrase action are distinct. Additional factors are typically required to excise the plasmid after integration. This makes integrases ideal for gene insertion.
A variety of integrases have also been the subject of protein engineering. Notably, using a series of chimeras between the related integrases from λ and HK022 phages, Yagil et al. showed that just five key residues were largely responsible for a substrate specificity difference, and that subsets of the key amino acids identified from screening yielded more relaxed specificity (241). Recently, the specificity of λ integrase was altered to target a sequence from the human genome, attH, which differs from the original AttB site at four positions, and the λ integrase activity was also enhanced using a novel selection scheme involving β-lactamase and a fused β-lactamase inhibitor protein that was removed upon recombination (242). As in most of the recombinase work cited here, genetic variability was introduced by error prone PCR, with staggered extension process (StEP) PCR (243) (rather than DNA shuffling) used to combine the mutations within the surviving clones.
The activity and specificity of the serine recombinase φC31 integrase, which naturally catalyzes unidirectional integration of phage DNA, has also been enhanced. This was accomplished through random PCR mutagenesis combined with alanine scanning of charged amino acids in the N-terminal domain (200). Mutations that allow φC31 integrase to target a human sequence on chromosome 8 were identified using a blue/white selection scheme (244). Other serine integrases, including Tn3, Bin, Tn21, have also been engineered for genome editing. To make them better suited for this task, they have had their activity enhanced and their requirement for accessory factors has been overcome (199,201,203). These molecules differ in their target sequences, and as with the Cre and Flp homologs mentioned above, this work expands the repertoire of well-characterized starting points for future recombinase design studies.
An important series of different protein engineering studies have capitalized on the modular nature of serine recombinases to alter their specificity as well. Notably, the specificity of Tn3 resolvase was changed by fusing an engineered version of its catalytic domain to Zif268, a mouse transcription factor with a zinc finger fold (245). This recombinase has been studied in some detail to better understand the linker lengths and DNA sequence requirements for efficient catalysis (246). Barbas and coworkers developed their own versions of zinc-finger fusions with Gin and Tn3, and they showed that these molecules are useful for transferring genes into mouse and human genomes (247). Based on the crystal structure of the γδ resolvase with DNA (248), and the structures of Sin and Gin recombinases without DNA (249,250) they also identified residues that, when mutated, allow heterodimer formation (251,252). In addition, using overlap extension PCR to randomize the sidechains of five key amino acids and an assay that relies on recombinase-based assembly of an antibiotic resistance gene, Barbas and coworkers developed a collection of Gin recombinase catalytic domains capable to recognizing and recombining millions of 20 bp ‘core sequences’ (253).
To extend the utility of these fused recombinase molecules even further, the same investigators used directed evolution to identify variants of Sin and β recombinases that target core sequences that the Gin variants cannot act on. In this case, variants of the catalytic domain were generated through error-prone PCR with approximately three mutations per selection cycle. After four rounds of selection, sequencing revealed that some key mutations were present in over 70% of the recombination-competent plasmids (254). Procedures for developing novel zinc-finger recombinases as well as expressing, purifying, and assaying them have been clearly summarized in two detailed methods papers (255,256). This class of recombinases has been designed to target safe harbor sites in the human genome (203), the bovine β-casein gene (for transgenic protein production in milk) (257), and a variety of other human sequences (254). Many other applications are sure to follow.
Collectively, the recombinase engineering efforts described here clearly demonstrate that a broad range of selection schemes and approaches can be effectively employed to identify specificity-altering recombinase mutations. The work to date clearly demonstrates that it is possible to engineer enzymes that act on sequences that bear little, if any, resemblance to the natural targets of the wild-type enzymes. It is notable that despite the availability of high resolution structures and/or detailed molecular models, with few exceptions, the engineering efforts thus far have relied primarily on screens rather than carefully designed mutations to alter DNA recognition. Given the wealth of data regarding the effects of specific mutations on DNA specificity, it is likely that a hybrid approach will prove most effective going forward. Such an approach might use existing, unbiased screening data to identify the specific residues that should be mutated given the target sequence and then use experimental structures and computational models to determine what subsets of residues are most likely to yield results at each mutated site.
A second important development involves the large and growing number of enzymes that are suitable for use in heterologous systems. The diversity of natural targets provides numerous starting points for protein engineering and synthetic evolution efforts. Some recombinases (i.e. Cre) are naturally better suited to excising DNA fragments than introducing new ones. Wild-type versions of others (i.e. the serine integrases) have the opposite activity. Thus, the desired genome modification, along with the specific sequences to be targeted will help decide the optimal structural platform and starting point. Recombinase engineering is not as easy as CRISPR-based gene targeting, but the efficiency of gene integration, combined with the precision of the molecular editing will likely continue to make recombinase-based genome engineering highly attractive for many applications.
Restriction endonuclease engineering
Overview
Restriction endonuclease (REases) are one component of genomic defense systems encoded within bacteria and archaea. REases provide a form of ‘innate immunity’ for their bacterial hosts, by recognizing and cleaving short DNA target sequences that are found randomly within phage genomes and other forms of mobilized invasive DNA. When coupled with the activity of corresponding methyltransferases (MTases), such restriction-modification (‘RM’) systems confer resistance to phage infection and transformation by foreign DNA, while protecting the host genome from similar enzymatic degradation.
Since their discovery (258) restriction endonucleases have been developed and used as workhorse tools for molecular and cell biology research. Their ability to recognize and cleave defined target sequences with exceptional fidelity, and to generate a wide variety of DNA products (including 5′ or 3′ ‘sticky ends’ of defined sequence) allows various REases to be employed for routine cloning purposes, analyses of methylation status (259), SNP detection (260,261), serial analyses of gene expression (‘SAGE’) (262,263), and preparation of DNA for high-throughput DNA sequencing (264).
REases vary greatly with respect to their DNA target specificity, catalytic mechanism, structural organization, protein sequence and size. These differences are the basis for their classification into four major groups, or ‘Types’, each with multiple sub-classes (defined in (265) and organized into the restriction endonuclease database (‘REBASE’) as described in (266)). Types I and III R-M enzymes (reviewed in (267,268)) are multi-subunit assemblages that combine cleavage and DNA-modification together into large multifunctional molecular machines. Type II systems (reviewed in (269)) are generally simpler, and for the most part comprise separate endonuclease and methyltransferase enzymes, each with all the elements needed for independent sequence-recognition and catalysis acting at the same DNA target. Despite their simplicity, Type II endonucleases are highly diverse, having many different folds for DNA recognition integrated with several different folds and catalytic motifs that employ distinct DNA-hydrolysis mechanisms. They display a wide variety of structural organizations and are often embellished with additional structural domains. They assemble into various quaternary arrangements that can lead to complex cooperative and allosteric behaviors.
Whereas the specificity of the nucleic acid binding proteins and enzymes described above have become amenable (with various and often significant investments of time and effort) to reprogramming, REases have generally proven to be highly recalcitrant to such efforts. This difference may be in large part attributable to the underlying biological function and purpose of these various DNA binding protein systems. The protein families described in the prior sections have biological functions that might reasonably lead one to expect that they can be reprogrammed during evolution: two (meganucleases, recombinases) are largely associated with the mobilization and transfer of their own coding sequences, another (TAL effectors) is responsible for the hostile takeover of a gene's expression and activity, and one (zinc fingers) are found to be employed in a highly combinatorial and ubiquitous manner to dictate DNA binding specificity for a wide variety of factors involved in disparate biological and genetic processes. In contrast, many restriction endonucleases (particularly the classic Type II endonucleases which operate as ‘stand-alone’ enzymes) cannot readily alter their recognition and cleavage specificity, due to the resulting toxicity that would likely result from cleavage at the new, unprotected target sites throughout the host bacterial genome absent a simultaneous alteration of their companion MTase to the same new specificity. Thus REases from R-M systems having separate DNA recognition moieties for modification and restriction are under evolutionary pressure to avoid changing DNA target specificity, as a change in either REase or MTase is likely to be lethal to the host.
Initial engineering efforts
A number of attempts have been reported to change the specificity of type II REases, including in particular work conducted using the EcoRI and EcoRV enzymes (which were two of the first REases to be visualized bound to their DNA target sites). Those experiments (270–272) involved both the substitution of individual amino acids observed to form interactions with individual nucleotide bases in the enzyme's target site, and the addition of structural elements and residues to attempt to increase the length of target site read-out. The amino acid substitutions were largely generated based on suggestions of ‘canonical’ complementarity between certain combinations of residues and bases (for example, asparagine or glutamine versus adenine, or arginine versus guanine). Investigators now know that such preferences have very little predictive power for unique enzyme-target site combinations, due to the complexities associated with protein–DNA interfaces and contacts. Not surprisingly (in retrospect) these experiments largely resulted in enzyme variants with greatly reduced catalytic power, and little to no shift in target specificity. Similar attempts to alter BamHI specificity have also been described, using an in vivo selection for binding with an inactive BamHI construct was employed in an attempt to change the BamHI recognition sequence (273). No variants that could cleave a new target sequence were created, although one that requires that a methylated adenine base was identified.
A different approach, using directed evolution, was subsequently described to attempt the alteration of BstYI specificity (5′-RGATCY-3′) to recognize only 5′ - AGATCT-3′. An REase construct that no longer cut GGATCC, and that displayed moderate fidelity of recognition (preferring the target site AGATCT 12-fold over AGATCC) was obtained, but a complete change in specificity was not accomplished (274). In another study, an approach that used random mutagenesis coupled with a genetic screen was used to to alter the specificity of NotI (GCGGCCGC). Similar to the prior studies with BstYI, constructs that cleaved the wild-type sequence plus several miscognate sequences (that differ at one base) were identified, however a specific new DNA target was not generated (275).
Like the meganucleases, certain types of ‘unorthodox’ type II REases that recognize split DNA target sequences have been amenable to the structural recombination of their half-site recognition domains into new combinations. This had previously been exploited to generate new specificities for certain type I R-M systems (276), and was then extended to type II REs that recognize split sequences (277).
A different strategy was used to alter the recognition specificity of the Eco57I REase (which recognizes 5′-CTGAAG-3′) (278). Eco57I is a variant of the ‘type IIG’ enzyme subtype, wherein a single polypeptide harbors both an endonuclease and a DNA methyltransferase, each targeted to the same site by a common target recognition domain (TRD). Here an alteration of target specificity was generated by using a nuclease-deficient construct and selecting a randomly mutated library for altered methylase activity (indicated by protection against cleavage by an unrelated endonuclease) to thereby create a corresponding altered endonuclease specificity (at the same target site) having altered recognition at the fourth position: 5′-CTGRAG-3′. These moderate successes were made in R-M systems that use a common DNA recognition domain to target both protective modification and REase activities.
Finally, rational engineering of new Type II REase enzyme variants that recognize and cut at predictable new DNA sequences, while maintaining activity and fidelity comparable to the wild-type enzymes, was achieved in a large family of Type IIG REases related to MmeI (Figure 6) (21). MmeI is an unusual type II endonuclease that cuts DNA two turns of the helix away from its asymmetric recognition sequence and possesses both DNA methyltransferase and endonuclease activities in the same polypeptide (279–282). The enzyme was found to have many homologs that share considerable protein sequence and structural similarity yet recognize different DNA target site. Investigators realized that the strong overall conservation of sequence and function in this REase subgroup, considered jointly with their highly diverse substrate recognition (Figure 6A), suggested that DNA specificity in this family is undergoing rapid evolution, promoted by limited numbers of protein substitutions. The protein positions that contact and determine DNA recognition for each base pair within the DNA targets recognized were identified through covariation analysis between the aligned DNA target sequences and the aligned protein sequences. Generally, the amino acid residues at a pair of positions were found to make direct contact to a base pair within the DNA target to specify recognition. By identifying the amino acid combinations specifying recognition for each base pair, the residue positions correlated with each base pair could be reliably mutated to produce a desired new recognition specificity. The subsequent determination of the crystal structure of MmeI in complex with its DNA target site (Figure 6B) (283) demonstrated the interactions that underlie DNA recognition (Figure 6C) and explained the basis for the results of the engineering study described above. The same covariation analysis approach has been successfully applied to rationally alter specificity in other families of Type IIG REases having TRD domains that differ from MmeI, as well as to classic Type I and Type I-SP systems (Morgan, R.D. unpublished observations, (284)).
Figure 6.

Engineering altered DNA specificity of the MmeI restriction endonuclease via a bioinformatics-driven approach. Panel A:Sequence alignment of target sites and key specificity determining region of C-terminal domain of MmeI and 19 homologues that have known specificities. The positions of base pair 6 in each enzyme's target site, and the residues in the enzyme that display significant covariation against that target position, are highlighted. Panel B:Ribbon diagram of the crystal structure of Mme bound to its DNA target, demonstrating the distribution and separation of the enzyme's endonuclease catalytic site (‘REase’), methyltransferase active site (‘MTase’) and target recognition domain (‘TRD’). Panel C:Close ups illustrating (left) the experimentally observed positions of residues E806 and R808 that were found to display direct contacts to base pair 6 in the wild-type DNA-bound crystal structure of MmeI, and (right) a corresponding model of the same two residues, after introduction of mutations (to K and D, respectively) that were predicted and later found to alter specificity from a G:C to a C:G base pair. The ability to systematically alter the specificity of this enzyme is facilitated by the availability of a large number of sequenced enzyme homologues with corresponding known target sites and by a protein architecture in which endonuclease catalytic activity is less intimately coupled to target recognition and binding.
The REase engineering efforts described here clearly demonstrate that those Type II REase enzymes that utilize a single DNA recognition domain to direct both their protective MTase and restrictive REase activities have proved amenable to specificity engineering. However, the more familiar Type II restriction REases, which have separate REase and MTase proteins, are quite resistant to specificity alteration for clear functional and evolutionary reasons. RM systems that recognize split DNA target sequences, such as Type I or Type IIB enzymes, can be altered by exchanging one or other half site TRD, with new target specificities limited to combinations of the naturally occuring half site targets. REases that recognize contiguous DNA targets using a single TRD, such as the Type IIG or Type ISP enzymes, appear to have evolved the ability to readily alter their recognition specificity, often through subtle mutation involving just one or two residues. When these REases can be grouped into families having highly similar protein sequences yet diverged DNA targets, analysis of the correlation between amino acid position and residues with varying DNA target recognition can be used to direct rational engineering of REases with new specificity. This approach generally allows the alteration of one or two base pair positions within the native 5 to 8 base recognition site. Having a diversity of natural targets provides numerous starting points for such protein engineering efforts and expands the number of DNA targets that can be recognized. While it is not currently possible to engineer an REase to recognize all possible DNA target sequences, the engineering described can be used to expand the number of available Type II REase DNA targets by several orders of magnitude to provide an expanded toolkit for these molecular biology workhorses.
CONCLUSIONS
As mentioned at the beginning of this review, the field of protein engineering is experiencing a rapid increase in its ability to create new types of protein folds, topologies and assemblages. This discipline is now poised to address the critical issue of incorporating novel functions, such as ligand recognition, catalytic behaviors, and predictable structural and functional responses to changes in environmental conditions or to the addition of effector molecules. The notable accomplishments within this field over the past 15 years can be attributed to (i) the development of increasingly powerful and accurate computational algorithms to create and refine ab initio atomic models of protein folds and interactions, and (ii) the simultaneous development of reliable methods to generate and screen extremely large and complex protein libraries. This latter capability has benefitted from new and improved in cellulo and cell-free protein expression systems, enhanced methods for combining surface display platforms with robust flow cytometric screening approaches, and the advent of high-throughput sequencing strategies (which can be used to accurately determine patterns of sequence co-variation and context dependence that dictate form and function during the course of protein selection experiments). The fact that each these technologies have matured over the same relative time-frame has led to a current state of the art for protein engineering that might reasonably be described as almost limitless in its potential for creating novel biomolecules.
The capabilities of protein engineering have been further enhanced by the rapid accumulation of genomic sequence information for the types of protein folds and scaffolds summarized above. Protein engineering for each of these systems has been greatly facilitated by the identification of large collections of homologous proteins, in numbers sufficient to derive significant predictive understanding of their structure-function relationships. Such analyses were important for the rapid development of gene targeting proteins using both zinc fingers and TAL effectors. Similar types of analyses are now becoming possible for more complex DNA binding protein families such as meganucleases (for which many hundreds of proteins can be found in microbial sequence databases) and restriction endonucleases (which provided the information that enabled investigators to engineer new specificities onto the Mme family of REase enzymes). While it is difficult to predict when and how the engineering of novel protein–DNA recognition properties might become significantly automated and reliable across multiple protein folds and families, given the pace of discovery and development in these fields it is not unreasonable to believe that such abilities will arrive in the not-too-distant future.
While the development of improved methods and approaches for protein engineering is an important and useful area of research in general, the explosion of activity and results involving CRISPR-based, RNA-guided DNA targeting technologies might very well raise the question of whether further research and development in engineering recognition specificity of DNA-binding proteins in particular is justifiable. In addressing this question, it is useful to consider the example of DNA targeting for genome editing. The development of therapeutic targeted nucleases that display an ideal combination of activity, specificity, deliverability, and gene modification outcomes is not a fully solved problem, and each of the current platforms (certainly including CRISPR) offers unique advantages for such applications, offset by behaviors and properties requiring further study and development. While CRISPR offers the advantages of speed and scale for experimentation, important questions for its therapeutic use (such as its specificity, packaging, delivery, and controllability of repair outcomes) remain outstanding. The same questions exist for each of the protein-based DNA targeting systems described above, but in each case those questions appear to yield different answers, reflecting the distinct properties and unique advantages of the different platforms. The most salient of these are highlighted below:
Zinc finger nucleases have been subjected to an extensive program of research to optimize their activity and specificity, and currently have the longest track record of therapeutic use in patients, both ex vivo and in vivo.
Meganucleases and MegaTALs are the most difficult of gene targeting nucleases to engineer. However, they exhibit small size, single-chain structures, generation of uniquely reactive 3′ DNA product overhangs, and specificity profiles that appear highly desirable for certain genome editing applications.
TAL effectors offer remarkable potential for fine-tuned targeting specificity at individual DNA base pairs, even non-uniformly across the target site, afforded by the variation in specificity profiles and affinity contributions of individual RVDs, the influences of the repeat backbone residues, and the effects of the anchoring cryptic repeats. This promises the ability to engineer the proteins to recruit enzymatic activities to a range of targets, from large sets of related sequences that vary across the genome, to a single specified target within such a set.
Site specific recombinases offer the potential for genome editing activities and outcomes that are solely the result of enzymatic function, without the need to invoke and control cellular DSB repair processes.
As a result, different biotechnology and gene therapy companies and their partners are aggressively pursuing different or multiple platforms for DNA targeting in medicine, agriculture, and industry. Given the importance and breadth of these applications, and the unique properties and advantages of the different platforms, continued research and development in engineering altered protein–DNA recognition specificity seems likely and well justified.
ACKNOWLEDGEMENTS
The authors of this review are supported for research in this area by the National Institutes of Health (B.L.S., A.B.), the National Science Foundation (A.J.B.), the Bill and Melinda Gates Foundation (B.L.S.), bluebird bio inc. (B.L.S.), Sangamo Inc. (J.C.M.) and New England Biolabs (R.D.M.). The authors thank their many colleagues, past and present, for the work and insights that are presented in this review article.
FUNDING
National Institute of General Medical Sciences [R01 GM105691]. Funding for open access charge: Discretionary and Endowment funds provided by the Fred Hutchinson Cancer Research Center.
Conflict of interest statement. J.C.M. and R.D.M. are employees of Sangamo, Inc. and New England Biolabs, Inc., respectively; those companies create engineered nucleases for potential commercial and therapeutic applications. The B.L.S. laboratory is funded in part by bluebird bio Inc., which also creates engineered gene targeting nucleases for commercial use; B.L.S. receives royalties from them as an inventor on licensed meganuclease patents. A.J.B. receives royalties from Cellectis, Inc., as an inventor on TALEN patents licensed to that company. B.L.S. is Senior Executive Editor of Nucleic Acids Research.
REFERENCES
- 1. Anderson J.E., Ptashne M., Harrison S.C.. Structure of the repressor-operator complex of bacteriophage 434. Nature. 1987; 326:846–852. [DOI] [PubMed] [Google Scholar]
- 2. Jordan S.R., Whitcombe T.V., Berg J.M., Pabo C.O.. Systematic variation in DNA length yields highly ordered repressor-operator cocrystals. Science. 1985; 230:1383–1385. [DOI] [PubMed] [Google Scholar]
- 3. Otwinowski Z., Schevitz R.W., Zhang R.G., Lawson C.L., Joachimiak A., Marmorstein R.Q., Luisi B.F., Sigler P.B.. Crystal structure of trp repressor/operator complex at atomic resolution. Nature. 1988; 335:321–329. [DOI] [PubMed] [Google Scholar]
- 4. Schultz S.C., Shields G.C., Steitz T.A.. Crystallization of Escherichia coli catabolite gene activator protein with its DNA binding site. The use of modular DNA. J. Mol. Biol. 1990; 213:159–166. [DOI] [PubMed] [Google Scholar]
- 5. Luscombe N.M., Laskowski R.A., Thornton J.M.. Amino acid-base interactions: a three-dimensional analysis of protein–DNA interactions at an atomic level. Nucleic Acids Res. 2001; 29:2860–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rohs R., Jin X., West S.M., Joshi R., Honig B., Mann R.S.. Origins of specificity in protein–DNA recognition. Annu. Rev. Biochem. 2010; 79:233–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Smith N.C., Matthews J.M.. Mechanisms of DNA-binding specificity and functional gene regulation by transcription factors. Curr. Opin. Struct. Biol. 2016; 38:68–74. [DOI] [PubMed] [Google Scholar]
- 8. Joshi R., Passner J.M., Rohs R., Jain R., Sosinsky A., Crickmore M.A., Jacob V., Aggarwal A.K., Honig B., Mann R.S.. Functional specificity of a Hox protein mediated by the recognition of minor groove structure. Cell. 2007; 131:530–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rohs R., West S.M., Sosinsky A., Liu P., Mann R.S., Honig B.. The role of DNA shape in protein–DNA recognition. Nature. 2009; 461:1248–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lazarovici A., Zhou T., Shafer A., Dantas Machado A.C., Riley T.R., Sandstrom R., Sabo P.J., Lu Y., Rohs R., Stamatoyannopoulos J.A. et al. Probing DNA shape and methylation state on a genomic scale with DNase I. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:6376–6381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kitayner M., Rozenberg H., Rohs R., Suad O., Rabinovich D., Honig B., Shakked Z.. Diversity in DNA recognition by p53 revealed by crystal structures with Hoogsteen base pairs. Nat. Struct. Mol. Biol. 2010; 17:423–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gordan R., Shen N., Dror I., Zhou T., Horton J., Rohs R., Bulyk M.L.. Genomic regions flanking E-box binding sites influence DNA binding specificity of bHLH transcription factors through DNA shape. Cell Rep. 2013; 3:1093–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Slattery M., Zhou T., Yang L., Dantas Machado A.C., Gordan R., Rohs R.. Absence of a simple code: how transcription factors read the genome. Trends Biochem. Sci. 2014; 39:381–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lavery R. Recognizing DNA. Q. Rev. Biophys. 2005; 38:339–344. [DOI] [PubMed] [Google Scholar]
- 15. Boch J., Bonas U.. Xanthomonas AvrBs3 family-type III effectors: discovery and function. Annu. Rev. Phytopathol. 2010; 48:419–436. [DOI] [PubMed] [Google Scholar]
- 16. Moscou M.J., Bogdanove A.J.. A simple cipher governs DNA recognition by TAL effectors. Science. 2009; 326:1501. [DOI] [PubMed] [Google Scholar]
- 17. Kuhlman B., Dantas G., Ireton G.C., Varani G., Stoddard B.L., Baker D.. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003; 302:1364–1368. [DOI] [PubMed] [Google Scholar]
- 18. Procko E., Berguig G.Y., Shen B.W., Song Y., Frayo S., Convertine A.J., Margineantu D., Booth G., Correia B.E., Cheng Y. et al. A computationally designed inhibitor of an Epstein-Barr viral Bcl-2 protein induces apoptosis in infected cells. Cell. 2014; 157:1644–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thyme S., Baker D.. Redesigning the specificity of protein–DNA interactions with Rosetta. Methods Mol. Biol. 2014; 1123:265–282. [DOI] [PubMed] [Google Scholar]
- 20. Karpinski J., Hauber I., Chemnitz J., Schafer C., Paszkowski-Rogacz M., Chakraborty D., Beschorner N., Hofmann-Sieber H., Lange U.C., Grundhoff A. et al. Directed evolution of a recombinase that excises the provirus of most HIV-1 primary isolates with high specificity. Nat. Biotechnol. 2016; 34:401–409. [DOI] [PubMed] [Google Scholar]
- 21. Morgan R.D., Luyten Y.A.. Rational engineering of type II restriction endonuclease DNA binding and cleavage specificity. Nucleic Acids Res. 2009; 37:5222–5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Urnov F.D., Miller J.C., Lee Y.-L., Beausejour C.M., Rock J.M., Augustus S., Jamieson A.C., Porteus M.H., Gregory P.D., Holmes M.C.. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005; 435:646–651. [DOI] [PubMed] [Google Scholar]
- 23. Werther R., Hallinan J.P., Lambert A.R., Havens K., Pogson M., Jarjour J., Galizi R., Windbichler N., Crisanti A., Nolan T. et al. Crystallographic analyses illustrate significant plasticity and efficient recoding of meganuclease target specificity. Nucleic Acids Res. 2017; 45:8621–8634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tupler R., Perini G., Green M.R.. Expressing the human genome. Nature. 2001; 409:832–833. [DOI] [PubMed] [Google Scholar]
- 25. Pabo C.O., Peisach E., Grant R.A.. Design and selection of novel Cys2His2 zinc finger proteins. Annu. Rev. Biochem. 2001; 70:313–340. [DOI] [PubMed] [Google Scholar]
- 26. Pavletich N.P., Pabo C.O.. Zinc finger-DNA recognition: crystal structure of a Zif268-DNA complex at 2.1 A. Science. 1991; 252:809–817. [DOI] [PubMed] [Google Scholar]
- 27. Kim Y.G., Cha J., Chandrasegaran S.. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:1156–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Smith J., Berg J.M., Chandrasegaran S.. A detailed study of the substrate specificity of a chimeric restriction enzyme. Nucleic Acids Res. 1999; 27:674–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rouet P., Smih F., Jasin M.. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol. Cell. Biol. 1994; 14:8096–8106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rouet P., Smih F., Jasin M.. Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:6064–6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bibikova M., Beumer K., Trautman J.K., Carroll D.. Enhancing gene targeting with designed zinc finger nucleases. Science. 2003; 300:764. [DOI] [PubMed] [Google Scholar]
- 32. Bibikova M., Golic M., Golic K.G., Carroll D.. Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics. 2002; 161:1169–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Osakabe K., Osakabe Y., Toki S.. Site-directed mutagenesis in Arabidopsis using custom-designed zinc finger nucleases. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:12034–12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang F., Maeder M.L., Unger-Wallace E., Hoshaw J.P., Reyon D., Christian M., Li X., Pierick C.J., Dobbs D., Peterson T. et al. High frequency targeted mutagenesis in Arabidopsis thaliana using zinc finger nucleases. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:12028–12033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morton J., Davis M.W., Jorgensen E.M., Carroll D.. Induction and repair of zinc-finger nuclease-targeted double-strand breaks in Caenorhabditis elegans somatic cells. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:16370–16375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Doyon Y., McCammon J.M., Miller J.C., Faraji F., Ngo C., Katibah G.E., Amora R., Hocking T.D., Zhang L., Rebar E.J. et al. Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nat. Biotechnol. 2008; 26:702–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Foley J.E., Yeh J.R., Maeder M.L., Reyon D., Sander J.D., Peterson R.T., Joung J.K.. Rapid mutation of endogenous zebrafish genes using zinc finger nucleases made by Oligomerized Pool ENgineering (OPEN). PLoS One. 2009; 4:e4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Meng X., Noyes M.B., Zhu L.J., Lawson N.D., Wolfe S.A.. Targeted gene inactivation in zebrafish using engineered zinc-finger nucleases. Nat. Biotechnol. 2008; 26:695–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Goldberg A.D., Banaszynski L.A., Noh K.M., Lewis P.W., Elsaesser S.J., Stadler S., Dewell S., Law M., Guo X., Li X. et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010; 140:678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Flisikowska T., Thorey I.S., Offner S., Ros F., Lifke V., Zeitler B., Rottmann O., Vincent A., Zhang L., Jenkins S. et al. Efficient immunoglobulin gene disruption and targeted replacement in rabbit using zinc finger nucleases. PLoS One. 2011; 6:e21045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Geurts A.M., Cost G.J., Freyvert Y., Zeitler B., Miller J.C., Choi V.M., Jenkins S.S., Wood A., Cui X., Meng X. et al. Knockout rats via embryo microinjection of zinc-finger nucleases. Science. 2009; 325:433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mashimo T., Takizawa A., Voigt B., Yoshimi K., Hiai H., Kuramoto T., Serikawa T.. Generation of knockout rats with X-linked severe combined immunodeficiency (X-SCID) using zinc-finger nucleases. PLoS One. 2010; 5:e8870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shukla V.K., Doyon Y., Miller J.C., DeKelver R.C., Moehle E.A., Worden S.E., Mitchell J.C., Arnold N.L., Gopalan S., Meng X. et al. Precise genome modification in the crop species Zea mays using zinc-finger nucleases. Nature. 2009; 459:437–441. [DOI] [PubMed] [Google Scholar]
- 44. Curtin S.J., Zhang F., Sander J.D., Haun W.J., Starker C., Baltes N.J., Reyon D., Dahlborg E.J., Goodwin M.J., Coffman A.P. et al. Targeted mutagenesis of duplicated genes in soybean with zinc-finger nucleases. Plant Physiol. 2011; 156:466–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hauschild J., Petersen B., Santiago Y., Queisser A.L., Carnwath J.W., Lucas-Hahn A., Zhang L., Meng X., Gregory P.D., Schwinzer R. et al. Efficient generation of a biallelic knockout in pigs using zinc-finger nucleases. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:12013–12017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yu S., Luo J., Song Z., Ding F., Dai Y., Li N.. Highly efficient modification of beta-lactoglobulin (BLG) gene via zinc-finger nucleases in cattle. Cell Res. 2011; 21:1638–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tebas P., Stein D., Tang W.W., Frank I., Wang S.Q., Lee G., Spratt S.K., Surosky R.T., Giedlin M.A., Nichol G. et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 2014; 370:901–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wolfe S.A., Grant R.A., Elrod-Erickson M., Pabo C.O.. Beyond the “recognition code”: structures of two Cys2His2 zinc finger/TATA box complexes. Structure. 2001; 9:717–723. [DOI] [PubMed] [Google Scholar]
- 49. Desjarlais J.R., Berg J.M.. Redesigning the DNA-binding specificity of a zinc finger protein: a data base-guided approach. Proteins. 1992; 13:272. [DOI] [PubMed] [Google Scholar]
- 50. Desjarlais J.R., Berg J.M.. Toward rules relating zinc finger protein sequences and DNA binding site preferences. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:7345–7349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Desjarlais J.R., Berg J.M.. Use of a zinc-finger consensus sequence framework and specificity rules to design specific DNA binding proteins. Proc. Natl. Acad. Sci. U.S.A. 1993; 90:2256–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Choo Y., Klug A.. Selection of DNA binding sites for zinc fingers using rationally randomized DNA reveals coded interactions. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:11168–11172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jamieson A.C., Kim S.H., Wells J.A.. In vitro selection of zinc fingers with altered DNA-binding specificity. Biochemistry. 1994; 33:5689–5695. [DOI] [PubMed] [Google Scholar]
- 54. Rebar E.J., Pabo C.O.. Zinc finger phage: affinity selection of fingers with new DNA-binding specificities. Science. 1994; 263:671–673. [DOI] [PubMed] [Google Scholar]
- 55. Wu H., Yang W.P., Barbas C.F. 3rd. Building zinc fingers by selection: toward a therapeutic application. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:344–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Segal D.J., Dreier B., Beerli R.R., Barbas C.F. 3rd. Toward controlling gene expression at will: selection and design of zinc finger domains recognizing each of the 5′-GNN-3′ DNA target sequences. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:2758–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dreier B., Beerli R.R., Segal D.J., Flippin J.D., Barbas C.F.. Development of zinc finger domains for recognition of the 5′-ANN-3′ family of DNA sequences and their use in the construction of artificial transcription factors. J. Biol. Chem. 2001; 276:29466–29478. [DOI] [PubMed] [Google Scholar]
- 58. Dreier B., Fuller R.P., Segal D.J., Lund C.V., Blancafort P., Huber A., Koksch B., Barbas C.F. 3rd. Development of zinc finger domains for recognition of the 5′-CNN-3′ family DNA sequences and their use in the construction of artificial transcription factors. J. Biol. Chem. 2005; 280:34488–35597. [DOI] [PubMed] [Google Scholar]
- 59. Ramirez C.L., Foley J.E., Wright D.A., Muller-Lerch F., Rahman S.H., Cornu T.I., Winfrey R.J., Sander J.D., Fu F., Townsend J.A. et al. Unexpected failure rates for modular assembly of engineered zinc fingers. Nat. Methods. 2008; 5:374–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Isalan M., Klug A., Choo Y.. A rapid, generally applicable method to engineer zinc fingers illustrated by targeting the HIV-1 promoter. Nat. Biotechnol. 2001; 19:656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Joung J.K., Ramm E.I., Pabo C.O.. A bacterial two-hybrid selection system for studying protein–DNA and protein–protein interactions. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:7382–7387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Maeder M.L., Thibodeau-Beganny S., Osiak A., Wright D.A., Anthony R.M., Eichtinger M., Jiang T., Foley J.E., Winfrey R.J., Townsend J.A. et al. Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol. Cell. 2008; 31:294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sander J.D., Dahlborg E.J., Goodwin M.J., Cade L., Zhang F., Cifuentes D., Curtin S.J., Blackburn J.S., Thibodeau-Beganny S., Qi Y. et al. Selection-free zinc-finger-nuclease engineering by context-dependent assembly (CoDA). Nat. Methods. 2011; 8:67–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Moore M., Klug A., Choo Y.. Improved DNA binding specificity from polyzinc finger peptides by using strings of two-finger units. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:1437–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Carroll D. Genome engineering with zinc-finger nucleases. Genetics. 2011; 188:773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Urnov F.D., Rebar E.J., Holmes M.C., Zhang H.S., Gregory P.D.. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010; 11:636–646. [DOI] [PubMed] [Google Scholar]
- 67. Perez E.E., Wang J., Miller J.C., Jouvenot Y., Kim K.A., Liu O., Wang N., Lee G., Bartsevich V.V., Lee Y.L. et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotechnol. 2008; 26:808–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chang K.H., Smith S.E., Sullivan T., Chen K., Zhou Q., West J.A., Liu M., Liu Y., Vieira B.F., Sun C. et al. Long-Term engraftment and fetal globin induction upon BCL11A gene editing in Bone-Marrow-Derived CD34(+) hematopoietic stem and progenitor cells. Mol. Ther. Methods Clin. Dev. 2017; 4:137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yusa K., Rashid S.T., Strick-Marchand H., Varela I., Liu P.Q., Paschon D.E., Miranda E., Ordonez A., Hannan N.R., Rouhani F.J. et al. Targeted gene correction of alpha1-antitrypsin deficiency in induced pluripotent stem cells. Nature. 2011; 478:391–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bitinaite J., Wah D.A., Aggarwal A.K., Schildkraut I.. FokI dimerization is required for DNA cleavage. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:10570–10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Smith J., Bibikova M., Whitby F.G., Reddy A.R., Chandrasegaran S., Carroll D.. Requirements for double-strand cleavage by chimeric restriction enzymes with zinc finger DNA-recognition domains. Nucleic Acids Res. 2000; 28:3361–3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bibikova M., Carroll D., Segal D.J., Trautman J.K., Smith J., Kim Y.G., Chandrasegaran S.. Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Mol. Cell. Biol. 2001; 21:289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Porteus M.H., Baltimore D.. Chimeric nucleases stimulate gene targeting in human cells. Science. 2003; 300:763. [DOI] [PubMed] [Google Scholar]
- 74. Handel E.M., Alwin S., Cathomen T.. Expanding or restricting the target site repertoire of zinc-finger nucleases: the inter-domain linker as a major determinant of target site selectivity. Mol. Ther. 2009; 17:104–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Shimizu Y., Bhakta M.S., Segal D.J.. Restricted spacer tolerance of a zinc finger nuclease with a six amino acid linker. Bioorg. Med. Chem. Lett. 2009; 19:3970–3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Guo J., Gaj T., Barbas C.F. 3rd. Directed evolution of an enhanced and highly efficient FokI cleavage domain for zinc finger nucleases. J. Mol. Biol. 2010; 400:96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Doyon Y., Vo T.D., Mendel M.C., Greenberg S.G., Wang J., Xia D.F., Miller J.C., Urnov F.D., Gregory P.D., Holmes M.C.. Enhancing zinc-finger-nuclease activity with improved obligate heterodimeric architectures. Nat. Methods. 2011; 8:74–79. [DOI] [PubMed] [Google Scholar]
- 78. Miller J.C., Holmes M.C., Wang J., Guschin D.Y., Lee Y.L., Rupniewski I., Beausejour C.M., Waite A.J., Wang N.S., Kim K.A. et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat. Biotechnol. 2007; 25:778–785. [DOI] [PubMed] [Google Scholar]
- 79. Szczepek M., Brondani V., Buchel J., Serrano L., Segal D.J., Cathomen T.. Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat. Biotechnol. 2007; 25:786–793. [DOI] [PubMed] [Google Scholar]
- 80. Ramirez C.L., Certo M.T., Mussolino C., Goodwin M.J., Cradick T.J., McCaffrey A.P., Cathomen T., Scharenberg A.M., Joung J.K.. Engineered zinc finger nickases induce homology-directed repair with reduced mutagenic effects. Nucleic Acids Res. 2012; 40:5560–5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wang J., Friedman G., Doyon Y., Wang N.S., Li C.J., Miller J.C., Hua K.L., Yan J.J., Babiarz J.E., Gregory P.D. et al. Targeted gene addition to a predetermined site in the human genome using a ZFN-based nicking enzyme. Genome Res. 2012; 22:1316–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Pattanayak V., Ramirez C.L., Joung J.K., Liu D.R.. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat. Methods. 2011; 8:765–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gabriel R., Lombardo A., Arens A., Miller J.C., Genovese P., Kaeppel C., Nowrouzi A., Bartholomae C.C., Wang J., Friedman G. et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat. Biotechnol. 2011; 29:816–823. [DOI] [PubMed] [Google Scholar]
- 84. Wood A.J., Lo T.W., Zeitler B., Pickle C.S., Ralston E.J., Lee A.H., Amora R., Miller J.C., Leung E., Meng X. et al. Targeted genome editing across species using ZFNs and TALENs. Science. 2011; 333:307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Choulika A., Perrin A., Dujon B., Nicolas J.F.. Induction of homologous recombination in mammalian chromosomes by using the I-SceI system of Saccharomyces cerevisiae. Mol. Cell. Biol. 1995; 15:1968–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Scalley-Kim M., McConnell-Smith A., Stoddard B.L.. Coevolution of homing endonuclease specificity and its host target sequence. J. Mol. Biol. 2007; 372:1305–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Chevalier B., Turmel M., Lemieux C., Monnat R.J. Jr, Stoddard B.L.. Flexible DNA target site recognition by divergent homing endonuclease isoschizomers I-CreI and I-MsoI. J. Mol. Biol. 2003; 329:253–269. [DOI] [PubMed] [Google Scholar]
- 88. Lambert A.R., Hallinan J.P., Shen B.W., Chik J.K., Bolduc J.M., Kulshina N., Robins L.I., Kaiser B.K., Jarjour J., Havens K. et al. Indirect DNA sequence recognition and its impact on nuclease cleavage activity. Structure. 2016; 24:862–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Thyme S.B., Jarjour J., Takeuchi R., Havranek J.J., Ashworth J., Scharenberg A.M., Stoddard B.L., Baker D.. Exploitation of binding energy for catalysis and design. Nature. 2009; 461:1300–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Choulika A., Perrin A., Dujon B., Nicolas J.F.. The yeast I-Sce I meganuclease induces site-directed chromosomal recombination in mammalian cells. C. R. Acad. Sci. III. 1994; 317:1013–1019. [PubMed] [Google Scholar]
- 91. Riviere J., Hauer J., Poirot L., Brochet J., Souque P., Mollier K., Gouble A., Charneau P., Fischer A., Paques F. et al. Variable correction of Artemis deficiency by I-Sce1-meganuclease-assisted homologous recombination in murine hematopoietic stem cells. Gene Ther. 2014; 21:529–532. [DOI] [PubMed] [Google Scholar]
- 92. Gouble A., Smith J., Bruneau S., Perez C., Guyot V., Cabaniols J.-P., Leduc S., Fiette L., Ave P., Micheau B. et al. Efficient in toto targeted recombination in mouse liver by meganuclease-induced double-strand break. J. Gene Med. 2006; 8:616–622. [DOI] [PubMed] [Google Scholar]
- 93. Flick K.E., Jurica M.S., Monnat R.J. Jr, Stoddard B.L.. DNA binding and cleavage by the nuclear intron-encoded homing endonuclease I-PpoI. Nature. 1998; 394:96–101. [DOI] [PubMed] [Google Scholar]
- 94. Jurica M.S., Monnat R.J. Jr, Stoddard B.L.. DNA recognition and cleavage by the LAGLIDADG homing endonuclease I-CreI. Mol. Cell. 1998; 2:469–476. [DOI] [PubMed] [Google Scholar]
- 95. Moure C.M., Gimble F.S., Quiocho F.A.. The crystal structure of the gene targeting homing endonuclease I-SceI reveals the origins of its target site specificity. J. Mol. Biol. 2003; 334:685–695. [DOI] [PubMed] [Google Scholar]
- 96. Gimble F.S., Moure C.M., Posey K.L.. Assessing the plasticity of DNA target site recognition of the PI-SceI homing endonuclease using a bacterial two-hybrid selection system. J. Mol. Biol. 2003; 334:993–1008. [DOI] [PubMed] [Google Scholar]
- 97. Seligman L., Chisholm K.M., Chevalier B.S., Chadsey M.S., Edwards S.T., Savage J.H., Veillet A.L.. Mutations altering the cleavage specificity of a homing endonuclease. Nucleic Acids Res. 2002; 30:3870–3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Gruen M., Chang K., Serbanescu I., Liu D.R.. An in vivo selection system for homing endonuclease activity. Nucleic Acids Res. 2002; 30:e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Ashworth J., Havranek J.J., Duarte C.M., Sussman D., Monnat R.J. Jr, Stoddard B.L., Baker D.. Computational redesign of endonuclease DNA binding and cleavage specificity. Nature. 2006; 441:656–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Rosen L.E., Morrison H.A., Masri S., Brown M.J., Springstubb B., Sussman D., Stoddard B.L., Seligman L.M.. Homing endonuclease I-CreI derivatives with novel DNA target specificities. Nucleic Acids Res. 2006; 34:4791–4800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Chames P., Epinat J.C., Guillier S., Patin A., Lacroix E., Paques F.. In vivo selection of engineered homing endonucleases using double-strand break induced homologous recombination. Nucleic Acids Res. 2005; 33:e178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Arnould S., Chames P., Perez C., Lacroix E., Duclert A., Epinat J.C., Stricher F., Petit A.S., Patin A., Guillier S. et al. Engineering of large numbers of highly specific homing endonucleases that induce recombination on novel DNA targets. J. Mol. Biol. 2006; 355:443–458. [DOI] [PubMed] [Google Scholar]
- 103. Smith J., Grizot S., Arnould S., Duclert A., Epinat J.C., Chames P., Prieto J., Redondo P., Blanco F.J., Bravo J. et al. A combinatorial approach to create artificial homing endonucleases cleaving chosen sequences. Nucleic Acids Res. 2006; 34:e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Paques F., Duchateau P.. Meganucleases and DNA double-strand break-induced recombination: perspectives for gene therapy. Curr. Gene Ther. 2007; 7:49–66. [DOI] [PubMed] [Google Scholar]
- 105. Ashworth J., Taylor G.K., Havranek J.J., Quadri S.A., Stoddard B.L., Baker D.. Computational reprogramming of homing endonuclease specificity at multiple adjacent base pairs. Nucleic Acids Res. 2010; 38:5601–5608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Thyme S.B., Baker D., Bradley P.. Improved modeling of side-chain. base interactions and plasticity in protein–DNA. interface design. J. Mol. Biol. 2012; 419:255–274. [Google Scholar]
- 107. Wang Y., Khan I.F., Boissel S., Jarjour J., Pangallo J., Thyme S., Baker D., Scharenberg A.M., Rawlings D.J.. Progressive engineering of a homing endonuclease genome editing reagent for the murine X-linked immunodeficiency locus. Nucleic Acids Res. 2014; 42:6463–6475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Windbichler N., Menichelli M., Papathanos P.A., Thyme S.B., Li H., Ulge U.Y., Hovde B.T., Baker D., Monnat R.J. Jr, Burt A. et al. A synthetic homing endonuclease-based gene drive system in the human malaria mosquito. Nature. 2011; 473:212–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Baxter S.K., Scharenberg A.M., Lambert A.R.. Engineering and flow-cytometric analysis of chimeric LAGLIDADG homing endonucleases from homologous I-OnuI-family enzymes. Methods Mol Biol. 2014; 1123:191–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Chevalier B.S., Kortemme T., Chadsey M.S., Baker D., Monnat R.J., Stoddard B.L.. Design, activity and structure of a highly specific artificial endonuclease. Mol. Cell. 2002; 10:895–905. [DOI] [PubMed] [Google Scholar]
- 111. Epinat J.C., Arnould S., Chames P., Rochaix P., Desfontaines D., Puzin C., Patin A., Zanghellini A., Paques F., Lacroix E.. A novel engineered meganuclease induces homologous recombination in yeast and mammalian cells. Nucleic Acids Res. 2003; 31:2952–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Silva G.H., Belfort M., Wende W., Pingoud A.. From monomeric to homodimeric endonucleases and back: engineering novel specificity of LAGLIDADG enzymes. J. Mol. Biol. 2006; 361:744–754. [DOI] [PubMed] [Google Scholar]
- 113. Gao H., Smith J., Yang M., Jones S., Djukanovic V., Nicholson M.G., West A., Bidney D., Falco S.C., Jantz D. et al. Heritable targeted mutagenesis in maize using a designed endonuclease. Plant J. 2010; 61:176–187. [DOI] [PubMed] [Google Scholar]
- 114. Li H., Pellenz S., Ulge U., Stoddard B.L., Monnat R.J. Jr. Generation of single-chain LAGLIDADG homing endonucleases from native homodimeric precursor proteins. Nucleic Acids Res. 2009; 37:1650–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Boissel S., Jarjour J., Astrakhan A., Adey A., Gouble A., Duchateau P., Shendure J., Stoddard B.L., Certo M.T., Baker D. et al. megaTALs: a rare-cleaving nuclease architecture for therapeutic genome engineering. Nucleic Acids Res. 2014; 42:2591–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Sather B.D., Romano Ibarra G.S., Sommer K., Curinga G., Hale M., Khan I.F., Singh S., Song Y., Gwiazda K., Sahni J. et al. Efficient modification of CCR5 in primary human hematopoietic cells using a megaTAL nuclease and AAV donor template. Sci. Transl. Med. 2015; 7:307ra156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Arnould S., Perez C., Cabaniols J.-P., Smith J., Gouble A., Grizot S., Epinat J.-C., Duclert A., Duchateau P., Paques F.. Engineered I-CreI derivatives cleaving sequences from the human XPC gene can induce highly efficient gene correction in mammalian cells. J. Mol. Biol. 2007; 371:49–65. [DOI] [PubMed] [Google Scholar]
- 118. Dupuy A., Valton J., Leduc S., Armier J., Galetto R., Gouble A., Lebuhotel C., Stary A., Paques F., Duchateau P. et al. Targeted gene therapy of xeroderma pigmentosum cells using meganuclease and TALEN. PLoS One. 2013; 8:e78678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Redondo P., Prieto J., Munoz I.G., Alibes A., Stricher F., Serrano L., Cabaniols J.P., Daboussi F., Arnould S., Perez C. et al. Molecular basis of xeroderma pigmentosum group C DNA recognition by engineered meganucleases. Nature. 2008; 456:107–111. [DOI] [PubMed] [Google Scholar]
- 120. Cabaniols J.P., Ouvry C., Lamamy V., Fery I., Craplet M.L., Moulharat N., Guenin S.P., Bedut S., Nosjean O., Ferry G. et al. Meganuclease-driven targeted integration in CHO-K1 cells for the fast generation of HTS-compatible cell-based assays. J. Biomol. Screen. 2010; 15:956–967. [DOI] [PubMed] [Google Scholar]
- 121. Cabaniols J.P., Paques F.. Robust cell line development using meganucleases. Methods Mol. Biol. 2008; 435:31–45. [DOI] [PubMed] [Google Scholar]
- 122. Djukanovic V., Smith J., Lowe K., Yang M., Gao H., Jones S., Nicholson M.G., West A., Lape J., Bidney D. et al. Male-sterile maize plants produced by targeted mutagenesis of the cytochrome P450-like gene (MS26) using a re-designed I-CreI homing endonuclease. Plant J. 2013; 76:888–899. [DOI] [PubMed] [Google Scholar]
- 123. Antunes M.S., Smith J.J., Jantz D., Medford J.I.. Targeted DNA excision in Arabidopsis by a re-engineered homing endonuclease. BMC Biotechnol. 2012; 12:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. D’Halluin K., Vanderstraeten C., Van Hulle J., Rosolowska J., Van Den Brande I., Pennewaert A., D’Hont K., Bossut M., Jantz D., Ruiter R. et al. Targeted molecular trait stacking in cotton through targeted double-strand break induction. Plant Biotechnol. J. 2013; 11:933–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Grizot S., Smith J., Daboussi F., Prieto J., Redondo P., Merino N., Villate M., Thomas S., Lemaire L., Montoya G. et al. Efficient targeting of a SCID gene by an engineered single-chain homing endonuclease. Nucleic Acids Res. 2009; 37:5405–5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Munoz I.G., Prieto J., Subramanian S., Coloma J., Redondo P., Villate M., Merino N., Marenchino M., D’Abramo M., Gervasio F.L. et al. Molecular basis of engineered meganuclease targeting of the endogenous human RAG1 locus. Nucleic Acids Res. 2011; 39:729–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Menoret S., Fontaniere S., Jantz D., Tesson L., Thinard R., Remy S., Usal C., Ouisse L.H., Fraichard A., Anegon I.. Generation of Rag1-knockout immunodeficient rats and mice using engineered meganucleases. FASEB J. 2013; 27:703–711. [DOI] [PubMed] [Google Scholar]
- 128. Grosse S., Huot N., Mahiet C., Arnould S., Barradeau S., Clerre D.L., Chion-Sotinel I., Jacqmarcq C., Chapellier B., Ergani A. et al. Meganuclease-mediated Inhibition of HSV1 infection in cultured cells. Mol. Ther. 2011; 19:694–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Popplewell L., Koo T., Leclerc X., Duclert A., Mamchaoui K., Gouble A., Mouly V., Voit T., Paques F., Cedrone F. et al. Gene correction of a duchenne muscular dystrophy mutation by meganuclease-enhanced exon knock-in. Hum. Gene Ther. 2013; 24:692–701. [DOI] [PubMed] [Google Scholar]
- 130. Jarjour J., West-Foyle H., Certo M.T., Hubert C.G., Doyle L., Getz M.M., Stoddard B.L., Scharenberg A.M.. High-resolution profiling of homing endonuclease binding and catalytic specificity using yeast surface display. Nucleic Acids Res. 2009; 37:6871–6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Chan Y.S., Takeuchi R., Jarjour J., Huen D.S., Stoddard B.L., Russell S.. The design and in vivo evaluation of engineered I-OnuI-Based enzymes for HEG gene drive. PLoS One. 2013; 8:e74254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Baxter S.K., Lambert A.R., Scharenberg A.M., Jarjour J.. Flow cytometric assays for interrogating LAGLIDADG homing endonuclease DNA-binding and cleavage properties. Methods Mol. Biol. 2013; 978:45–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Takeuchi R., Choi M., Stoddard B.L.. Redesign of extensive protein–DNA interfaces of meganucleases using iterative cycles of in vitro compartmentalization. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:4061–4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Takeuchi R., Choi M., Stoddard B.L.. Engineering of customized meganucleases via in vitro compartmentalization and in cellulo optimization. Methods Mol. Biol. 2015; 1239:105–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Hutin M., Perez-Quintero A.L., Lopez C., Szurek B.. MorTAL Kombat: the story of defense against TAL effectors through loss-of-susceptibility. Front. Plant Sci. 2015; 6:535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Boch J., Scholze H., Schornack S., Landgraf A., Hahn S., Kay S., Lahaye T., Nickstadt A., Bonas U.. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009; 326:1509–1512. [DOI] [PubMed] [Google Scholar]
- 137. Mak A.N., Bradley P., Cernadas R.A., Bogdanove A.J., Stoddard B.L.. The crystal structure of TAL effector PthXo1 bound to its DNA target. Science. 2012; 335:716–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Deng D., Yan C., Pan X., Mahfouz M., Wang J., Zhu J.K., Shi Y., Yan N.. Structural basis for sequence-specific recognition of DNA by TAL effectors. Science. 2012; 335:720–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Falahi Charkhabi N., Booher N.J., Peng Z., Wang L., Rahimian H., Shams-Bakhsh M., Liu Z., Liu S., White F.F., Bogdanove A.J.. Complete genome sequencing and targeted mutagenesis reveal virulence contributions of Tal2 and Tal4b of Xanthomonas translucens pv. undulosa ICMP11055 in bacterial leaf streak of wheat. Front. Microbiol. 2017; 8:1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. de Lange O., Schreiber T., Schandry N., Radeck J., Braun K.H., Koszinowski J., Heuer H., Strauss A., Lahaye T.. Breaking the DNA-binding code of Ralstonia solanacearum TAL effectors provides new possibilities to generate plant resistance genes against bacterial wilt disease. New Phytol. 2013; 199:773–786. [DOI] [PubMed] [Google Scholar]
- 141. Streubel J., Blucher C., Landgraf A., Boch J.. TAL effector RVD specificities and efficiencies. Nat. Biotechnol. 2012; 30:593–595. [DOI] [PubMed] [Google Scholar]
- 142. Christian M.L., Demorest Z.L., Starker C.G., Osborn M.J., Nyquist M.D., Zhang Y., Carlson D.F., Bradley P., Bogdanove A.J., Voytas D.F.. Targeting G with TAL effectors: a comparison of activities of TALENs constructed with NN and NK repeat variable di-residues. PLoS One. 2012; 7:e45383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Cong L., Zhou R., Kuo Y.C., Cunniff M., Zhang F.. Comprehensive interrogation of natural TALE DNA-binding modules and transcriptional repressor domains. Nat. Commun. 2012; 3:968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Valton J., Dupuy A., Daboussi F., Thomas S., Marechal A., Macmaster R., Melliand K., Juillerat A., Duchateau P.. Overcoming transcription activator-like effector (TALE) DNA binding domain sensitivity to cytosine methylation. J. Biol. Chem. 2012; 287:38427–38432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Nakagawa S., Gisselbrecht S.S., Rogers J.M., Hartl D.L., Bulyk M.L.. DNA-binding specificity changes in the evolution of forkhead transcription factors. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:12349–12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Gao H., Wu X., Chai J., Han Z.. Crystal structure of a TALE protein reveals an extended N-terminal DNA binding region. Cell Res. 2012; 22:1716–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Cuculis L., Abil Z., Zhao H., Schroeder C.M.. Direct observation of TALE protein dynamics reveals a two-state search mechanism. Nat. Commun. 2015; 6:7277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Cuculis L., Abil Z., Zhao H., Schroeder C.M.. TALE proteins search DNA using a rotationally decoupled mechanism. Nat. Chem. Biol. 2016; 12:831–837. [DOI] [PubMed] [Google Scholar]
- 149. Doyle E.L., Hummel A.W., Demorest Z.L., Starker C.G., Voytas D.F., Bradley P., Bogdanove A.J.. TAL effector specificity for base 0 of the DNA target is altered in a complex, effector- and assay-dependent manner by substitutions for the tryptophan in cryptic repeat -1. PLoS One. 2013; 8:e82120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Schreiber T., Bonas U.. Repeat 1 of TAL effectors affects target specificity for the base at position zero. Nucleic Acids Res. 2014; 42:7160–7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Garg A., Lohmueller J.J., Silver P.A., Armel T.Z.. Engineering synthetic TAL effectors with orthogonal target sites. Nucleic Acids Res. 2012; 40:7584–7595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Meckler J.F., Bhakta M.S., Kim M.S., Ovadia R., Habrian C.H., Zykovich A., Yu A., Lockwood S.H., Morbitzer R., Elsaesser J. et al. Quantitative analysis of TALE-DNA interactions suggests polarity effects. Nucleic Acids Res. 2013; 41:4118–4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Rinaldi F.C., Doyle L.A., Stoddard B.L., Bogdanove A.J.. The effect of increasing numbers of repeats on TAL effector DNA binding specificity. Nucleic Acids Res. 2017; 45:6960–6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Stella S., Molina R., Bertonatti C., Juillerrat A., Montoya G.. Expression, purification, crystallization and preliminary X-ray diffraction analysis of the novel modular DNA-binding protein BurrH in its apo form and in complex with its target DNA. Acta Crystallogr. F Struct. Biol. Commun. 2014; 70:87–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Engler C., Gruetzner R., Kandzia R., Marillonnet S.. Golden gate shuffling: a one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS One. 2009; 4:e5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Cermak T., Doyle E.L., Christian M., Wang L., Zhang Y., Schmidt C., Baller J.A., Somia N.V., Bogdanove A.J., Voytas D.F.. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011; 39:e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Li T., Huang S., Zhao X., Wright D.A., Carpenter S., Spalding M.H., Weeks D.P., Yang B.. Modularly assembled designer TAL effector nucleases for targeted gene knockout and gene replacement in eukaryotes. Nucleic Acids Res. 2011; 39:6315–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Weber E., Gruetzner R., Werner S., Engler C., Marillonnet S.. Assembly of designer TAL effectors by Golden Gate cloning. PLoS One. 2011; 6:e19722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Morbitzer R., Elsaesser J., Hausner J., Lahaye T.. Assembly of custom TALE-type DNA binding domains by modular cloning. Nucleic Acids Res. 2011; 39:5790–5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Schmid-Burgk J.L., Schmidt T., Kaiser V., Honing K., Hornung V.. A ligation-independent cloning technique for high-throughput assembly of transcription activator-like effector genes. Nat. Biotechnol. 2013; 31:76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Briggs A.W., Rios X., Chari R., Yang L., Zhang F., Mali P., Church G.M.. Iterative capped assembly: rapid and scalable synthesis of repeat-module DNA such as TAL effectors from individual monomers. Nucleic Acids Res. 2012; 40:e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Reyon D., Maeder M.L., Khayter C., Tsai S.Q., Foley J.E., Sander J.D., Joung J.K.. Engineering customized TALE nucleases (TALENs) and TALE transcription factors by fast ligation-based automatable solid-phase high-throughput (FLASH) assembly. Curr. Protoc. Mol. Biol. 2013; doi:10.1002/0471142727.mb1216s103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Sakuma T., Yamamoto T.. Current Overview of TALEN Construction Systems. Methods Mol. Biol. 2017; 1630:25–36. [DOI] [PubMed] [Google Scholar]
- 164. Booher N.J., Bogdanove A.J.. Tools for TAL effector design and target prediction. Methods. 2014; 69:121–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Bogdanove A.J., Booher N.J.. Kühn R, Wurst W, Wefers B. TALENs: Methods and Protocols. 2016; 1338:NY: Humana Press Springer; 43–47. [Google Scholar]
- 166. Doyle E.L., Booher N.J., Standage D.S., Voytas D.F., Brendel V.P., Vandyk J.K., Bogdanove A.J.. TAL effector-nucleotide targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acids Res. 2012; 40:W117–W122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Perez-Quintero A.L., Rodriguez R.L., Dereeper A., Lopez C., Koebnik R., Szurek B., Cunnac S.. An improved method for TAL effectors DNA-binding sites prediction reveals functional convergence in TAL repertoires of Xanthomonas oryzae strains. PLoS One. 2013; 8:e68464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Rogers J.M., Barrera L.A., Reyon D., Sander J.D., Kellis M., Joung J.K., Bulyk M.L.. Context influences on TALE-DNA binding revealed by quantitative profiling. Nat. Commun. 2015; 6:7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Cernadas R.A., Doyle E.L., Nino-Liu D.O., Wilkins K.E., Bancroft T., Wang L., Schmidt C.L., Caldo R., Yang B., White F.F. et al. Code-assisted discovery of TAL effector targets in bacterial leaf streak of rice reveals contrast with bacterial blight and a novel susceptibility gene. PLoS Pathogens. 2014; 10:e1003972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Streubel J., Pesce C., Hutin M., Koebnik R., Boch J., Szurek B.. Five phylogenetically close rice SWEET genes confer TAL effector-mediated susceptibility to Xanthomonas oryzae pv. oryzae. New Phytol. 2013; 200:808–819. [DOI] [PubMed] [Google Scholar]
- 171. Christian M., Cermak T., Doyle E.L., Schmidt C., Zhang F., Hummel A., Bogdanove A.J., Voytas D.F.. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics. 2010; 186:757–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Li T., Huang S., Jiang W.Z., Wright D., Spalding M.H., Weeks D.P., Yang B.. TAL nucleases (TALNs): hybrid proteins composed of TAL effectors and FokI DNA-cleavage domain. Nucleic Acids Res. 2011; 39:359–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Miller J.C., Tan S., Qiao G., Barlow K.A., Wang J., Xia D.F., Meng X., Paschon D.E., Leung E., Hinkley S.J. et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011; 29:143–148. [DOI] [PubMed] [Google Scholar]
- 174. de Lange O., Binder A., Lahaye T.. From dead leaf, to new life: TAL effectors as tools for synthetic biology. Plant J. 2014; 78:753–771. [DOI] [PubMed] [Google Scholar]
- 175. Yanik M., Alzubi J., Lahaye T., Cathomen T., Pingoud A., Wende W.. TALE-PvuII fusion proteins - novel tools for gene targeting. PLoS One. 2013; 8:e82539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Kleinstiver B.P., Wang L., Wolfs J.M., Kolaczyk T., McDowell B., Wang X., Schild-Poulter C., Bogdanove A.J., Edgell D.R.. The I-TevI nuclease and linker domains contribute to the specificity of monomeric TALENs. G3 (Bethesda). 2014; 4:1155–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Bolukbasi M.F., Gupta A., Oikemus S., Derr A.G., Garber M., Brodsky M.H., Zhu L.J., Wolfe S.A.. DNA-binding-domain fusions enhance the targeting range and precision of Cas9. Nat. Methods. 2015; 12:1150–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Christian M., Qi Y.P., Zhang Y., Voytas D.F.. Targeted mutagenesis of Arabidopsis thaliana using engineered TAL effector nucleases. G3-Genes Genom. Genet. 2013; 3:1697–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Gil-Humanes J., Wang Y., Liang Z., Shan Q., Ozuna C.V., Sanchez-Leon S., Baltes N.J., Starker C., Barro F., Gao C. et al. High-efficiency gene targeting in hexaploid wheat using DNA replicons and CRISPR/Cas9. Plant J. 2017; 89:1251–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Xiao A., Wang Z., Hu Y., Wu Y., Luo Z., Yang Z., Zu Y., Li W., Huang P., Tong X. et al. Chromosomal deletions and inversions mediated by TALENs and CRISPR/Cas in zebrafish. Nucleic Acids Res. 2013; 41:e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Kim Y., Kweon J., Kim A., Chon J.K., Yoo J.Y., Kim H.J., Kim S., Lee C., Jeong E., Chung E. et al. A library of TAL effector nucleases spanning the human genome. Nat. Biotechnol. 2013; 31:251–258. [DOI] [PubMed] [Google Scholar]
- 182. Blount B.A., Weenink T., Vasylechko S., Ellis T.. Rational diversification of a promoter providing fine-tuned expression and orthogonal regulation for synthetic biology. PLoS One. 2012; 7:e33279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Carlson D.F., Tan W., Lillico S.G., Stverakova D., Proudfoot C., Christian M., Voytas D.F., Long C.R., Whitelaw C.B., Fahrenkrug S.C.. Efficient TALEN-mediated gene knockout in livestock. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:17382–17387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Qasim W., Zhan H., Samarasinghe S., Adams S., Amrolia P., Stafford S., Butler K., Rivat C., Wright G., Somana K. et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017; 9:eaaj2013. [DOI] [PubMed] [Google Scholar]
- 185. Miller J.C., Zhang L., Xia D.F., Campo J.J., Ankoudinova I.V., Guschin D.Y., Babiarz J.E., Meng X., Hinkley S.J., Lam S.C. et al. Improved specificity of TALE-based genome editing using an expanded RVD repertoire. Nat. Methods. 2015; 12:465–471. [DOI] [PubMed] [Google Scholar]
- 186. Yang J., Zhang Y., Yuan P., Zhou Y., Cai C., Ren Q., Wen D., Chu C., Qi H., Wei W.. Complete decoding of TAL effectors for DNA recognition. Cell Res. 2014; 24:628–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. de Lange O., Wolf C., Thiel P., Kruger J., Kleusch C., Kohlbacher O., Lahaye T.. DNA-binding proteins from marine bacteria expand the known sequence diversity of TALE-like repeats. Nucleic Acids Res. 2015; 43:10065–10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. de Lange O., Schandry N., Wunderlich M., Berendzen K.W., Lahaye T.. Exploiting the sequence diversity of TALE-like repeats to vary the strength of dTALE-promoter interactions. Synth.Biol. 2017; 2:ysx004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Tochio N., Umehara K., Uewaki J.I., Flechsig H., Kondo M., Dewa T., Sakuma T., Yamamoto T., Saitoh T., Togashi Y. et al. Non-RVD mutations that enhance the dynamics of the TAL repeat array along the superhelical axis improve TALEN genome editing efficacy. Sci. Rep. 2016; 6:37887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Holkers M., Maggio I., Liu J., Janssen J.M., Miselli F., Mussolino C., Recchia A., Cathomen T., Goncalves M.A.. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res. 2013; 41:e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Richter A., Streubel J., Blucher C., Szurek B., Reschke M., Grau J., Boch J.. A TAL effector repeat architecture for frameshift binding. Nat. Commun. 2014; 5:3447. [DOI] [PubMed] [Google Scholar]
- 192. Grindley N.D., Whiteson K.L., Rice P.A.. Mechanisms of site-specific recombination. Annu. Rev. Biochem. 2006; 75:567–605. [DOI] [PubMed] [Google Scholar]
- 193. Gaj T., Sirk S.J., Barbas C.F. 3rd. Expanding the scope of site-specific recombinases for genetic and metabolic engineering. Biotechnol. Bioeng. 2014; 111:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Meinke G., Bohm A., Hauber J., Pisabarro M.T., Buchholz F.. Cre Recombinase and other tyrosine recombinases. Chem. Rev. 2016; 116:12785–12820. [DOI] [PubMed] [Google Scholar]
- 195. Anastassiadis K., Fu J., Patsch C., Hu S., Weidlich S., Duerschke K., Buchholz F., Edenhofer F., Stewart A.F.. Dre recombinase, like Cre, is a highly efficient site-specific recombinase in E. coli, mammalian cells and mice. Disease Models Mech. 2009; 2:508–515. [DOI] [PubMed] [Google Scholar]
- 196. Karimova M., Abi-Ghanem J., Berger N., Surendranath V., Pisabarro M.T., Buchholz F.. Vika/vox, a novel efficient and specific Cre/loxP-like site-specific recombination system. Nucleic Acids Res. 2013; 41:e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Karimova M., Splith V., Karpinski J., Pisabarro M.T., Buchholz F.. Discovery of Nigri/nox and Panto/pox site-specific recombinase systems facilitates advanced genome engineering. Sci. Rep. 2016; 6:30130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Suzuki E., Nakayama M.. VCre/VloxP and SCre/SloxP: new site-specific recombination systems for genome engineering. Nucleic Acids Res. 2011; 39:e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Arnold P.H., Blake D.G., Grindley N.D., Boocock M.R., Stark W.M.. Mutants of Tn3 resolvase which do not require accessory binding sites for recombination activity. EMBO J. 1999; 18:1407–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Keravala A., Lee S., Thyagarajan B., Olivares E.C., Gabrovsky V.E., Woodard L.E., Calos M.P.. Mutational derivatives of PhiC31 integrase with increased efficiency and specificity. Mol. Ther. 2009; 17:112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Klippel A., Cloppenborg K., Kahmann R.. Isolation and characterization of unusual gin mutants. EMBO J. 1988; 7:3983–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Lorbach E., Christ N., Schwikardi M., Droge P.. Site-specific recombination in human cells catalyzed by phage lambda integrase mutants. J. Mol. Biol. 2000; 296:1175–1181. [DOI] [PubMed] [Google Scholar]
- 203. Wallen M.C., Gaj T., Barbas C.F. 3rd. Redesigning recombinase specificity for safe harbor sites in the human genome. PLoS One. 2015; 10:e0139123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Wallace H.A., Marques-Kranc F., Richardson M., Luna-Crespo F., Sharpe J.A., Hughes J., Wood W.G., Higgs D.R., Smith A.J.. Manipulating the mouse genome to engineer precise functional syntenic replacements with human sequence. Cell. 2007; 128:197–209. [DOI] [PubMed] [Google Scholar]
- 205. Hirano N., Muroi T., Takahashi H., Haruki M.. Site-specific recombinases as tools for heterologous gene integration. Appl. Microbiol. Biotechnol. 2011; 92:227–239. [DOI] [PubMed] [Google Scholar]
- 206. Voziyanova E., Malchin N., Anderson R.P., Yagil E., Kolot M., Voziyanov Y.. Efficient Flp-Int HK022 dual RMCE in mammalian cells. Nucleic Acids Res. 2013; 41:e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Van Duyne G.D. Cre Recombinase. Microbiol. Spectrum. 2015; 3:doi:10.1128/microbiolspec.MDNA3-0014-2014. [DOI] [PubMed] [Google Scholar]
- 208. Senecoff J.F., Cox M.M.. Directionality in FLP protein-promoted site-specific recombination is mediated by DNA-DNA pairing. J. Biol. Chem. 1986; 261:7380–7386. [PubMed] [Google Scholar]
- 209. Baldwin E.P., Martin S.S., Abel J., Gelato K.A., Kim H., Schultz P.G., Santoro S.W.. A specificity switch in selected cre recombinase variants is mediated by macromolecular plasticity and water. Chem. Biol. 2003; 10:1085–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Rufer A.W., Sauer B.. Non-contact positions impose site selectivity on Cre recombinase. Nucleic Acids Res. 2002; 30:2764–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Missirlis P.I., Smailus D.E., Holt R.A.. A high-throughput screen identifying sequence and promiscuity characteristics of the loxP spacer region in Cre-mediated recombination. BMC Genomics. 2006; 7:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Sheren J., Langer S.J., Leinwand L.A.. A randomized library approach to identifying functional lox site domains for the Cre recombinase. Nucleic Acids Res. 2007; 35:5464–5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Janbandhu V.C., Moik D., Fassler R.. Cre recombinase induces DNA damage and tetraploidy in the absence of loxP sites. Cell cycle (Georgetown, Tex.). 2014; 13:462–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Santoro S.W., Schultz P.G.. Directed evolution of the substrate specificities of a site-specific recombinase and an aminoacyl-tRNA synthetase using fluorescence-activated cell sorting (FACS). Methods Mol. Biol. 2003; 230:291–312. [DOI] [PubMed] [Google Scholar]
- 215. Santoro S.W., Schultz P.G.. Directed evolution of the site specificity of Cre recombinase. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:4185–4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Stemmer W.P. Rapid evolution of a protein in vitro by DNA shuffling. Nature. 1994; 370:389–391. [DOI] [PubMed] [Google Scholar]
- 217. Buchholz F., Stewart A.F.. Alteration of Cre recombinase site specificity by substrate-linked protein evolution. Nat. Biotechnol. 2001; 19:1047–1052. [DOI] [PubMed] [Google Scholar]
- 218. Buchholz F. Molecular evolution of the tre recombinase. J. Visual. Exp.: JoVE. 2008; 29:791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Buchholz F., Hauber J.. In vitro evolution and analysis of HIV-1 LTR-specific recombinases. Methods. 2011; 53:102–109. [DOI] [PubMed] [Google Scholar]
- 220. Karpinski J., Chemnitz J., Hauber I., Abi-Ghanem J., Paszkowski-Rogacz M., Surendranath V., Chakrabort D., Hackmann K., Schrock E., Pisabarro M.T. et al. Universal Tre (uTre) recombinase specifically targets the majority of HIV-1 isolates. J. Int. AIDS Soc. 2014; 17:19706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Mariyanna L., Priyadarshini P., Hofmann-Sieber H., Krepstakies M., Walz N., Grundhoff A., Buchholz F., Hildt E., Hauber J.. Excision of HIV-1 proviral DNA by recombinant cell permeable tre-recombinase. PLoS One. 2012; 7:e31576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Sarkar I., Hauber I., Hauber J., Buchholz F.. HIV-1 proviral DNA excision using an evolved recombinase. Science. 2007; 316:1912–1915. [DOI] [PubMed] [Google Scholar]
- 223. Abi-Ghanem J., Chusainow J., Karimova M., Spiegel C., Hofmann-Sieber H., Hauber J., Buchholz F., Pisabarro M.T.. Engineering of a target site-specific recombinase by a combined evolution- and structure-guided approach. Nucleic Acids Res. 2013; 41:2394–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Meinke G., Karpinski J., Buchholz F., Bohm A.. Crystal structure of an engineered, HIV-specific recombinase for removal of integrated proviral DNA. Nucleic Acids Res. 2017; 45:9726–9740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Hauber I., Hofmann-Sieber H., Chemnitz J., Dubrau D., Chusainow J., Stucka R., Hartjen P., Schambach A., Ziegler P., Hackmann K. et al. Highly significant antiviral activity of HIV-1 LTR-specific tre-recombinase in humanized mice. PLoS Pathog. 2013; 9:e1003587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Castillo F., Benmohamed A., Szatmari G.. Xer site specific Recombination: Double and single recombinase systems. Front. Microbiol. 2017; 8:453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Saraf-Levy T., Santoro S.W., Volpin H., Kushnirsky T., Eyal Y., Schultz P.G., Gidoni D., Carmi N.. Site-specific recombination of asymmetric lox sites mediated by a heterotetrameric Cre recombinase complex. Bioorg. Med. Chem. 2006; 14:3081–3089. [DOI] [PubMed] [Google Scholar]
- 228. Gelato K.A., Martin S.S., Liu P.H., Saunders A.A., Baldwin E.P.. Spatially directed assembly of a heterotetrameric Cre-Lox synapse restricts recombination specificity. J. Mol. Biol. 2008; 378:653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Zhang C., Myers C.A., Qi Z., Mitra R.D., Corbo J.C., Havranek J.J.. Redesign of the monomer-monomer interface of Cre recombinase yields an obligate heterotetrameric complex. Nucleic Acids Res. 2015; 43:9076–9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Eroshenko N., Church G.M.. Mutants of Cre recombinase with improved accuracy. Nat. Commun. 2013; 4:2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Buchholz F., Angrand P.O., Stewart A.F.. Improved properties of FLP recombinase evolved by cycling mutagenesis. Nat. Biotechnol. 1998; 16:657–662. [DOI] [PubMed] [Google Scholar]
- 232. Konieczka J.H., Paek A., Jayaram M., Voziyanov Y.. Recombination of hybrid target sites by binary combinations of Flp variants: mutations that foster interprotomer collaboration and enlarge substrate tolerance. J. Mol. Biol. 2004; 339:365–378. [DOI] [PubMed] [Google Scholar]
- 233. Voziyanov Y., Konieczka J.H., Stewart A.F., Jayaram M.. Stepwise manipulation of DNA specificity in Flp recombinase: progressively adapting Flp to individual and combinatorial mutations in its target site. J. Mol. Biol. 2003; 326:65–76. [DOI] [PubMed] [Google Scholar]
- 234. Bolusani S., Ma C.H., Paek A., Konieczka J.H., Jayaram M., Voziyanov Y.. Evolution of variants of yeast site-specific recombinase Flp that utilize native genomic sequences as recombination target sites. Nucleic Acids Res. 2006; 34:5259–5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Shah R., Li F., Voziyanova E., Voziyanov Y.. Target-specific variants of Flp recombinase mediate genome engineering reactions in mammalian cells. FEBS J. 2015; 282:3323–3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Voziyanova E., Anderson R.P., Shah R., Li F., Voziyanov Y.. Efficient genome manipulation by variants of Site-Specific recombinases R and TD. J. Mol. Biol. 2016; 428:990–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Biswas T., Aihara H., Radman-Livaja M., Filman D., Landy A., Ellenberger T.. A structural basis for allosteric control of DNA recombination by lambda integrase. Nature. 2005; 435:1059–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Rice P.A. Serine resolvases. Microbiol. Spectrum. 2015; 3:doi:10.1128/microbiolspec.MDNA3-0045-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Smith M.C., Brown W.R., McEwan A.R., Rowley P.A.. Site-specific recombination by phiC31 integrase and other large serine recombinases. Biochem. Soc. Trans. 2010; 38:388–394. [DOI] [PubMed] [Google Scholar]
- 240. Van Duyne G.D. Lambda integrase: armed for recombination. Curr. Biol.: CB. 2005; 15:R658–R660. [DOI] [PubMed] [Google Scholar]
- 241. Yagil E., Dorgai L., Weisberg R.A.. Identifying determinants of recombination specificity: construction and characterization of chimeric bacteriophage integrases. J. Mol. Biol. 1995; 252:163–177. [DOI] [PubMed] [Google Scholar]
- 242. Siau J.W., Chee S., Makhija H., Wai C.M., Chandra S.H., Peter S., Droge P., Ghadessy F.J.. Directed evolution of lambda integrase activity and specificity by genetic derepression. Protein Eng. Des. Selection: PEDS. 2015; 28:211–220. [DOI] [PubMed] [Google Scholar]
- 243. Zhao H., Giver L., Shao Z., Affholter J.A., Arnold F.H.. Molecular evolution by staggered extension process (StEP) in vitro recombination. Nat. Biotechnol. 1998; 16:258–261. [DOI] [PubMed] [Google Scholar]
- 244. Sclimenti C.R., Thyagarajan B., Calos M.P.. Directed evolution of a recombinase for improved genomic integration at a native human sequence. Nucleic Acids Res. 2001; 29:5044–5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Akopian A., He J., Boocock M.R., Stark W.M.. Chimeric recombinases with designed DNA sequence recognition. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:8688–8691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Prorocic M.M., Wenlong D., Olorunniji F.J., Akopian A., Schloetel J.G., Hannigan A., McPherson A.L., Stark W.M.. Zinc-finger recombinase activities in vitro. Nucleic Acids Res. 2011; 39:9316–9328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Gersbach C.A., Gaj T., Gordley R.M., Mercer A.C., Barbas C.F. 3rd. Targeted plasmid integration into the human genome by an engineered zinc-finger recombinase. Nucleic Acids Res. 2011; 39:7868–7878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Yang W., Steitz T.A.. Crystal structure of the site-specific recombinase gamma delta resolvase complexed with a 34 bp cleavage site. Cell. 1995; 82:193–207. [DOI] [PubMed] [Google Scholar]
- 249. Keenholtz R.A., Rowland S.J., Boocock M.R., Stark W.M., Rice P.A.. Structural basis for catalytic activation of a serine recombinase. Structure. 2011; 19:799–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Ritacco C.J., Kamtekar S., Wang J., Steitz T.A.. Crystal structure of an intermediate of rotating dimers within the synaptic tetramer of the G-segment invertase. Nucleic Acids Res. 2013; 41:2673–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Gaj T., Mercer A.C., Gersbach C.A., Gordley R.M., Barbas C.F. 3rd. Structure-guided reprogramming of serine recombinase DNA sequence specificity. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Gaj T., Sirk S.J., Tingle R.D., Mercer A.C., Wallen M.C., Barbas C.F. 3rd. Enhancing the specificity of recombinase-mediated genome engineering through dimer interface redesign. J. Am. Chem. Soc. 2014; 136:5047–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Gaj T., Mercer A.C., Sirk S.J., Smith H.L., Barbas C.F. 3rd. A comprehensive approach to zinc-finger recombinase customization enables genomic targeting in human cells. Nucleic Acids Res. 2013; 41:3937–3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Sirk S.J., Gaj T., Jonsson A., Mercer A.C., Barbas C.F. 3rd. Expanding the zinc-finger recombinase repertoire: directed evolution and mutational analysis of serine recombinase specificity determinants. Nucleic Acids Res. 2014; 42:4755–4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Gaj T., Barbas C.F. 3rd. Genome engineering with custom recombinases. Methods Enzymol. 2014; 546:79–91. [DOI] [PubMed] [Google Scholar]
- 256. Olorunniji F.J., Rosser S.J., Marshall Stark W.. Purification and In Vitro Characterization of Zinc Finger Recombinases. Methods Mol. Biol. 2017; 1642:229–245. [DOI] [PubMed] [Google Scholar]
- 257. Proudfoot C., McPherson A.L., Kolb A.F., Stark W.M.. Zinc finger recombinases with adaptable DNA sequence specificity. PLoS One. 2011; 6:e19537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Kelly T.J.J., O. S.H.. A restriction enzyme from Haemophilus influenzae. J. Mol. Biol. 1970; 51:393–409. [DOI] [PubMed] [Google Scholar]
- 259. Zilberman D., Henikoff S.. Genome-wide analysis of DNA methylation patterns. Development. 2007; 134:3959–3965. [DOI] [PubMed] [Google Scholar]
- 260. Chang H.-W., Yang C.-H., Chang P.-L., Cheng Y.-H., Chuang L.-Y.. SNP-RFLPing: restriction enzyme mining for SNPs in genomes. BMC Genomics. 2006; 7:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Chuang L.-Y., Yang C.-H., Tsui K.-H., Cheng Y.-H., Chang P.-L., Wen C.-H., Chang H.-W.. Restriction enzyme mining for SNPs in genomes. Anticancer Res. 2008; 28:2001–2007. [PubMed] [Google Scholar]
- 262. Porter D., Yao J., Polyak K.. SAGE and related approaches for cancer target identificatio. Drug Discov. Today. 2006; 11:110–118. [DOI] [PubMed] [Google Scholar]
- 263. Nielsen K.L. DeepSAGE: higher sensitivity and multiplexing of samples using a simpler experimental protocol. Methods Mol. Biol. 2008; 387:81–94. [DOI] [PubMed] [Google Scholar]
- 264. Tanaka K., Ohtake R., Yoshida S., Shinohara T.. Effective DNA fragmentation technique for simple sequence repeat detection with a microsatellite-enriched library and high-throughput sequencing. Biotechniques. 2017; 62:180–182. [DOI] [PubMed] [Google Scholar]
- 265. Roberts R.J., Belfort M., Bestor T., Bhagwat A.S., Bickle T.A., Bitinaite J., Blumenthal R.M., Degtyarev S., Dryden D.T., Dybvig K. et al. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res. 2003; 31:1805–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Roberts R.J., Vincze T., Posfai J., Macelis D.. REBASE–a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2015; 43:D298–D299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Rao D.N., Dryden D.T., Bheemanaik S.. Type III restriction-modification enzymes: a historical perspective. Nucleic Acids Res. 2014; 42:45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Loenen W.A., Dryden D.T., Raleigh E.A., Wilson G.G.. Type I restriction enzymes and their relatives. Nucleic Acids Res. 2014; 42:20–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Pingoud A., Wilson G.G., Wende W.. Type II restriction endonucleases–a historical perspective and more. Nucleic Acids Res. 2014; 42:7489–7527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Rosenberg J., Wang B., Frederick C., Reich N., Greene P., Grable J., McClarin J., Oxender D.L., Fox F.. Development of a protein design strategy for EcoRI endonuclease. Protein Eng. 1987; 13:237–250. [Google Scholar]
- 271. Jeltsch A., Wenz C., Wende W., Selent U., Pingoud A.. Engineering novel restriction endonucleases: principles and applications. Trends Biotechnol. 1996; 14:235–238. [DOI] [PubMed] [Google Scholar]
- 272. Lanio T., Jeltsch A., Pingoud A.. On the possibilities and limitations of rational protein design to expand the specificity of restriction enzymes: a case study employing EcoRV as the target. Protein Eng. 2000; 13:275–281. [DOI] [PubMed] [Google Scholar]
- 273. Dorner L.F., Bitinaite J., Whitaker R.D., Schildkraut I.. Genetic analysis of the base-specific contacts of BamHI restriction endonuclease. J. Mol. Biol. 1999; 285:1515–1523. [DOI] [PubMed] [Google Scholar]
- 274. Samuelson J.C., Xu S.-y.. Directed evolution of restriction endonuclease BstYI to achieve increased substrate specificity. J. Mol. Biol. 2002; 319:673–683. [DOI] [PubMed] [Google Scholar]
- 275. Samuelson J.C., Morgan R.D., Benner J.S., Claus T.E., Packard S.L., Xu S.-y.. Engineering a rare-cutting restriction enzyme: genetic screening and selection of NotI variants. Nucleic Acids Res. 2006; 34:796–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Gubler M., Braguglia D., Meyer J., Piekarowicz A., Bickle T.A.. Recombination of constant and variable modules alters DNA sequence recognition by type IC restriction-modification enzymes. EMBO J. 1992; 11:233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Jurenaite-Urbanaviciene S., Serksnaite J., Kriukiene E., Giedriene J., Venclovas C., Lubys A.. Generation of DNA cleavage specificities of type II restriction endonucleases by reassortment of target recognition domains. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:10358–10363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Rimseliene R., Maneliene Z., Lubys A., Janulaitis A.. Engineering of restriction endonucleases: using methylation activity of the bifunctional endonuclease Eco571 to select the mutant with a novel sequence specificity. J. Mol. Biol. 2003; 327:383–391. [DOI] [PubMed] [Google Scholar]
- 279. Boyd A.C., Charles I.G., Keyte J.W., Brammar W.J.. Isolation and computer-aided characterization of MmeI, a type II restriction endonuclease from Methylophilus methylotrophus. Nucleic Acids Res. 1986; 14:5255–5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Morgan R.D., Bhatia T.K., Lovasco L., Davis T.B.. MmeI: a minimal Type II restriction-modification system that only modifies one DNA strand for host protection. Nucleic Acids Res. 2008; 36:6558–6570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Morgan R.D., Dwinell E.A., Bhatia T.K., Lang E.M., Luyten Y.A.. The MmeI family: type II restriction-modification enzymes that employ single-strand modification for host protection. Nucleic Acids Res. 2009; 37:5208–5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Nakonieczna J., Kaczorowski T., Obarska-Kosinska A., Bujnicki J.M.. Functional analysis of MmeI from methanol utilizer Methylophilus methylotrophus, a subtype IIC restriction-modification enzyme related to type I enzymes. Appl. Environ. Microbiol. 2009; 75:212–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Callahan S.J., Luyten Y.A., Gupta Y.K., Wilson G.G., Roberts R.J., Morgan R.D., Aggarwal A.K.. Structure of type IIL Restriction-Modification enzyme MmeI in complex with DNA has implications for engineering new specificities. PLoS Biol. 2016; 14:e1002442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Kulkarni M., Nirwan N., van Aelst K., Szczelkun M.D., Saikrishnan K.. Structural insights into DNA sequence recognition by Type ISP restriction-modification enzymes. Nucleic Acids Res. 2016; 44:4396–4408. [DOI] [PMC free article] [PubMed] [Google Scholar]