


Abstract
Chromatin remodelers are ATP-dependent motors that reorganize DNA packaging by disrupting canonical histone–DNA contacts within the nucleosome. Here, we show that the Chd1 chromatin remodeler stimulates DNA unwrapping from the edge of the nucleosome in a nucleotide-dependent and DNA sequence-sensitive fashion. Nucleosome binding, monitored by stopped flow, was complex and sensitive to nucleotide, with AMP–PNP promoting faster binding than ADP·BeF3–. Nucleosome unwrapping by Chd1, examined by bulk FRET, occurred in the presence and absence of nucleotide and did not require the Chd1 DNA-binding domain. In AMP–PNP conditions, Chd1 unwrapped one side of the Widom 601 DNA more easily than the other, consistent with previous observations of 601 asymmetry and indicating that Chd1 amplifies intrinsic sequence properties of nucleosomal DNA. Using small angle X-ray scattering (SAXS) with contrast variation, we found distinct DNA conformations depending on the nucleotide analog bound to Chd1: with AMP–PNP, DNA primarily unwrapped in-plane with the nucleosomal disk, whereas with ADP·BeF3–, a significant fraction showed distinctive out-of-plane unwrapping as well. Taken together, our findings show tight coupling between entry/exit DNA of the nucleosome and the Chd1 ATPase motor, suggesting that dynamic nucleosome unwrapping is coupled to nucleosome binding and remodeling by Chd1.
INTRODUCTION
Nucleosomes are the fundamental genomic packaging unit of eukaryotes, with approximately 146 bp of DNA tightly wrapped around a histone octamer (1). In addition to compacting the genome into the small space of the nucleus, nucleosomes provide a platform for storing epigenetic information while also limiting access to DNA. Chromatin remodelers are an essential class of enzymes that regulate DNA accessibility by disrupting the canonical histone–DNA interactions that occur in the nucleosome. Typically, remodelers alter the availability of DNA by assembling nucleosomes from histones and naked DNA, moving existing nucleosomes along DNA, or removing histones from DNA (2). Different families of remodelers appear to be specialized to achieve distinct biochemical outcomes, which correlate with their roles in vivo. Here, we focus on the Chd1 remodeler, which is involved in gene transcription and is required for stem cell pluripotency and suppression of cryptic transcription (3,4).
Chd1 has been found to act both in gene bodies and promoters, and directly interacts with a number of key factors involved in transcription. Chd1 contributes to nucleosome spacing in coding regions and directly interacts with several elongation factors such as Spt4-Spt5, the FACT complex, and the PAF complex (5–9). In the absence of Chd1, chromatin appears to be perturbed by passage of RNA polymerase II (Pol II), which in turn leads to cryptic transcription (4). Thus, an important role of Chd1 is to help re-establish the chromatin barrier after Pol II travels through the coding region.
Two primary biochemical activities have previously been demonstrated for Chd1: an ability to assemble nucleosomes and to reposition nucleosomes along DNA (10,11). Nucleosomes often fail to properly form when histones are rapidly deposited on naked DNA, and instead produce prenucleosomes, which lack the canonical superhelical wrapping of DNA around the histone core (12). Chd1 catalyzes nucleosome assembly when histones are deposited on DNA by chaperones (10), which coincides with the ATP- and remodeler-dependent maturation of nucleosomes from prenucleosomes (12). How nucleosome assembly is achieved is not yet understood, but has been shown to be a characteristic of ISWI-type remodelers as well.
The ability to reposition nucleosomes, also referred to as nucleosome sliding, is common to several remodeling families. For Chd1 and other remodelers such as SWI/SNF and ISWI, sliding is achieved by the ATPase motor translocating on nucleosomal DNA at an internal site called superhelix location 2 (SHL2) (13–16). Due to the inherent symmetry of the histone octamer and thus the nucleosome, there are two such SHL2 sites where ATPase motors can act, and two Chd1 ATPases can simultaneously bind to these two sites (17). In addition to the ATPase motor, Chd1 possesses an N-terminal pair of chromodomains and a C-terminal DNA-binding domain (DBD) (18). The chromodomains contact nucleosomal DNA at SHL1, adjacent to the ATPase motor, whereas the DBD binds DNA exiting the nucleosome on the opposite gyre (17,19). Interestingly, negative stain and cryoEM revealed that Chd1 binding is coupled to DNA unwrapping from the nucleosome edge (17,19,20). Using single molecule FRET, DNA unwrapping was shown to occur dynamically with Chd1 binding in the presence and absence of nucleotide analogs (20). As seen in a high resolution cryoEM structure (19), the chromodomains, ATPase motor and DBD make a tripartite unit, with unwrapped DNA extending from the nucleosome appearing to be held by the DBD.
Here, we use fluorescence resonance energy transfer (FRET) and small angle X-ray scattering (SAXS) to study how Chd1 alters the organization of nucleosomal DNA. As shown by stopped-flow binding experiments, the association of Chd1 with the nucleosome is strongly nucleotide dependent, suggesting distinct conformational changes occur in the nucleosome and/or remodeler upon binding. As shown by FRET, DNA unwrapping by Chd1 can also be accomplished when the DBD is absent. Thus, the ATPase motor of Chd1 appears to be capable of disrupting entry/exit DNA on its own. Unexpectedly, SAXS analysis suggested that the conformations of nucleosomes unwrapped by Chd1 are strongly dependent on the nature of the bound nucleotide, with in-plane unwrapping favored by AMP–PNP and distinct out-of-plane unwrapping stimulated by ADP·BeF3–. Additionally, both SAXS and FRET indicated a strong asymmetry in unwrapping, consistent with the unwrapping characteristics previously demonstrated for the Widom 601 positioning sequence (21). Taken together, our findings suggest a tight coupling between placement of DNA exiting the nucleosome and the conformational state of the ATPase motor of Chd1. We propose that Chd1-dependent unwrapping represents a previously unappreciated mechanism for altering histone–DNA interactions and DNA accessibility that may influence transcriptional elongation.
MATERIALS AND METHODS
Protein expression and purification
The Chd1 proteins used in this study were from Saccharomyces cerevisiae and contained the chromodomains, ATPase motor and DBD (residues 118–1274, referred to throughout as Chd1) or only the chromodomains, ATPase motor, and the linker prior to the DBD (residues 118–1014, referred to as Chd1ΔDBD). Expression and purification of Chd1 proteins were carried out as previously described (17,22). Histone protein sequences were those of Xenopus laevis, and were expressed and purified as described (23). For fluorescence experiments using 12N80 or 80N12 nucleosomes, a single cysteine was introduced H3(V35C) and labeled with Cy5-maleimide. Histones were prepared by refolding H2A and H2B into dimers and H3 and H4 into tetramers, purifying each by size exclusion chromatography.
DNA and nucleosome purification
Nucleosomal DNA was generated by large-scale PCR using the Widom 601 sequence (24) as the template, and purified by native acrylamide gel using a BioRad Prep Cell as previously described (23). The DNA constructs are given in Supplementary Figure S1. For FRET experiments, PCR reactions used DNA oligos containing Cy3 or Cy5 at the 5′ end (IDT). Nucleosomes were reconstituted by using salt gradient dialysis to deposit purified histone octamer onto 601-containing DNA, mixed in a 1:1 ratio, as previously described (23).
Stopped-flow experiments
The on-rates of Chd1 binding to nucleosomes were measured using an SX20 Stopped-Flow Spectrophotometer (Applied Photophysics). For these experiments, samples from two syringes were rapidly mixed. One syringe contained Chd1 at the specified concentrations, the other contained 20 nM 12N12 nucleosome. Both samples were suspended in 1X binding buffer (20 mM Hepes pH 7.5, 10 mM MgCl2, 0.1 mM EDTA, 5% sucrose, 1 mM DTT, 0.02% Nonidet P-40, 0.1 mg/ml BSA, 100 mM KCl without or with 1 mM AMP–PNP or ADP·BeF3– [1 mM ADP, 1.2 mM BeCl2, 6 mM NaF]). The nucleosome was Cy3-labeled on H4(A15C). The dye was excited at 510 nm and the emission intensity collected using a 570 nm long-pass filter. For each set of syringes, progress curves from multiple injections were recorded at 25°C. Typically, three to six traces (technical replicates) were averaged together for each experiment. Fitting was carried out in Mathematica using the NonLinearModelFit function with the triple exponential form, 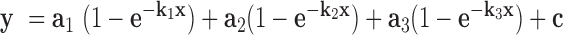 . Here an were the amplitudes (fractions of total fluorescence range), kn were the rates (s− 1) and c was a constant. The observed rates plotted versus Chd1 concentration provide the apparent kon rates (M−1 ·s−1), shown in Figure 1.
. Here an were the amplitudes (fractions of total fluorescence range), kn were the rates (s− 1) and c was a constant. The observed rates plotted versus Chd1 concentration provide the apparent kon rates (M−1 ·s−1), shown in Figure 1.
Figure 1.

The bound nucleotide affects the kinetics of Chd1 binding to nucleosomes. (A) Progress curves from stopped-flow binding experiments of Chd1 with 10 nM 12N12 nucleosomes in the presences of ADP·BeF3–. Each curve is an average of three to six technical replicates. Shown is a representative titration of three independent experiments. (B) Plots of observed rate constants versus Chd1 concentration. For both AMP–PNP and ADP·BeF3– conditions, the two slower rates (right) were plotted separately from the fastest rate (left) to more clearly show the concentration dependence and rate differences. The calculated on-rates (slopes) show concentration dependence for all three observed rates in ADP·BeF3– conditions, whereas under AMP–PNP conditions the middle rate failed to show a clear concentration dependence. Error bars show the standard deviation from three independent experiments.
FRET and fluorescence-based unwrapping assays
Fluorescently labeled nucleosomes possessed both Cy3 and Cy5 dyes, and emission spectra were collected after excitation at either 510 nm (for Cy3) or 645 nm (for Cy5) on a Fluorolog 3 fluorometer (Horiba). For 16N16 nucleosomes, experiments were performed with a 2 ml cuvette (Starna 3-Q-10). Emission spectra were collected with excitation at 510 nm for 10 nM nucleosome, first in the presence of 1× binding buffer (20 mM Hepes pH 7.5, 10 mM MgCl2, 0.1 mM EDTA, 5% sucrose, 1 mM DTT, 0.02% Nonidet P-40, 0.1 mg/ml BSA, 100 mM KCl) and either 1 mM AMP–PNP or 1 mM ADP·BeF3–, and then again after addition of 30 nM Chd1.
Two nucleosome constructs, called 12N80 and 80N12, were also dual labeled for FRET experiments. These each had a single Cy3 label on the 12 end, and Cy5 labels on H3(V35C). As described in the main text, in addition to FRET, Cy5 fluorescence was also affected by DNA unwrapping in a Cy3-independent manner. Therefore, rather than FRET, we describe the differences in 12N80 versus 80N12 unwrapping by plotting changes in peak Cy3 emission (Figure 2). An advantage of this approach is that Cy3 should report only on the 12 bp DNA end, and therefore allows us to separately study the two sides of the Widom 601 sequence, using 12N80 from 80N12 constructs. A disadvantage of using Cy3 emission only is that the measurements were much more sensitive to instrument fluctuations and differences in nucleosome concentrations. Thus, the Cy3 data were noisier than the FRET data calculated from the same experiments.
Figure 2.

Using FRET to monitor nucleosome unwrapping by Chd1. (A) Schematic illustration of Cy3/Cy5 FRET pair positioning on the 16N16 nucleosome, with one dye on the 5′ end of each DNA strand. Chd1 is colored by domains: chromodomains (yellow), ATPase motor (red/blue), and DNA-binding domain (green). (B) Wavelength emission scans for 10 nM 16N16 nucleosomes upon excitation at 510 nm (black, gray). As indicated by arrows, addition of 30 nM Chd1 promoted an increase in Cy3 emissions (564 nm) and a corresponding decrease in Cy5 emissions (664 nm) in the presence of AMP–PNP (green) and ADP·BeF3– (magenta). (C) Nucleosomes made with the Widom 601 positioning sequence are unwrapped by Chd1 more readily on the TA poor side. Cartoon representations of 12N80 (top) and 80N12 (bottom) are shown above wavelength emission scans (with 510 nm excitation) with increasing amounts of Chd1 in the presence of 1 mM AMP–PNP. (D) Nucleosome unwrapping monitored by Cy3 emission with excitation at 510 nm. Chd1 titrations were performed in the presence of 20 nM 12N80 (top) or 20 nM 80N12 (bottom) nucleosomes in various conditions: apo (gray), 1 mM AMP–PNP (green), and 1 mM ADP·BeF3– (magenta). Insets highlight the stoichiometric unwrapping response in ADP·BeF3– at low Chd1 concentrations. Error bars represent standard deviation from three or more replicates. Each dataset was fit to a binding isotherm, with the calculated averages shown by black lines. (E) The Chd1 DNA-binding domain (DBD) is not required for nucleosome unwrapping. Titrations of Chd1ΔDBD performed in the presence of 12N80 (top) and 80N12 (bottom) nucleosomes under ADP·BeF3– conditions.
Experiments with 12N80 from 80N12 nucleosomes were performed with a 100 μl cuvette (Hellma 105-250-15-40). Emission spectra were collected from 530–720 nm with excitation at 510 nm using a 5 nm slit width. These experiments used 20 nM nucleosomes and, when present, 1 mM nucleotide in a 140 μl reaction. For AMP–PNP and nucleotide-free (apo) conditions, nucleosomes were equilibrated to 25°C in binding buffer with or without AMP–PNP. Chd1 was added, mixed, and allowed to equilibrate for 6 minutes before collecting another spectrum. For a given experiment, successive 2 μl additions of Chd1 to the nucleosome sample resulted in Chd1 concentrations of 0, 39, 78, 153, 303, 597, 1179, 1745, 2297, 2835, 3360 nM for apo and 0, 5, 10, 19, 38, 75, 147, 291, 574, 1134, 1680, 2212, 2732 nM for AMP–PNP. For ADP·BeF3– conditions, reactions containing 20 nM nucleosome, 1 mM ADP·BeF3– and varying amounts of Chd1 (0, 0.625, 1.25, 2.5, 5, 10, 20, 40, 80, 160, 320, 640 nM for Chd1 WT and 0, 10, 20, 40, 80, 160, 320, 640 and 1280 nM for Chd1ΔDBD) were assembled at room temperature. Samples containing Chd1 with ADP·BeF3– were allowed a 2–4 h pre-incubation before collecting spectra. After pre-incubation, each reaction was placed in the cuvette and allowed to equilibrate in the fluorometer chamber for one minute before collecting spectra. For salt titrations, 1× binding buffer containing 4 M NaCl was added in 5 μl increments to 140 μl reactions containing 20 nM nucleosome and 1 mM AMP–PNP, yielding final NaCl concentrations of 100, 234, 360, 477, 588, 691, 788, 880, 967, 1049, 1126 and 1200 mM. Identical titrations were performed adding 1× binding buffer alone to assess the impact of dilution.
For Chd1 titrations performed with ADP·BeF3–, the responses were stoichiometric; therefore linear fits were made to the ascending portion, and we report the point of intersection with the saturated signal. For binding titrations of Chd1 in apo and AMP–PNP conditions, and Chd1ΔDBD in ADP·BeF3– conditions, data were fit to binding isotherms of the form: 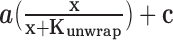 , with the Chd1 concentration x (nM), amplitude a, apparent unwrapping constant Kunwrap (nM) and constant c.
, with the Chd1 concentration x (nM), amplitude a, apparent unwrapping constant Kunwrap (nM) and constant c.
SAXS data collection and modeling
SAXS data were collected at G1 station at Cornell High Energy Synchrotron Source. The X-ray energy was 11.18 keV and sample-to-detector distance was 2.06 m (measured using a silver-behenate standard). Scattering intensities were collected on a Pilatus 200K detector over a q-range from ≈0.007–0.26 Å−1. After azimuthal integration, SAXS profiles were normalized by the intensity of the primary beam, which was imaged directly on the detector, after attenuation by a 200 μm molybdenum beamstop. Samples were oscillated in a 2 mm quartz capillary with 10 μm thick walls. Multiple, 10 s exposures were collected and averaged. Profiles were carefully monitored for signs of radiation damage. 12N12 nucleosomes (5 μM) were incubated with 10 μM Chd1 for greater than 60 minutes (ADP·BeF3– conditions) or at least 10 min (all other conditions) before being measured. Buffers contained 10 mM Tris pH 7.8, 100 mM NaCl, 2 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 60% (w/v) sucrose and ±0.5 mM AMP–PNP or ADP·BeF3– (0.5 mM ADP, 4 mM NaF, 0.6 mM BeCl2). For each condition, three to five separate measurements were made and analyzed independently to confirm reproducibility.
Radius of gyration (Rg) values were calculated using GNOM (25). To carry out ensemble modeling, a pool containing 14 807 DNA structures was generated and theoretical SAXS profiles for each structure were calculated using CRYSOL (26) (CRYSOL parameters: number of harmonics = 50, maximum s-value = 0.25, number of points = 201). To select representative ensembles, we applied an ensemble optimization method (27–29) which exploits a genetic algorithm (GAJOE) to optimize the selection of models from a pool whose summed scattering profiles best represents the experimentally measured profile, over the q range from 0.016–0.16 Å−1 (GAJOE parameters: number of generations = 10 000; number of ensembles = 50; ensemble size fixed = no; maximum/minimum number of curves per ensemble = 20/5; curve repetition allowed = yes; constant subtraction = yes; number of times genetic algorithm repeated = 10). For the Chd1–12N12 complex measured in the presence of ADP·BeF3– and AMP–PNP, the final best fitting ensembles for each of the 10 iterations of the genetic algorithm were pooled together.
RESULTS
The rate that Chd1 binds to nucleosomes is nucleotide-dependent
Chd1 forms a high-affinity complex with the nucleosome in the presence of both AMP–PNP and ADP·BeF3–, yet these nucleotide analogs confer distinct properties to Chd1–nucleosome complexes. In the presence of ADP·BeF3–, nucleosome binding by Chd1 is less sensitive to the presence of flanking DNA and more resistant to increased ionic strength, with higher affinity Chd1-nucleosome complexes observed than with AMP–PNP (17). We hypothesize that these two nucleotide states favor distinct conformations of Chd1 on the nucleosome. To explore potential differences in these states, we measured the kinetics of nucleosome binding using stopped-flow. Previous work with the ISWI remodeler showed that nucleosomes labeled with Cy3 on the histone H4 tail (A15C) yield higher fluorescence in the bound state (30). We discovered that for Chd1, Cy3 at this position increased binding affinity compared with unlabeled nucleosomes (Supplementary Figure S2), and therefore previously reported measurements using H4(A15C-Cy3) nucleosomes likely overestimated affinities (17,22). Interestingly, reactions performed with and without the Cy3 label typically required a substantially longer time (>20 min) to equilibrate in ADP·BeF3– compared with AMP–PNP (<5 min), suggestive of slow conformational changes. We reasoned that the higher affinity toward H4(A15C-Cy3) nucleosomes should increase the on-rate or decrease the off-rate, and therefore rapid kinetics of binding could still be informative for detecting significant barriers in forming a stable Chd1-nucleosome complex.
To monitor Chd1 binding, we designed a nucleosome with 12 bp of DNA on either side of the Widom 601 strong nucleosome positioning sequence (12N12) containing H4(A15C-Cy3). To determine on-rates, rapid mixing experiments with 10 nM nucleosomes were carried out with increasing concentrations of Chd1 (Figure 1A). Binding reactions for both AMP–PNP and ADP·BeF3– proceeded in several stages and were best fit with triple exponential equations. Plotting the observed rates with respect to concentration yielded apparent on-rates for both nucleotide conditions (Figure 1B). For ADP·BeF3–, all three rates were concentration dependent, whereas for AMP–PNP, only the fastest rate showed a clear concentration dependence. Interestingly, despite a higher apparent affinity for ADP·BeF3–, the fastest on-rate (2.7 ± 0.3 × 106 M–1 s–1) was ∼5-fold slower than that of AMP–PNP (1.3 ± 0.1 × 107 M–1 s–1). These different binding characteristics suggest that Chd1 adopts distinct conformations in each nucleotide state, possibly leading to specific, nucleotide-dependent rearrangements of the nucleosome.
FRET reveals asymmetric unwrapping by Chd1
Reorganization of histone–DNA contacts within the nucleosome is believed to be principally driven by DNA translocation of the Chd1 ATPase motor, which requires turnover of ATP. However, recent work suggests that Chd1 binding unwraps DNA from the edge of the nucleosome in the presence of the non-hydrolyzable ATP mimics, independently from active ATP hydrolysis (17,19,20). We hypothesized that the nucleotide dependent binding dynamics we observed may correlate with altered conformations of nucleosomal DNA. To investigate this idea, we designed a FRET-labeled nucleosome having 16 bp of flanking DNA on each side of the strong Widom 601 positioning sequence (16N16), with Cy3 and Cy5 dyes at the DNA ends (Figure 2A). This labeling scheme yielded average FRET efficiencies of 0.45 ± 0.02 for the nucleosome alone, consistent with trajectories of entry/exit DNA of nucleosome crystal structures and indicative of a fully wrapped state. Addition of Chd1 in the presence of AMP–PNP or ADP·BeF3– markedly reduced FRET efficiencies to 0.088 ± 0.005 and 0.065 ± 0.015, respectively, consistent with DNA unwrapping by Chd1 (Figure 2B). A decrease in FRET was also observed with Chd1 in the absence of nucleotide (apo state), but required the addition of considerably more Chd1 (1 μM for apo versus 30 nM for nucleotide analogs) to achieve a smaller change in FRET (from 0.45 to 0.32) (Supplementary Figure S3). This suggests that Chd1 is capable of unwrapping DNA in the apo state, but does so more readily in the presence of nucleotide, possibly due to more stable binding or by adopting conformations favorable for unwrapping when bound to ATP-mimicking analogs.
One disadvantage of monitoring DNA unwrapping with the 16N16 construct is its sensitivity to small DNA movements. By labeling both flanking DNA segments, FRET changes can be amplified by the 2-fold symmetry of the nucleosome, as simultaneous unwrapping on both sides would double the change in dye separation. Additionally, due to the superhelical geometry of the nucleosome, the DNA ends experience the greatest changes in position upon unwrapping, resulting in high FRET sensitivity for small changes in the angle of entry/exit DNA. Another drawback of this construct is the inability to distinguish between unwrapping from either side of the nucleosome. To resolve these issues, we designed different nucleosomes with Cy3/Cy5 FRET pairs that would only respond to unwrapping on one side and would require a greater degree of unwrapping to separate the dye pair. Following the original FRET design by Widom (31), Cy5 maleimide was attached to histone H3(V35C) adjacent to DNA entering/exiting the nucleosome, and Cy3 was attached to the DNA end, 12 bp outside the nucleosome on 12N80 and 80N12 constructs (Figure 2C). Using these two constructs enabled the comparison of unwrapping from either side of the Widom 601, which is asymmetric with respect to the number of phased TpA dinucleotides (TA steps) located where the minor groove faces the octamer. These TA steps are thought to facilitate the bending of DNA around the histone octamer, and previous force spectroscopy studies have shown that higher force is required to disrupt histone-DNA contacts from the TA-rich side of the Widom 601 (21,32).
To monitor FRET, we collected emission spectra while exciting Cy3 at 510 nm. Titrations showed that increasing amounts of Chd1 yielded progressively more intense Cy3 emission peaks and less intense Cy5 emission peaks (Figure 2C). While the dependence of Cy5 fluorescence on Cy3 excitation indicates FRET, some of the decrease in Cy5 intensity might have resulted from Chd1 binding rather than separation from the Cy3 donor. To investigate this possibility, emission spectra were also collected with direct excitation of Cy5 (645 nm). As shown in Supplementary Figure S4A, the Cy5 emission peak did in fact decrease with addition of Chd1, indicating that, in addition to FRET, environmental changes surrounding Cy5 also decreased fluorescence intensity. To determine whether the change in Cy5 intensity was due to Chd1 binding directly, or was an indirect effect due to unwrapping, we monitored fluorescence with alternating Cy3 and Cy5 excitation during salt-induced disassembly of the nucleosome (Supplementary Figure S5). The salt titrations revealed dramatic drops in Cy5 fluorescence during excitation of either Cy3 or Cy5, indicating that the Cy3-independent changes in Cy5 fluorescence likely arose from unwrapping.
In comparing Chd1 titrations for 12N80 and 80N12 nucleosomes in AMP–PNP conditions, emission peaks from Cy5 direct excitation were generally quite similar, whereas the profiles of Cy3 emission peaks differed between these two nucleosomes (Supplementary Figure S4B). This discrepancy may be explained by the labeling scheme: Cy3 was uniquely on one DNA end for each nucleosome, whereas Cy5 was in two symmetrically related positions on the H3 tail. We therefore focused on changes in Cy3 emission as a more appropriate reporter for how Chd1 titrations affected unwrapping of the two sides of the nucleosome. Each titration was fit to a binding isotherm to calculate Kunwrap, corresponding to the Chd1 concentration required to achieve half of the maximum signal change. In AMP–PNP conditions, Kunwrap was dramatically different for these two nucleosome constructs, with a much larger value of 700 ± 200 nM for 12N80 compared with 13 ± 2 nM for 80N12 (Figure 2D, green symbols). Interpreting these differences with respect to the Widom 601 sequence, it appears that the TA poor side unwraps more readily by Chd1 than the TA rich side, consistent with previously published work (21).
Chd1-dependent changes in Cy3 and Cy5 fluorescence were also observed in apo and ADP·BeF3– conditions. Consistent with the 16N16 experiments, higher amounts of Chd1 were required in apo conditions to elicit changes in Cy3, corresponding to apparent Kunwrap values of 800± 400 nM for 12N80 and 700 ± 200 nM for 80N12 (gray symbols). In sharp contrast, ADP·BeF3– conditions promoted maximal changes in Cy3 fluorescence with stoichiometric amounts of Chd1 (magenta symbols). The saturation point was 51 ± 5 nM, close to 2:1, for the 12N80 and 22 ± 2 nM, or roughly 1:1, for the 80N12, again consistent with the TA-poor edge of the Widom 601 more easily unwrapping at low concentrations of Chd1. Thus, DNA unwrapping by Chd1 is strongly affected by both the sequence of the DNA and the nucleotide state of the remodeler.
Recent cross-linking and EM data suggest that all three domains of Chd1 work in concert to span both gyres of DNA and unwrap DNA from the nucleosome edge (17,19,20). We suspected that a specific, nucleotide-dependent organization of Chd1 domains held the DBD in a position that favored an unwrapped trajectory of DNA exiting the nucleosome. To test this idea, we examined the extent that increases in Cy3 intensity could be stimulated by a Chd1 construct lacking the DBD. Binding was performed in the presence of ADP·BeF3–, since this nucleotide state appeared to yield the tightest Chd1-nucleosome complex. Titrating Chd1ΔDBD in the presence of ADP·BeF3– gave a strong change in Cy3 fluorescence, indicative of unwrapping (Figure 2E). The amplitude of the fluorescence changes were comparable to titrations with Chd1, yet the Chd1ΔDBD titrations showed that a much higher protein concentration was required to approach saturation (Supplementary Figure S6). While these data show the same trends, individual experiments unfortunately were noisier (Supplementary Figure S6), resulting in greater variation in Kunwrap values, which were 500 ± 300 nM for the 12N80 and 300 ± 200 nM for the 80N12. Despite the poorer fits, these data indicate that the chromo-ATPase portion of Chd1 is sufficient for unwrapping DNA from the nucleosome. While unwrapping can be achieved by binding of a transcription factor to the outer DNA gyre of the nucleosome (31), it seems unlikely that binding to the outer DNA gyre could explain the unwrapping observed with Chd1ΔDBD. In previous work, we found that a chromo-ATPase construct was unable to stably associate with naked DNA (33). Thus, even the sub-μM concentrations of Chd1ΔDBD used here would likely be insufficient for significant unwrapping by mass action. These results are consistent with the idea that the DBD is required for high affinity binding, yet not necessary for stimulating changes in DNA trajectory. Given the direct contact that the ATPase motor makes with the opposite gyre DNA as seen by cryoEM (19), as well as the sensitivity of unwrapping to bound nucleotide (Figure 2D), we favor the notion that the ATPase motor can disrupt wrapping at the edge of the nucleosome when bound at the internal SHL2 site.
The structural distributions of unwrapped nucleosome states are sensitive to the nucleotide analog bound by Chd1
While the sensitivity of fluorescent probes provides a powerful tool to detect perturbations of nucleosomal DNA, we sought to obtain additional insight into what an altered structure of the nucleosome might look like. We therefore turned to SAXS, which can provide insight into the global structures of individual macromolecules in solution. For large complexes containing multiple components with different electron densities (e.g. proteins and DNA), however, interpretation of SAXS data is limited due to the difficulty in resolving each component's contribution to the measured SAXS profile (34). Therefore, we applied contrast variation small angle x-ray scattering (CV-SAXS) to resolve the nucleotide-dependent conformation(s) of nucleosomal DNA changes upon Chd1 binding. In CV-SAXS, sucrose is added to the bulk solvent until its electron density equals that of the lower electron density component, in this case the proteins. Under this matched condition, the protein components no longer contribute to the signal and details of the DNA conformation can be resolved. We previously used CV-SAXS to study DNA unwrapping during the salt-induced disassembly of nucleosomes (35,36). Here, we applied this technique to determine whether Chd1 altered the canonically wrapped conformation of nucleosomal DNA when bound in different nucleotide states.
The concentration of sucrose required to mask the protein signals was experimentally determined. Initially, SAXS profiles of the DNA (5 μM), Chd1 (15 μM), and histones (5 μM) were separately measured in solutions containing varying percentages of sucrose (Figure 3A). In 60% sucrose, the signals from protein components (Chd1 and histones) were effectively eliminated, but, due to its higher electron density, sufficient signal remained from the DNA to measure its conformation in the presence (or absence) of protein partners.
Figure 3.

Protein contrast matching was achieved with 60% sucrose. (A) SAXS signals are shown for DNA, Chd1, and histone octamer measured separately with 0% and 60% sucrose. In 0% sucrose, DNA (solid black), Chd1 (solid blue), and histone (solid red) have distinct scattering profiles. In 60% sucrose, the DNA signal (dashed black) is reduced by a factor of ≈4, but scattering signals from Chd1 (dashed blue) and histone (dashed red) vanish. (B) SAXS profiles measured in 60% sucrose for 12N12 alone (in ADP·BeF3– conditions, black), and Chd1–12N12 complex in apo (blue), ADP·BeF3– (red) and AMP–PNP (green).
Figure 3B shows SAXS profiles measured in 60% sucrose for 12N12 nucleosomes (5 μM), with and without Chd1 (10 μM). All measured SAXS profiles are qualitatively similar, displaying the characteristic supercoiled shape of DNA around the histone octamer, suggesting that most of the DNA remains wrapped in nucleosomal structures. As a control, we measured SAXS profiles for nucleosomes alone in the presence of different nucleotides, but without the remodeler. These profiles are identical (Supplementary Figure S7A and B). Small but significant differences were detected when Chd1 was present, dependent on the presence of ADP·BeF3– or AMP–PNP, or the absence of nucleotide (apo) (Supplementary Figure S7C and D). The radius of gyration (Rg) for the 12N12 nucleosome alone was 49.4 ± 0.3 Å, consistent with expectations for a fully wrapped structure. In the presence of Chd1, the Rg increased to 56.6 ± 0.7 Å and 55.3 ± 0.5 Å with the addition of ADP·BeF3– and AMP–PNP, respectively, consistent with the partial unwrapping of the DNA ends observed using FRET (Figure 2D). The complex (Chd1–12N12) in the apo state had an Rg of 50.4 ± 0.2 Å. This decreased response is consistent with the weaker fluorescence changes and a requirement for higher Chd1 concentrations in the apo state (Figure 2D and Supplementary Figure S3).
To gain structural insights that go beyond the average, measured Rg values, we applied an ensemble optimization method (EOM) to identify potential DNA structures present in the various complexes. Starting from a large pool of possible structures, EOM finds the subset of structures (‘ensemble’) that best recapitulates the SAXS data. Following the strategies in ref (35,36), we manually unwrapped DNA to generate a pool of structures. These structures were based on the nucleosome crystal structure 1KX5 (37) with varying amounts of DNA removed and replaced with straight segments (linear B-form DNA). This procedure produced 9182 structures with the DNA unwrapped along the natural trajectory of the nucleosomal DNA. To further diversify the DNA pool, we generated 5625 variations of unwrapped nucleosomal DNAs that contained kinks directing DNA out-of-plane with the nucleosome disk (Supplementary Figure S8). These kinks were generated by introducing various combinations of bends into 12N12 nucleosomal DNA using 3D-DART (38,39). From this pool of 14 807 total structures, a genetic algorithm (27–29) selected ensembles of DNA structures using the SAXS data collected for 12N12 nucleosomes with and without Chd1 in 60% sucrose. The resulting fits to the data are shown in Figure 4A–D.
Figure 4.

Representative ensembles for Chd1–12N12 complexes in different nucleotide states. (A–D) SAXS data and EOM fits for nucleosome alone (12N12, in ADP·BeF3– buffer), and the Chd1–12N12 complex in apo, AMP–PNP, and ADP·BeF3– conditions, respectively. (E–F) Rg distributions for the DNA pool (E) and the selected ensembles (F) for nucleosome alone (12N12, black) and Chd1–12N12 complex in the apo (blue), ADP·BeF3– (red), and AMP–PNP (green) conditions. (G–J) DNA structures selected by EOM for 12N12 alone and Chd1–12N12 complexes in different nucleotide states. Structures were organized by color according to characteristic features. Unwrapping was classified according to the predominantly unwrapped side. In each cluster, the DNA is shaded according to its weight in the EOM-selected pools. (K) Most partially unwrapped models selected by EOM are asymmetric. The RMSD for backbone phosphate atoms was calculated for each half of each model compared to an idealized and symmetrically wrapped nucleosome. In this representation, each model is shown as a line connecting two points, with the left side reporting the lesser deviation and right side the greater deviation from the wrapped standard. The slopes of the connecting lines highlight the degree of asymmetry for each model. Shading reflects the weighting of each model in the EOM pools.
To characterize the ensembles, we calculated Rg distributions for the structures selected by EOM (Figure 4E and F). Although the pool contained structures that ranged in size from 45–165 Å (Figure 4E), the structures within the selected ensembles cluster in size between 45–70 Å (Figure 4F), consistent with DNA in 12N12 nucleosomes being mostly wrapped in the presence of Chd1 regardless of the nucleotide state. The nucleosomes alone are represented by the peak centered around 49 Å. For Chd1–12N12 complexes in the presence of AMP–PNP and ADP·BeF3–, the Rg distributions showed a prominent peak centered around 55 Å, reflecting partially unwrapped nucleosome structures. For Chd1–12N12 in the apo state, the Rg distribution suggests a mixture between mostly wrapped nucleosomes and a small population of partially unwrapped structures. The EOM pools therefore suggest subtle but measurable differences in DNA conformation in response to nucleotide-dependent conformations of Chd1.
Ensembles for each condition are shown and compared in Figure 4G–J. The structures in each ensemble were assigned colors according to shared characteristic features. Under all conditions, at least some DNA remained mostly wrapped (green in Figure 4G–J). These wrapped states are most prevalent for the nucleosomes without Chd1 (100%) and Chd1–12N12 in the apo state (64%). Between a quarter and a third of the structures remain mostly wrapped for Chd1–12N12 in AMP–PNP (27%) and ADP·BeF3– (31%). This distribution is consistent with dynamic unwrapping, as has been illustrated for Chd1-nucleosome complexes in apo and AMP–PNP states (20).
The partially unwrapped structures observed in the presence of Chd1 showed distinct trajectories (blue, yellow, and red in Figure 4G–J) that were present with varying weights depending on the nucleotide state. In the unwrapped structures, the DNA end typically peeled away from the histone at a common internal site, up to 25 bp from the nucleosome edge, which is consistent with the cryoEM structure of Chd1 partially unwrapping one side of a nucleosome (19). While most structures showed some degree of unwrapping on both sides, unwrapping was typically more prominent on one side than the other. To visualize the extent of asymmetry, we calculated the extent that each half of each model deviated from an idealized, symmetrically wrapped model (Figure 4K). Models that were mostly wrapped, which in this analysis showed deviations up to but not beyond ∼10 Å, showed the least asymmetry. Most of the AMP–PNP and ADP·BeF3– models with greater deviations were asymmetric, with more unwrapping on one side than the other. An exception was one of the ADP·BeF3– models, which showed a similar out-of-plane unwrapping on both sides. Although a 2-fold molar equivalent of Chd1 was present relative to nucleosomes, some asymmetry may have arisen from incomplete saturation of nucleosomes, resulting in 1:1 (Chd1:nucleosome) complexes. Since SAXS is not sensitive to chirality, this analysis cannot distinguish unwrapping from a particular side of the nucleosome, though we would expect the unwrapping of the TA-poor side to be favored, based on the FRET experiments with 12N80 and 80N12 nucleosomes in AMP–PNP conditions (Figure 2D). With ADP·BeF3–, FRET showed that both sides of the nucleosome were highly sensitive to Chd1, with apparent saturation at 2:1. It was therefore unexpected to observe significant asymmetry for most EOM models for ADP·BeF3– SAXS data.
Comparison of the EOM-selected models to the Chd1-nucleosome complex solved by cryoEM (19) revealed interesting nucleotide-specific trends. In the cryoEM structure, which was solved in an ADP·BeF3– state, the nucleosomal DNA adjacent to where Chd1 is bound is unwrapped by ∼2 helical turns and is slightly out-of-plane (19). Alignment to this structure shows that the SAXS-derived, AMP–PNP models have a wider range of unwrapping, yet always remain more in-plane than DNA in the cryoEM complex (Figure 5A). In contrast, the SAXS-derived, ADP·BeF3– models show approximately the same magnitude of in-plane unwrapping as the cryoEM structure, yet more significant unwrapping in the out-of-plane direction (Figure 5B). To evaluate how critical these unique features were for fitting the SAXS data, an additional round of EOM selections was performed where the starting pool was limited to the selected models of the other nucleotide state. In both cases, the fits between the available models and data were worse: ADP·BeF3– data fit with AMP–PNP models yielded a χ2 = 0.97 (compared to χ2 = 0.89 for ADP·BeF3– models, Figure 4D), and AMP–PNP data fit with ADP·BeF3– models yielded a χ2 = 1.28 (compared to χ2 = 0.87 for AMP–PNP models, Figure 4C). The EOM-based interpretation of the SAXS data therefore supports the conclusion that the distributions of DNA shapes are distinct. These differences in unwrapping geometry imply that the nucleotide bound by Chd1 significantly alters the dynamics or conformational state of the ATPase motor on the nucleosome, which in turn influences both the extent and direction of unwrapping.
Figure 5.

EOM modeling of CV-SAXS data reveals distinct structural features of Chd1–12N12 complexes in AMP–PNP and ADP·BeF3– conditions. (A) Shown are partially wrapped (green) and unwrapped (blue) models that matched CV-SAXS data of Chd1–12N12 complexes in AMP–PNP conditions. These two structural classes represented 97% of the weighted models obtained to match SAXS curves. The intensity of the colors reflects the model weighting, with lighter shades of blue/green indicating models with combined weights of <5%, whereas stronger colors are >5%. Shown as a white molecular surface is the DNA from a nucleosome unwrapped by Chd1 in the ADP·BeF3– state (19). An idealized, symmetrically wrapped model based on the 1KX5 crystal structure (37) is shown in black for reference. (B) Models obtained for 12N12–Chd1 complexes in ADP·BeF3– conditions, colored as for (A). Note the distinct out-of-plane unwrapping (red) that is absent in models for AMP–PNP conditions.
DISCUSSION
This work explores the ability of the Chd1 chromatin remodeler to unwrap DNA from the edge of the nucleosome. Recent studies have revealed that the ATPase motor and DBD of Chd1 interact across the gyres of the nucleosome, with the ATPase motor bound to the internal SHL2 site and the DBD engaged with DNA exiting the nucleosome (17,19). While both of these domains appear to stabilize an unwrapped state of the nucleosome in a recent cryoEM structure (19), we show here that the DBD is not required for unwrapping (Figure 2E). We therefore propose that the Chd1 ATPase motor is sufficient for destabilizing histone–DNA contacts at the nucleosome edge. Consistent with this idea, we observed that unwrapping was strongly correlated to the nature of the bound nucleotide, with significant differences for ADP·BeF3– compared to AMP–PNP. FRET experiments showed that ADP·BeF3– promoted Chd1-dependent unwrapping of the 601 nucleosome more readily than AMP–PNP (Figure 2D). More striking, however, was the finding that these two nucleotide-bound states favored distinct conformations of unwrapped Chd1-nucleosome complexes. As determined by EOM analysis of CV-SAXS data, Chd1 loaded with AMP–PNP unwrapped DNA at a wider angle but essentially in-plane with the nucleosome disk, whereas a significant fraction of DNA unwrapped more distinctly out-of-plane with ADP·BeF3– (Figures 4 and 5).
As for all ATPase motors, Chd1 is expected to be structurally sensitive to its nucleotide-bound state. Accordingly, we observed distinct kinetics of nucleosome binding in AMP–PNP versus ADP·BeF3– conditions (Figure 1), which we interpret as reflecting conformational changes in Chd1 domains and/or the nucleosome upon binding. Together with our findings that these nucleotide analogs produce structurally unique distributions of unwrapped nucleosomes, these results indicate that DNA unwrapping is tightly coupled to the dynamics and/or conformational states of the ATPase motor. Although it is unclear whether each cycle of ATP hydrolysis produces a specific in-plane to out-of-plane pattern of unwrapping, this tight coupling suggests that the position of exit DNA on the opposite gyre could influence ATPase activity. We speculate that particular conformations of flanking DNA—whether biased or stabilized via the Chd1 DBD, DNA sequence, or other factors—would affect the conformational landscape of the ATPase motor during the ATP binding and hydrolysis cycle, and thus could provide an external means of regulation.
Nucleosome repositioning and assembly are two defining characteristics of Chd1 (10,11), but it seems unlikely that the DNA unwrapping described here is required for either of these processes. When bound at SHL2, the ATPase motor of Chd1 directly contacts the DNA being unwrapped from the opposite gyre (19). It has been suggested that contact between the ATPase motor and opposite gyre DNA may be a common characteristic of chromatin remodelers: a cryoEM structure of the SWI/SNF ATPase bound to a nucleosome identified conserved ATPase residues in contact with opposite gyre DNA, and mutation of these residues reduced sliding activity (40). The SWI/SNF-nucleosome complex, however, showed no unwrapping, suggesting that those ATPase contacts are insufficient for the unpeeling of DNA observed in the Chd1–nucleosome complex. Although it is not yet known whether ISWI remodelers also stimulate nucleosome unwrapping, it is interesting that both Chd1 and ISWI remodelers slide and assemble nucleosomes yet are differently affected by the presence of linker histones, which stabilize a wrapped form of nucleosomes (41). Chd1 cannot incorporate histone H1 during nucleosome assembly (10), and the presence of H1 and H5 interfere with nucleosome sliding by Chd1 (42). In contrast, ISWI remodelers can both assemble and slide nucleosomes in the presence of linker histones (10,42).
Notably, in the Chd1–nucleosome complex visualized by cryoEM, a loop protruding from the ATPase motor packs against the phosphate backbone where DNA unwraps from the histone core (19) (Supplementary Figure S9A). Sequence alignment of different remodeler families reveals that this loop has conserved basic character and is longer in Chd1 orthologs than SWI/SNF or ISWI remodelers (Supplementary Figure S9B). Based on its location in the cryoEM structure and positively charged character, we propose that this family-specific loop would assist in stimulating and/or stabilizing DNA unwrapping. This loop is longer in S. cerevisiae than human Chd1, and therefore the propensity to unwrap may also differ among Chd1 orthologs. It is also interesting to note that the two other CHD subfamilies also have a pronounced loop in this region, with a significantly longer and highly charged segment for the CHD3/CHD4/CHD5 group. Future work will be needed to determine the extent that this ATPase loop is involved in nucleosome unwrapping.
Nucleosomal DNA can unwrap spontaneously and transiently, which is important for allowing access to transcription factor binding sites at the nucleosome edge (31,43–45). DNA sequence determines the intrinsic stability of histone–DNA contacts, and the most favored sequences, such as the widely used Widom 601, contain phased motifs (such as TA steps) that bias DNA bending in one direction (24,46). Despite its high affinity, however, the strength of histone–DNA contacts are not symmetrically distributed for the Widom 601 (32), and under tension, 601 nucleosomes preferentially unwrap on the less flexible side, which has fewer TA steps (21). Asymmetric unwrapping was prevalent in our SAXS experiments (Figure 4), and FRET indicated that with AMP–PNP, Chd1 more easily unwrapped nucleosomes on the TA poor side (Figure 2). These findings indicate that Chd1 amplifies the natural unwrapping tendencies of the underlying sequence. Such amplification of nucleosome dynamics should likewise increase the sequence-dependent accessibility for chromatin-associated factors. In addition to transcription factors, histone modifying enzymes are also likely to be affected. As observed in the first and many subsequent nucleosome crystal structures, residues 39–42 of histone H3 pass between the DNA gyres of the nucleosome at the DNA entry/exit site (47). DNA unwrapping by Chd1 exposes this N-terminal segment of histone H3 (19), which is consistent with our observation of unwrapping-coupled environmental changes of Cy5-labeled H3(V35C) (Supplementary Figure S5). Unwrapping would therefore increase accessibility for post-translational modifications such as H3R42 methylation and H3Y41 and H3T45 phosphorylation (48–51). In addition, methylation and acetylation of the nearby H3K36 is strongly coupled to transcription (52–54), and unwrapping may also favor deposition or recognition of these marks.
In addition to increasing DNA and histone accessibility, nucleosome unwrapping by Chd1 may potentially influence transcriptional elongation. Nucleosomes present a barrier to elongating polymerases such as RNA polymerase II (Pol II), causing transcriptional pausing (55,56). Studitsky and coworkers have shown that DNA unwrapping from the upstream edge of the nucleosome facilitates transcription of Pol II through the outermost turn of nucleosomal DNA, whereas unwrapping from the downstream edge of the nucleosome greatly diminishes the major Pol II pause site at the upstream SHL2 (57). Engagement of Chd1 with the downstream SHL2 would unwrap the upstream edge of the nucleosome and therefore potentially help Pol II entry. However, the presence of Pol II at the upstream SHL2, a major site of pausing, would sterically block Chd1 from unwrapping the downstream edge. Interestingly, Pol II has more difficulty transcribing through the upstream SHL2 site of 601 nucleosomes if the upstream DNA immediately behind the polymerase does not rewrap (57). DNA unwrapping, if maintained by Chd1 on the entry side, therefore could be antagonistic to passage of Pol II through nucleosomes. Pol II transcription through nucleosomes is assisted by several elongation factors, and the FACT complex both reduces polymerase pausing and stabilizes histone–DNA contacts during transcription (58). Although its mechanism of action is not presently clear, FACT appears to act in opposition to Chd1, with FACT mutant phenotypes in budding yeast rescued by Chd1 defects (59). It has been shown that two polymerases traveling together evict nucleosomes (60), and therefore while speculative, controlled pausing may help keep Pol II complexes separated from each other. Determining whether DNA unwrapping by Chd1 contributes to transcription-related processes is an exciting new direction for future studies.
DATA AVAILABILITY
SAXS data and the EOM-selected models described here have been deposited in SASBDB (https://www.sasbdb.org/) with the following accession numbers: SASDCT6 (12N12 nucleosome alone in 60% sucrose, ADP·BeF3– buffer conditions); SASDCU6 (Chd1–12N12 complex in 60% sucrose, nucleotide-free buffer); SASDCV6 (Chd1–12N12 complex in 60% sucrose, ADP·BeF3– buffer); and SASDCW6 (Chd1–12N12 complex in 60% sucrose, AMP–PNP buffer).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Sarah Woodson for use of her stopped-flow and steady state fluorometers. We thank Arthur Woll for technical assistance at the CHESS G1 beamline.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R35-GM122514 to L.P., R01-GM084192 and R01-GM113240 to G.D.B., T32-GM007231 to R.J.T.]; National Science Foundation (NSF) & National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS) via NSF award [DMR-0936384 to CHESS]. Funding for open access charge: National Institutes of Health [R01-GM113240].
Conflict of interest statement. None declared.
REFERENCES
- 1. McGinty R.K., Tan S.. Recognition of the nucleosome by chromatin factors and enzymes. Curr. Opin. Struct. Biol. 2016; 37:54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Narlikar G.J., Sundaramoorthy R., Owen-Hughes T.. Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes. Cell. 2013; 154:490–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gaspar-Maia A., Alajem A., Polesso F., Sridharan R., Mason M.J., Heidersbach A., Ramalho-Santos J., McManus M.T., Plath K., Meshorer E. et al. Chd1 regulates open chromatin and pluripotency of embryonic stem cells. Nature. 2009; 460:863–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Smolle M., Venkatesh S., Gogol M.M., Li H., Zhang Y., Florens L., Washburn M.P., Workman J.L.. Chromatin remodelers Isw1 and Chd1 maintain chromatin structure during transcription by preventing histone exchange. Nat. Struct. Mol. Biol. 2012; 19:884–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Simic R., Lindstrom D.L., Tran H.G., Roinick K.L., Costa P.J., Johnson A.D., Hartzog G.A., Arndt K.M.. Chromatin remodeling protein Chd1 interacts with transcription elongation factors and localizes to transcribed genes. EMBO J. 2003; 22:1846–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Krogan N.J., Kim M., Ahn S.H., Zhong G., Kobor M.S., Cagney G., Emili A., Shilatifard A., Buratowski S., Greenblatt J.F.. RNA polymerase II elongation factors of Saccharomyces cerevisiae: a targeted proteomics approach. Society. 2002; 22:6979–6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kelley D.E., Stokes D.G., Perry R.P.. CHD1 interacts with SSRP1 and depends on both its chromodomain and its ATPase/helicase-like domain for proper association with chromatin. Chromosoma. 1999; 108:10–25. [DOI] [PubMed] [Google Scholar]
- 8. Hughes A.L., Rando O.J.. Comparative genomics reveals Chd1 as a determinant of nucleosome spacing in vivo. G3 (Bethesda). 2015; 5:1889–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ocampo J., Chereji R. V., Eriksson P.R., Clark D.J.. The ISW1 and CHD1 ATP-dependent chromatin remodelers compete to set nucleosome spacing in vivo. Nucleic Acids Res. 2016; 44:4625–4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lusser A., Urwin D.L., Kadonaga J.T.. Distinct activities of CHD1 and ACF in ATP-dependent chromatin assembly. Nat. Struct. Mol. Biol. 2005; 12:160–166. [DOI] [PubMed] [Google Scholar]
- 11. Stockdale C., Flaus A., Ferreira H., Owen-Hughes T.. Analysis of nucleosome repositioning by yeast ISWI and Chd1 chromatin remodeling complexes. J. Biol. Chem. 2006; 281:16279–16288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fei J., Torigoe S.E., Brown C.R., Khuong M.T., Kassavetis G.A., Boeger H., Kadonaga J.T.. The prenucleosome, a stable conformational isomer of the nucleosome. Genes Dev. 2015; 29:2563–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schwanbeck R., Xiao H., Wu C.. Spatial contacts and nucleosome step movements induced by the NURF chromatin remodeling complex. J. Biol. Chem. 2004; 279:39933–39941. [DOI] [PubMed] [Google Scholar]
- 14. Saha A., Wittmeyer J., Cairns B.R.. Chromatin remodeling through directional DNA translocation from an internal nucleosomal site. Nat. Struct. Mol. Biol. 2005; 12:747–755. [DOI] [PubMed] [Google Scholar]
- 15. Zofall M., Persinger J., Kassabov S.R., Bartholomew B.. Chromatin remodeling by ISW2 and SWI/SNF requires DNA translocation inside the nucleosome. Nat. Struct. Mol. Biol. 2006; 13:339–346. [DOI] [PubMed] [Google Scholar]
- 16. McKnight J.N., Jenkins K.R., Nodelman I.M., Escobar T., Bowman G.D.. Extranucleosomal DNA binding directs nucleosome sliding by Chd1. Mol. Cell. Biol. 2011; 31:4746–4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nodelman I.M., Bleichert F., Patel A., Ren R., Horvath K.C., Berger J.M., Bowman G.D.. Interdomain communication of the Chd1 chromatin remodeler across the DNA gyres of the nucleosome. Mol. Cell. 2017; 65:447–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Delmas V., Stokes D.G., Perry R.P.. A mammalian DNA-binding protein that contains a chromodomain and an SNF2/SWI2-like helicase domain. Proc. Natl. Acad. Sci. U.S.A. 1993; 90:2414–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Farnung L., Vos S.M., Wigge C., Cramer P.. Nucleosome–Chd1 structure and implications for chromatin remodelling. Nature. 2017; 154:490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sundaramoorthy R., Hughes A.L., Singh V., Wiechens N., Ryan D.P., El-Mkami H., Petoukhov M., Svergun D.I., Treutlein B., Quack S. et al. Structural reorganization of the chromatin remodeling enzyme Chd1 upon engagement with nucleosomes. Elife. 2017; 6:1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ngo T.T.M., Zhang Q., Zhou R., Yodh J.G., Ha T.. Asymmetric unwrapping of nucleosomes under tension directed by DNA local flexibility. Cell. 2015; 160:1135–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nodelman I.M., Horvath K.C., Levendosky R.F., Winger J., Ren R., Patel A., Li M., Wang M.D., Roberts E., Bowman G.D.. The Chd1 chromatin remodeler can sense both entry and exit sides of the nucleosome. Nucleic Acids Res. 2016; 44:7580–7591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Luger K., Rechsteiner T.J., Richmond T.J.. Preparation of nucleosome core particle from recombinant histones. Methods Enzymol. 1999; 304:3–19. [DOI] [PubMed] [Google Scholar]
- 24. Lowary P.T., Widom J.. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998; 276:19–42. [DOI] [PubMed] [Google Scholar]
- 25. Svergun D.I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 1992; 25:495–503. [Google Scholar]
- 26. Svergun D., Barberato C., Koch M.H.. CRYSOL - a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 1995; 28:768–773. [Google Scholar]
- 27. Bernadó P., Mylonas E., Petoukhov M. V, Blackledge M., Svergun D.I.. Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 2007; 129:5656–5664. [DOI] [PubMed] [Google Scholar]
- 28. Bernadó P., Svergun D.I.. Structural analysis of intrinsically disordered proteins by small-angle X-ray scattering. Mol. BioSyst. 2012; 8:151–167. [DOI] [PubMed] [Google Scholar]
- 29. Tria G., Mertens H.D.T., Kachala M., Svergun D.I.. Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ. 2015; 2:207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Leonard J.D., Narlikar G.J.. A nucleotide-driven switch regulates flanking DNA length sensing by a dimeric chromatin remodeler. Mol. Cell. 2015; 57:850–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li G., Widom J.. Nucleosomes facilitate their own invasion. Nat. Struct. Mol. Biol. 2004; 11:763–769. [DOI] [PubMed] [Google Scholar]
- 32. Hall M.A., Shundrovsky A., Bai L., Fulbright R.M., Lis J.T., Wang M.D.. High-resolution dynamic mapping of histone–DNA interactions in a nucleosome. Nat. Struct. Mol. Biol. 2009; 16:124–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Qiu Y., Levendosky R.F., Chakravarthy S., Patel A., Bowman G.D., Myong S.. The Chd1 chromatin remodeler shifts nucleosomal DNA bidirectionally as a monomer. Mol. Cell. 2017; 68:76–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tokuda J.M., Pabit S.A., Pollack L.. Protein-DNA and ion-DNA interactions revealed through contrast variation SAXS. Biophys. Rev. 2016; 8:139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen Y., Tokuda J.M., Topping T., Sutton J.L., Meisburger S.P., Pabit S.a., Gloss L.M., Pollack L.. Revealing transient structures of nucleosomes as DNA unwinds. Nucleic Acids Res. 2014; 42:8767–8776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen Y., Tokuda J.M., Topping T., Meisburger S.P., Pabit S.A., Gloss L.M., Pollack L.. Asymmetric unwrapping of nucleosomal DNA propagates asymmetric opening and dissociation of the histone core. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:334–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Davey C.A., Sargent D.F., Luger K., Maeder A.W., Richmond T.J.. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9Å resolution. J. Mol. Biol. 2002; 319:1097–1113. [DOI] [PubMed] [Google Scholar]
- 38. van Dijk M., Bonvin A.M.J.J.. 3D-DART: A DNA structure modelling server. Nucleic Acids Res. 2009; 37:235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lu X.J., Olson W.K.. 3DNA: A software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003; 31:5108–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu X., Li M., Xia X., Li X., Chen Z.. Mechanism of chromatin remodelling revealed by the Snf2-nucleosome structure. Nature. 2017; 544:440–445. [DOI] [PubMed] [Google Scholar]
- 41. Zhou B.-R., Feng H., Kato H., Dai L., Yang Y., Zhou Y., Bai Y.. Structural insights into the histone H1-nucleosome complex. Proc. Natl. Acad. Sci. U.S.A. 2013; 48110:19390–19395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Maier V.K., Chioda M., Becker P.B.. ATP-dependent chromatosome remodeling. Biol. Chem. 2008; 389:345–352. [DOI] [PubMed] [Google Scholar]
- 43. Polach K.J., Widom J.. Mechanism of protein access to specific DNA sequences in chromatin: a dynamic equilibrium model for gene regulation. J. Mol. Biol. 1995; 254:130–149. [DOI] [PubMed] [Google Scholar]
- 44. North J.A., Shimko J.C., Javaid S., Mooney A.M., Shoffner M.A., Rose S.D., Bundschuh R., Fishel R., Ottesen J.J., Poirier M.G.. Regulation of the nucleosome unwrapping rate controls DNA accessibility. Nucleic Acids Res. 2012; 40:10215–10227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li G., Levitus M., Bustamante C., Widom J.. Rapid spontaneous accessibility of nucleosomal DNA. Nat. Struct. Mol. Biol. 2004; 12:46–53. [DOI] [PubMed] [Google Scholar]
- 46. Chua E.Y.D., Vasudevan D., Davey G.E., Wu B., Davey C.A.. The mechanics behind DNA sequence-dependent properties of the nucleosome. Nucleic Acids Res. 2012; 40:6338–6352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Luger K., Mäder A.W., Richmond R.K., Sargent D.F., Richmond T.J.. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997; 389:251–260. [DOI] [PubMed] [Google Scholar]
- 48. Casadio F., Lu X., Pollock S.B., LeRoy G., Garcia B. a, Muir T.W., Roeder R.G., Allis C.D.. H3R42me2a is a histone modification with positive transcriptional effects. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:14894–14899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Baker S.P., Phillips J., Anderson S., Qiu Q., Shabanowitz J., Smith M.M., Yates J.R., Hunt D.F., Grant P.A.. Histone H3 Thr 45 phosphorylation is a replication-associated post-translational modification in S. cerevisiae. Nat. Cell Biol. 2010; 12:294–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dawson M.A., Bannister A.J., Göttgens B., Foster S.D., Bartke T., Green A.R., Kouzarides T.. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature. 2009; 461:819–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jang S.M., Azebi S., Soubigou G., Muchardt C.. DYRK1A phosphorylates histone H3 to differentially regulate the binding of HP1 isoforms and antagonize HP1-mediated transcriptional repression. EMBO Rep. 2014; 15:686–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xiao T., Hall H., Kizer K.O., Shibata Y., Hall M.C., Borchers C.H., Strahl B.D.. Phosphorylation of RNA polymerase II CTD regulates H3 methylation in yeast. Genes Dev. 2003; 17:654–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bell O., Wirbelauer C., Hild M., Scharf A.N.D., Schwaiger M., MacAlpine D.M., Zilbermann F., van Leeuwen F., Bell S.P., Imhof A. et al. Localized H3K36 methylation states define histone H4K16 acetylation during transcriptional elongation in Drosophila. EMBO J. 2007; 26:4974–4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Morris S.A., Rao B., Garcia B.A., Hake S.B., Diaz R.L., Shabanowitz J., Hunt D.F., Allis C.D., Lieb J.D., Strahl B.D.. Identification of histone H3 lysine 36 acetylation as a highly conserved histone modification. J. Biol. Chem. 2007; 282:7632–7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kireeva M.L., Walter W., Tchernajenko V., Bondarenko V., Kashlev M., Studitsky V.M.. Nucleosome remodeling induced by RNA polymerase II. Mol. Cell. 2002; 9:541–552. [DOI] [PubMed] [Google Scholar]
- 56. Kireeva M.L., Hancock B., Cremona G.H., Walter W., Studitsky V.M., Kashlev M.. Nature of the nucleosomal barrier to RNA polymerase II. Mol. Cell. 2005; 18:97–108. [DOI] [PubMed] [Google Scholar]
- 57. Kulaeva O.I., Gaykalova D.A., Pestov N.A., Golovastov V. V, Vassylyev D.G., Artsimovitch I., Studitsky V.M.. Mechanism of chromatin remodeling and recovery during passage of RNA polymerase II. Nat. Struct. Mol. Biol. 2009; 16:1272–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hsieh F.-K., Kulaeva O.I., Patel S.S., Dyer P.N., Luger K., Reinberg D., Studitsky V.M.. Histone chaperone FACT action during transcription through chromatin by RNA polymerase II. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:7654–7659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Biswas D., Dutta-Biswas R., Stillman D.J.. Chd1 and yFACT act in opposition in regulating transcription. Mol. Cell. Biol. 2007; 27:6279–6287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kulaeva O.I., Hsieh F.-K., Studitsky V.M.. RNA polymerase complexes cooperate to relieve the nucleosomal barrier and evict histones. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:11325–11330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
SAXS data and the EOM-selected models described here have been deposited in SASBDB (https://www.sasbdb.org/) with the following accession numbers: SASDCT6 (12N12 nucleosome alone in 60% sucrose, ADP·BeF3– buffer conditions); SASDCU6 (Chd1–12N12 complex in 60% sucrose, nucleotide-free buffer); SASDCV6 (Chd1–12N12 complex in 60% sucrose, ADP·BeF3– buffer); and SASDCW6 (Chd1–12N12 complex in 60% sucrose, AMP–PNP buffer).