


Abstract
Inverted repeats (IRs) are abundant in genomes and frequently serve as substrates for chromosomal aberrations, including gene amplification. In the early stage of amplification, repeated cycles of chromosome breakage and rearrangement, called breakage-fusion-bridge (BFB), generate a large inverted structure, which evolves into highly-amplified, complex end products. However, it remains to be determined how IRs mediate chromosome rearrangements and promote subsequent hyper-amplification and amplicon evolutions. To dissect the complex processes, we constructed repetitive structures in a yeast chromosome and selected amplified cells using genetic markers with limited expression. The genomic architecture was associated with replication stress and produced extra-/intra-chromosomal amplification. Genetic analysis revealed structure-specific endonucleases, Mus81 and Rad27, and post-replication DNA repair protein, Rad18, suppress the amplification processes. Following BFB cycles, the intra-chromosomal products undergo intensive rearrangements, such as frequent inversions and deletions, indicative of rolling-circle replication. This study presents an integrated view linking BFB cycles to hyper-amplification driven by rolling-circle replication.
INTRODUCTION
Inverted repeats (IRs), two arms of repeated DNA sequences with one arm being reverse complemented relative to the other, are common in eukaryotic genomes. IRs and their subgroup, palindromes, in which two arms of IRs are separated by less than a few base pairs, can cause gross chromosomal rearrangements (GCRs), in particular, large inverted duplications of chromosomal segments. Large inverted duplications are abundant in human cancer genomes and are considered to be initial chromosomal structures that lead to the increase in gene copies at very high level (genomic amplification; (1)). Therefore, understanding how IRs promote the inverted chromosomal duplications and how inverted chromosomal duplication lead to high-copy genomic amplification has significant implications for both chromosome biology and disease etiology.
Several experimental systems have been developed to study how IRs promote large inverted duplications in yeasts (2–5). Narayanan et al. showed that the insertion of 320 bp inverted repeat into a chromosome in Saccharomyces cerevisiae induces GCRs, including inverted duplications of chromosomal segments (2). This duplication is likely initiated by the extrusion of 320 bp inverted repeats to form a cruciform-like structure, followed by the resolution of the cruciform. Replication of the resulting hairpin-capped chromosome completes inverted duplication. Mizuno et al. employed the inducible replication fork barrier next to an ectopic inverted repeat of a few-kb and showed that restarted replication forks frequently make U-turns at the inverted repeat and initiate inverted duplication in S. pombe (3,5). Inverted duplication can also be generated when a DNA double-strand break occurred next to an IR. This process occurs during developmentally-programmed chromosome rearrangements in Tetrahymena, in which a 42 bp IR is required for the formation of inverted dimer carrying ribosomal RNA genes. Following the 5′ to 3′ resection, a broken chromosome end anneals intra-molecularly between inverted repeat sequences and initiates DNA synthesis (foldback priming) to generate a hairpin-capped chromosome, the replication of which results in inverted duplications. Although this is a developmentally-programmed inverted duplication in Tetrahymena, somatic inverted duplication following foldback priming from either a natural or experimentally-induced chromosome break has been demonstrated in several yeasts and mammalian cell systems. Most recently, Deng et al. have shown that small inverted repeats (5–9 bp) are sufficient to initiate foldback priming and inverted duplication (6). In mammalian cells, IRs with either 229 or 79 bp of homologies between the arms promote inverted duplications more efficiently than small IRs (7).
Inverted duplication of a hairpin-capped chromosome can result in either isochromosome with two centromeres (dicentric chromosomes) or ones without centromeres (acentric chromosomes). Acentric chromosomes can accumulate in a cell by unequal segregation, as it is proposed for gene amplification by double minute chromosomes (DMs) in cancer cells. Dicentric chromosomes can lead to genomic amplification through breakage-fusion-bridge (BFB) cycles (8). During chromosome segregation, each centromere is pulled to opposite pole, resulting in a break somewhere between centromeres. This de novo break can be processed for subsequent foldback priming, followed by the formation of a hairpin-capped chromosome and inverted duplication. In each cycle, a random break between two centromeres creates the imbalance of genetic material between daughter cells: a segment close to the break will be duplicated in one daughter cell and deleted in the other daughter cell. If the segment harbors an oncogene, duplication of the segment would be beneficial to tumor cell growth and promote clonal expansion. BFB cycles are a prevalent mechanism for the amplification of ERBB2 oncogene that goes up to as many as 50 copies (9,10). However, detailed investigations would be required to determine whether BFB cycle alone can produce such a high-copy amplification. Broken ends generated during BFB cycles can be stabilized by invading into other chromosomes and initiating break-induced replication (BIR) toward the end of the chromosomes, where telomeres will be added to the broken ends. If telomere is added to the broken end after a few cycles, BFB cycles will cease and copy number gains would be limited.
To address the questions, we have focused on a rapid gene amplification mode, rolling-circle replication (RCR), and have developed RCR-based model systems in yeast or mammalian cells (11,12). These RCR-inducible systems could produce extra-/intra-chromosomal amplifications as observed in cancer cells and drug-resistant cultured cells. Furthermore, the RCR processes involved intensive rearrangements, such as inversions and deletions, and we demonstrated the direct association between RCR and intensive rearrangements using yeast 2μ plasmid (13). The RCR-dependent rearrangement was the first mechanistic description of the evolution of intra-chromosomal amplicon, strongly suggesting that RCR play an important role in the late-stage of gene amplification in mammalian cells.
These previous studies indicated that IRs play a critical role in both palindromic gene amplification and RCR-based amplification. Despite the role of IRs in both processes, little is known about the interaction between palindromic gene amplification and RCR. The synergy between the two processes would promote high-copy genomic amplification. To test the possibility, we placed complex inverted structures in a yeast chromosome and studied the natural amplification processes. We found that (i) the IRs are associated with replication stress; (ii) amplification is initiated by palindromic duplication via the IRs; (iii) the resulting repetitive structures cause RCR through tandem pairs of the repeat. Thus, IRs play multiple roles in genomic amplification.
MATERIALS AND METHODS
Yeast strains
Yeast strain LS20 was used as the parental host strain, whose genotype and genomic are described previously (14). Plasmids for creating complex repeat structures were constructed as indicated in Supplementary Figures S6 and S7. The engineered genomic region was used for gene amplification systems in our previous works (11,12). The 3.1-kb sequence partially overlaps only with the IRC7 gene, which is not deleted or mutated. Although the amplicons can express Irc7p (intra-/extra-chromosomal products) and a putative protein of unknown function, Yfr057w (extra-chromosomal), there is no apparent effect on growth, viability, and morphology in this work and our previous works (11,12). To construct mutant strains, yeast knockout clones were purchased from Open Biosystems and each kanMX cassette was PCR-amplified with primers listed in Supplementary Table S1. Cells were transformed with the PCR fragments for targeting using Frozen-EZ Yeast Transformation II Kit (Zymo Research) and selected with 200 or 300 μg/ml of G418.
Selection of yeast cells with gene amplification
The FRFR/FFFR cells were grown in synthetic medium containing 2% glucose, lacking uracil and lysine (nonselective medium) to mid-log phase, harvested by centrifugation, washed twice with sterile distilled water, and plated at various dilutions onto equivalent medium lacking leucine and tryptophan (selective medium). The nonselective and selective media for the FRF/FFF/RRF cells contain lysine and tryptophan, respectively. Cells were also plated onto the nonselective medium to measure the number of viable cells. Cells were grown at 25°C. Colonies were counted after 5 or 6 days of growth.
Gel electrophoresis and Southern analysis
Pulsed-field gel electrophoresis was performed as described previously (12). The auto-algorithm mode of the CHEF Mapper XA system was used with the size range of 150–800 kb (Figures 1B–D and 5, Supplementary Figure S3B and C) and 10–60 kb (Figure 4A, Supplementary Figures S2A, C and S3A). Two-dimensional gel electrophoresis was carried out as described previously (15). Southern analyses were performed with DIG labeling and detection systems (Roche) according to the manufacturer's instructions. Probes were labeled using PCR DIG Probe Synthesis Kit (Roche). Hybridization, washing, and detection were performed using DIG Easy Hyb and DIG Wash and Block Buffer Set (Roche).
Figure 1.
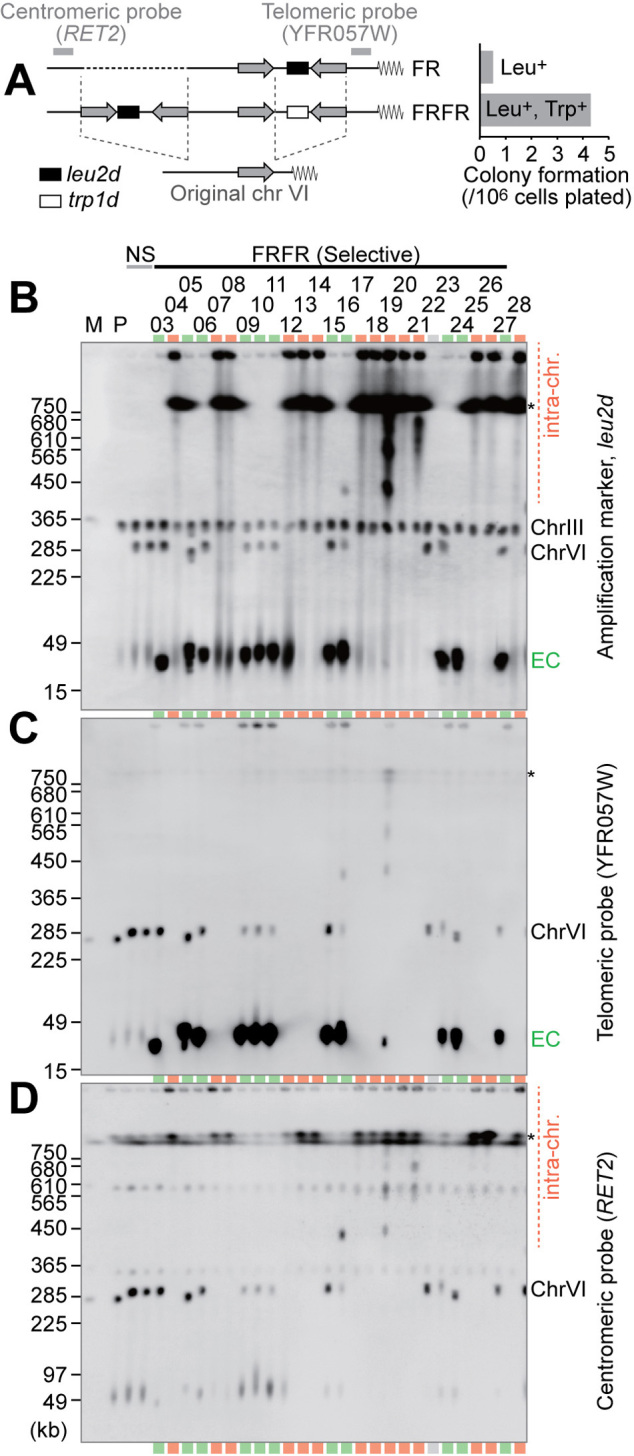
Intra-/extra-chromosomal amplification produced from a complex IR structure. (A) Schematic structures of FR and FRFR constructs. The leu2d and trp1d genes are amplification markers. A 3.1-kb genomic sequence (gray arrow) was PCR-amplified to construct IR structures. The frequency of Leu+ (for FR) or Leu+Trp+ (for FRFR) colony formation was plotted. (B–D) Southern blots of chromosomal DNA with the leu2d probe (B), telomeric probe (C), or centromeric probe (D). The samples marked in red and green indicate intra- and extra-chromosomal products, respectively. The gray lane showed no sign of amplification, suggesting Leu+ conversion between the leu2d marker and the mutated original leu2 allele on chr III. Black asterisks on the right side of panels indicate separation limit under the PFGE-condition. M: S. cerevisiae marker; P: the parental strain, LS20; NS: non-selective conditions; EC: extra-chromosomal products.
Figure 5.

Genetic analysis of gene amplification from the FRFR construct. (A) Frequencies of colony formation in indicated mutants with the FRFR construct. Mutations were introduced into the FRFR strain and evaluated the colony formation relative to the FRFR strain. Means and medians from three independent experiments were shown as box plots. A paired t-test is used to evaluate the mutations in the amplification events (*P< 0.05, **P< 0.01). (B) Types of amplification products in mutants. Representative colonies were analyzed by Southern hybridization with the leu2d probe as in Figure 1B. The numbers of identified products are indicated. (C) Repeat-associated replication stress assessed by 2D analysis. The analyzed region is derived from the original chromosome VI. The late-firing origin ARS609 is more efficient than the telomere-proximal origin ARS610. (D) A possible model generating acentric/dicentric chromosomes. (Top) Replication fork from origins on the telomeric (left) and centric (right) side. Inverted structures are shown above the replication forks. Replication fork slowdown could cause fork regression potentially, which can be resolved by the Mus81 endonuclease. The regressed forks could be resected by an exonuclease and invade into an ectopic inverted sequence. The intermediates of ectopic invasion may be resolved by Rad27 (yeast FEN1). Steps linking dicentric chromosomes to intra-chromosomal amplification require a recombinational pathway involving Mus81 and Rad52. (E) Colony formation and product type from a Δpol32 mutant. Means and standard deviations are indicated.
Figure 4.

Analysis of amplification products from variants of repeat structure. (A) Schematic structures of FFFR, FRF, FFF and FRFR constructs. The leu2d and trp1d genes are amplification markers. A 3.1-kb genomic sequence (gray arrow) was PCR-amplified to construct IR structures. The frequency of Leu+ (for FR) or Leu+Trp+ (for FRFR) colony formation was plotted. (B–E) Southern blots of chromosomal DNA from FFFR (B), FRF (C), FFF (D) and RRF (E). The samples marked in red and green indicate intra- and extra-chromosomal products, respectively. The gray lanes showed no sign of amplification, suggesting Leu+ recombination between the leu2d marker and the mutated original leu2 allele on chr III. The blue samples suggest moderate copy number increase of the leu2d gene likely through unequal sister chromatid exchange. The yellow samples possibly contain a fusion between chromosome VI and III, which cause Leu+ recombination between the leu2d marker and the mutated original leu2 allele on chr III. Black asterisks on the right side of panels indicate separation limit under the PFGE-condition. M: S. cerevisiae marker; P: the parental strain, LS20; NS: non-selective conditions; EC: extra-chromosomal products.
Array CGH analysis
Genomic DNA from colony #17 and the parental strain, LS20, was extracted using DNeasy Blood & Tissue Kit (Qiagen). The extracted DNA was fluorescently labeled, hybridized onto Nimblegen 385K array for S. cerevisiae, and scanned by Hokkaido System Science.
RESULTS
Inverted structures constructed in a yeast chromosome produced extra- and intra-chromosomal amplification
To examine the role of IRs in initiating gene amplification, we first constructed yeast strains that harbor IRs with amplification markers in chromosome VI (FR and FRFR haploid strains, Figure 1A, left). To effectively mimic inverted repeats formed during BFB cycles or found in eukaryotic genomes, we inferred that several kilobase-size fragments were required for the repeat structures. The repeat length of IRs is 3.1 kb, while the spacers between repeats are ∼3 kb in size. The amplification markers with truncated promoters, leu2d and trp1d, can complement leucine and tryptophan auxotrophy when amplified, respectively (16). Two strains were constructed. The FR strain has IRs with the leu2d marker between the repeats. The FRFR strain has two pairs of IRs; the pair on the centromeric side carries the leu2d marker, whereas the pair on the telomeric side carries the trp1d marker. There are no essential genes between the IRs and telomere (4.4 kb), so the genomic environment does not affect the rearrangement frequency.
The FRFR strain formed 4.3 ± 2.0 colonies per 106 plated cells on leucine/tryptophan-omitted plates, while the FR construct produced ∼8.0-fold fewer colonies under Leu- selection (5.4 colonies per 107 plated cells, Figure 1A, right). Next, the colonies from FRFR strain was analyzed by Southern hybridization of uncut DNA separated by pulse-field gel electrophoresis (PFGE, Figure 1B). Under non-selective conditions (NS, Leu+/Trp+ plates), the leu2d probe detects the modified chromosome VI (∼290-kb) in addition to chromosome III (∼360-kb) carrying leu2 fragments in the parental host strain, LS20 (14). In contrast, two types of products with increased signal intensities were seen under Leu-/Trp- selection. The samples with 30- to 40-kb leu2d products (green lanes) retained the modified chromosome VI (∼290-kb), suggesting that additional small chromosomes harboring leu2d accumulate extra-chromosomally in a cell (extra-chromosomal amplification). The samples marked in red showed strong leu2d signals above the separation limit and at the well position and lost the signals from the chromosome VI (∼290-kb), indicating extensive amplification within chromosome VI (intra-chromosomal amplification). The repeat structure also produced some variation of survivors. We assume that the clone 22 underwent gene conversion between the original leu2 on chromosome III and the leu2d gene in the constructed repeats. For clone 24, we speculate the extra-chromosomal amplification was accompanied by a subsequent deletion of the leu2d gene from chromosome VI.
Intra-chromosomal amplification by BFB cycles
To precisely map the amplified genomic segments (amplicons), we designed probes on the telomeric (YFR057W) and centromeric (RET2) side of the IRs to determine the co-amplification of the flanking genomic segments (Figure 1C and D). The extra-chromosomal products (green) exhibited signals with high intensity with the telomeric probe, indicating that the telomeric segment is co-amplified. In contrast, intra-chromosomal amplicons carried neither of probes at high intensity; the telomeric probe does not hybridize to any fragments, whereas weak signals were detected both at wells and at above the resolution limit with the centromeric probe. This suggests that the high-copy amplification in intra-chromosomal products would be limited within the segment flanked by two IRs.
To understand the mechanism underlying intra-chromosomal amplification, we used array comparative genome hybridization (CGH) and examined genome-wide copy number alterations (Figure 2A). From the genome-wide view, we noticed very high-level amplification of markers that are ectopically inserted between two IRs, leu2 (chr III), URA3 (chr V), LYS5 (chr VII) and ADH1 terminator (chr XV), along with the copy number gain of chromosome VI. By a closer look at chromosome VI, we noticed the terminal deletion between the IRs and the telomere, which is consistent with the results from Southern blotting. (Please note that any probes for chromosome VI do not hybridize with the ectopically-inserted metabolic markers.) Furthermore, the centromere-proximal regions showed copy number gains. The 350-kb region immediately centromeric to the IRs is amplified (green in Figure 2A, bottom), while the 500 kb region further centromeric to the amplified region suggests copy number gain in a small sub-population of cells (light green). The terminal deletion with the amplification of centromere-proximal segments has been attributed to palindromic duplication initiated by a chromosome break. Palindromic duplication of the rest of chromosome VI would generate a chromosome with two centromeres. Dicentric chromosome will cause a break during chromosome segregation, which could initiate another rearrangement. The centromeric boundary of the 350-kb region contains short palindromic sequences that potentially form a hairpin-capped end following a chromosome break under near-physiological conditions (Figure 2B). A chromosome with a hairpin-capped end becomes palindromic duplication after DNA replication. The repeated palindromic duplication within the chromosome VI strongly suggests the history of breakage-fusion-bridge (BFB) cycles. The lower copy number gain may result from an alternative or additional BFB event in a small subpopulation.
Figure 2.

An array CGH analysis of an intra-chromosomal product. (A) DNA from colony #17 was co-hybridized with DNA from the parental strain, LS20, to a S. cerevisiae CGH microarray. Genome-wide overview (top) and the enlarged chrVI (bottom) are shown. The FRFR structure is flanked by copy number gains and telomeric deletion. Note that TRP1 gene is deleted in the parental strain, LS20, so that no peak for the trp1d gene was detected. (B) A potential hairpin structure for chromatid fusion in BFB cycle. The dicentric chromosome is illustrated based on the array CGH profile. The 200 bp region around the boundary (red dotted line) contains a palindromic pair of sequences, which could form a hairpin structure (Tm = 32.2°C) after resection of the DSB end. The hairpin capped end and next round of DNA replication form a dicentric chromosome with copy number gain.
Very frequent recombination within amplicons indicates the involvement of rolling-circle replication
The markers located between IRs amplified at a much higher level than amplicons at chromosome VI. To understand the underlying mechanisms of additional amplification, we next analyzed the structure of amplified products in detail using Southern hybridization of BamHI-digested DNA (Figure 3A). Only one BamHI site is located within the IRs (Figure 3B, top), providing clear structural information.
Figure 3.

Characterization of amplified chromosomal structures. (A) Southern blot of BamHI-digested DNA with the leu2d probe. The samples in Figure 1B–D, are digested with BamHI. The samples marked in red and green indicate intra- and extra-chromosomal products, respectively. The sample marked in light gray showed no sign of amplification, suggesting Leu+ recombination between the leu2d marker and the mutated original leu2 allele on chr III. M: S. cerevisiae marker; P: the parental strain, LS20; NS: non-selective conditions. (B) A predicted process for intra-chromosomal amplification. The replication-based event forms a dicentric chromosome followed by BFB cycles, producing a chromosomal break that can initiate RCR. (C) Predicted structure of RCR-amplification products associated with intensive rearrangements. RCR-amplification could form an original repeat array that produces 24.4-kb BamHI-fragments. RCR-associated inversions and deletions produce rearranged BamHI-fragments (12.2, 15.3, 27.6, 36.8, 39.8 kb). IRs engaged in the inversions are marked with red bars. (D) A predicted process for extra-chromosomal amplification. The replication-based rearrangement event between IRs marked with a bracket forms a 43-kb extra-chromosome. The BamHI-map indicates a leu2d-containing fragment (black) and a non-hybridizing fragment (gray).
Unexpectedly, intra-chromosomal products showed several amplified BamHI fragments with distinct sizes (12, 15, 24, 28, 37, 40-kb), among which the 24 kb fragments are most commonly shared between clones (11/14 clones). Such variations of band pattern were observed in our previous amplification system based on rolling-circle replication (RCR; (11,12), and we demonstrated that RCR frequently induces recombination on yeast 2μ plasmids (13). From these facts, we hypothesize that a broken end generated during BFB cycles can initiate RCR by the invasion of repeat (a) into repeat (b) (Figure 3B). The resulting 24-kb repeat units contain a variety of direct or inverted repeats (Figure 3C). Recombination between repeats can cause deletion, duplication, or inversion, and thus the multimer of the 24-kb unit could be an ancestor of other amplified BamHI fragments.
As an example, an inversion between two distantly located repeats can create a 40-kb unit (Figure 3C, top right), and a deletion between two direct repeats crossing a BamHI site can create a 37-kb unit (top left). A 28-kb band can be efficiently produced by an inversion through nearby long homologous sequences in an inverted orientation (bottom left). Furthermore, another case of inversion between two distantly located repeats can create a 15-kb unit (bottom center), and a deletion occurring within the primary 24-kb unit can create a 12-kb unit (bottom right). This structural variation of amplicon among clones is also observed in RCR-amplified clones (12) and cancer cells (17), supporting the notion that the hyper-amplification and associated amplicon evolution are achieved by RCR following BFB cycles.
In clones 19 and 28, smaller BamHI fragments (15 and 12 kb) are more prominently amplified than the 24-kb fragment (Figure 3A). These clones underwent two independent events for leu2d and trp1d amplification. As shown in Supplementary Figure S1B and C, the invasion of repeat (a) into repeat (b) could generate 12-kb BamHI repeat units, and inversions across the units can generate 15-kb fragments. Independently of the leu2d amplification, linear (clone 19, described later) or circular (clone 28) extra-chromosomes were formed (Supplementary Figure S1B–D).
The majority of the extra-chromosomal amplicon (green) produces ∼23-kb BamHI fragments (Figure 3A). This product can arise through a U-turn of replication fork originated from the telomeric side (ARS610), generating 43-kb acentric mini-chromosomes. ((Figure 3D and Supplementary Figure S1E). The smaller (10 kb) BamHI fragment can arise from the 43-kb mini-chromosomes by the deletion between direct repeats (clone 24). Alternatively, a U-turn replication via the most-centromeric and most-telomeric repeats can produce the 30-kb extra-chromosomes (Supplementary Figure S1E, clone 3).
Complex inverted structures promote hyper-amplification process and shape amplicon structures in response to genetic selection
IRs are a potent trigger for the formation of dicentric chromosomes that can initiate BFB cycles, and thereafter hyper-amplification by RCR requires a direct repeat on the dicentric chromosome. Hence, forms of complex repeat structures could determine whether and how the hyper-amplification events proceed. To clarify the structural requirement for hyper-amplification, we constructed four additional strains (Figure 4A, left). The FFFR strain has both leu2d and trp1d markers, but leu2d is franked by direct repeats. Other three strains FRF, FFF and RRF only have the leu2d marker and three repeats. While FRF and RRF retain one pair of IR, three repeats are aligned as direct repeats in FFF.
These strains were selected for the amplification of leu2d and the structure of amplified leu2d and surrounding genomic regions were analyzed by PFGE-Southern analysis. First, the FFF strain produced few colonies in the plates lacking leucine, suggesting that at least one pair of IR is needed for amplification to occur efficiently (Figure 4A, right). Second, extra-chromosomal amplification was predominant and intra-chromosomal amplification became a minor class. In contrast to 54% (14/26) of colonies in FRFR strain, less than 23% of colonies harbored intra-chromosomal amplification: 5/26 colonies in FFFR, 4/35 colonies in FRF and 8/35 colonies in RRF strain (Figure 4B–E). The extra-chromosomal amplicons are small, <50 kb in size and have telomeric sequences, consistent with the linear dimer structure with telomere on both ends (Supplementary Figures S2 and S3A).
Intra-chromosomal amplicons are classified into three categories. First, the FRF strain showed hyper-amplification that generates 12- and 15-kb BamHI-fragments, strongly suggesting the involvement of RCR (Supplementary Figure S3B). Second, the FFF and RRF constructs produced intra-chromosomal amplification as light smear signals, indicating the lower stability of amplified regions. The 6-kb BamHI-fragment suggests that RCR formed the tandem repeats (Supplementary Figures S2A, S3C and D). The FFF would require a spontaneous chromosome break to initiate RCR, while the palindromic duplication by IRs in RRF promotes RCR initiation (Supplementary Figure S3C and S3D). Third, moderate level of intra-chromosomal amplification in FFFR showed heterogeneous patterns of BamHI-fragment and copy number gains of the centromeric region (RET2, Supplementary Figure S2C). This observation suggests that BFB cycles experienced fold-back priming at endogenous IRs in the centromeric side of inserted IRs. The BamHI-digestion patterns of intra-chromosomal FFFR products were distinct from those of RRF (Supplementary Figure S2A), although these strains have a structural similarity (direct repeats with a single IR). A key difference between these strains is the selection method; the FFFR strain was selected on Leu-/Trp- plates and the RRF on Leu- plates. Thus, complex repeat arrangements and genetic selection can generate a variety of amplicon. Furthermore, these results support the notion that multiple IRs facilitate efficient RCR and stable maintenance of amplified regions.
These repeat structures also produced other types of surviving colonies (Supplementary Figure S2D–G); probably through Leu+ conversion (gray lanes), moderate amplification via unequal sister chromatid exchange (blue lanes), and Leu+ recombination forming a fusion chromosome III-VI (yellow lanes). These strains, containing different sets of amplification markers, were selected in a different way; the RRF selected on Leu- plates and the FFFR on Leu-/Trp- plates. Thus, in response to the difference in genetic selection, complex inverted structures diversely shape amplicon structure.
Chromosomal rearrangements in the constructed repeats were independent of homologous recombination pathways
Similar types of amplification were produced in the systems we previously constructed, by using inducible chromosome breaks within repeats (11) or association between replication and inducible recombination (12). Additionally, other groups showed that fusions of nearby inverted repeats formed acentric and dicentric chromosomes, depending on homologous recombination proteins (3,5,18) or based on a model, faulty switching of replication templates (4,19,20). We here carried out genetic analysis to better understand molecular mechanisms for the amplification processes. We first examine the role of homologous recombination process, which engages in a wide range of genomic rearrangement between direct repeats, invited repeats and sister chromatids (21–23). Deletion of core homologous recombination (HR) factors, RAD52 or RAD51, had little impact on the frequency of colony formation (Figure 5A, green), but the products in the rad52 mutant were exclusively extra-chromosomal (Figure 5B). The lack of intra-chromosomal product suggests that Rad52 is required in any step in intra-chromosomal amplification, such as BFB cycles.
Complex inverted repeats were associated with replication stress
We next focused on another cause of chromosomal rearrangement, DNA replication stress. Repetitive sequences, such as triplet repeats and palindromic inverted Alu sequences, have been shown to stall replication forks (24–29). To monitor replication fork movement across the dispersed repeats with kilobase-sized spacers, we used two-dimensional (2D) agarose gel electrophoresis (15). There are two replication origins on each side of this region. The late-firing origin ARS609 is more efficient than the telomere-proximal origin ARS610 (30). The hybridization probe specifically detected the identical XhoI fragments from the FRFR and parental strain. The parental strain showed an even Y-arc signal, indicating that replication forks passed this segment without a problem (Figure 5C). In contrast, the large Y-arc was more intense than the small one in the FRFR strain, indicating replication stress within the FRFR repeats. Furthermore, the FFF strain also showed the accumulation of large-Y molecules (Figure S4A), suggesting that complex repeat structures, regardless of their orientation, could be associated with replication stress, leading to genome rearrangement or gene amplification.
Stalled forks are generally protected or stabilized by checkpoint proteins, mutations of which can cause collapsed forks and deleterious chromosomal rearrangements (4,19,31). Consistently, in the strain lacking a checkpoint kinase Chk1, we observed the 4-fold increase of colony formation (Figure 5A, blue). The distribution of intra- and extra-amplification was not affected by the CHK1 deletion (Figure 5B), suggesting that collapsed forks could become substrates for both types of amplification.
Repeat-mediated chromosomal rearrangements were suppressed by structure-specific nucleases, Mus81 and Rad27
Next, we tested the genes for processing stalled forks. One of the key molecules that can process stalled forks is a structure-specific nuclease, Mus81, which produces one-ended DNA breaks for a restart of productive replication (32–34). Surprisingly, the deletion of MUS81 dramatically enhanced the colony formation (∼70-fold), indicating Mus81p prevents the chromosomal rearrangement leading to amplification (Figure 5A, purple). Mus81 interacts with another structure-specific endonuclease, Rad27 (yeast FEN1) or Slx1-Slx4 (35–37). In our system, the mutation of RAD27 flap endonuclease dramatically increased Leu+/Trp+ colonies (∼60-fold), while slx1 mutation had no effect.
An E3 ubiquitin ligase, Rad18, is recruited to stalled forks and involved in other types of processing. Rad18 mono-ubiquitinates PCNA in response to fork stalling (38,39) and loads translesion synthesis (TLS) DNA polymerase η to stalled forks (40). Rad18 can also facilitate hemicatenane formation and promote gap filling in the lagging strand behind the replication fork (39,41). The ∼20-fold increase of colony formation in a rad18 mutant suggests that TLS pathway and/or hemicatenane formation contribute to suppression of the chromosomal rearrangements (Figure 5A, orange).
Based on these genetic analyses and earlier studies, we propose that reversal forks could be involved in the chromosomal rearrangements (Figure 5D). Indications of replication stress, such as fork slowdown or stalling, could accumulate positive supercoil ahead of the forks, and this can induce fork regression (42). Oncogene-induced replication stress can cause replication fork slowdown and induce fork reversal, which accumulates in MUS81-depleted cells (43). MUS81 depletion slows replication forks in human cells (44), and FEN1 is required for efficient re-initiation of stalled replication forks at telomeres in human cells (45). In our system, the deletion of MUS81 did not affect replication stress in FRFR repeats (Supplementary Figure S4A), inferring that Mus81p could act mainly in the processing of stalled forks and/or restarting forks in concert with Rad27p. Previously, Paek et al. proposed a model in which regressed forks cause chromosomal rearrangement mediated by endogenous long terminal repeat (LTR) sequences. Our model proposes that ectopic invasion from regressed forks generate acentric and dicentric chromosomes (Figure 5D and Supplementary Figure S4B).
The intra-chromosomal amplification relies on Rad52 and Mus81
The chromosome breaks formed in dicentric chromosomes would play important roles in initiating RCR. We here proposed that a resected broken end invades into a directly-oriented intra-chromosomal repeat and thereby initiates RCR, and tested the involvement of break-induced replication (BIR) in the RCR amplification. However, unexpectedly, a mutant of POL32, a subunit of DNA polymerase δ that is required for BIR, could produce intra-chromosomal amplification although the frequency of colony formation decreased moderately (Figure 5E and Supplementary Figure S5). The smear signals and small copy number gains in some Δpol32 colonies suggest that an efficient RCR-amplification requires Pol32 (Supplementary Figure S6, gray lanes). Consistently, previous studies showed Pol32-independent BIR can cause chromosomal rearrangements and Pol32 affects the efficiency (46,47). The distribution of amplification forms in this study indicates the dependency of RCR on Rad52 and Mus81 (Figure 5B and Supplementary Figure S5). Previously, the Pol32-independent rearrangement depends on recombination proteins including Rad52 (46,47). Mus81 is proposed to cleave D-loop after strand invasion to establish replication forks, and was shown to promote BIR (48). Collectively, it remains to be characterized how the broken end initiates RCR and which proteins are required for the process.
DISCUSSION
We here constructed complex inverted structures and thereby traced gene amplification events seen in tumorigenesis or drug-resistance; a palindromic duplication followed by BFB cycles, extra-/intra-chromosomal amplification, and hyper-amplification accompanied by intensive rearrangements (Figure 6).
Figure 6.

A schematic model for BFB–RCR amplification. Replication stress slows fork movement so that a part of replication forks can be regressed. If regressed forks are not rescued appropriately, ectopic DNA strand invasions can potentially result in dicentric chromosome formation. Dicentric chromosomes initiate BFB cycles, during which a chromosome break can trigger RCR hyper-amplification. RCR and its associated intensive rearrangements can generate a variety of amplicon according to a combination of pairs of repeats and genetic selection.
The RCR process is possible to highly amplify a genomic segment even in a single cell cycle, whereas BFB cycles require cell divisions and thus take a long time to achieve high-level amplification. This rapid and dynamic process can arise in a small population, providing heterogeneous hyper-amplified cells with amplicon variations. The genomic variation is the basis of intra-tumor heterogeneity, a key phenotype underlying tumor malignancy including metastasis, recurrence, and drug-resistance (49,50). It has been proposed that rolling-circle replication (RCR) possibly participates in amplification events (2,51). However, RCR is a difficult process to study due to the transient nature. To overcome this difficulty, we have developed inducible systems for RCR-type amplification in yeast or mammalian cells (11,12), and determined a characteristic feature of RCR, intensive rearrangement in amplified regions (13). In this study, the semi-natural inverted structure (not depending on inducible replication fork blocking or DNA double-strand breaks) was used as an effective approach to trace RCR-amplification and associated rearrangements, instead of analyzing complex end products formed endogenously. The semi-natural setting could cause replication stress, presenting the fork slowdown in the 2D analysis.
An elegant system of inducible fork blocking in S. pombe has been reported to produce acentric or dicentric chromosomes in concert with inverted repeats, depending on recombination proteins; Rhp51 (Rad51 in S. cerevisiae), Rad22 (Rad52), and Rhp54 (Rad54) (3,18). A tumor suppressor, BRCA1, is also engaged in homologous recombination at inducible fork stalling on a plasmid DNA in mammalian cells (52). In our genetic analysis, however, rad51/52 deletions did not affect the frequency of amplification, suggesting that the processes observed here would differ from those in the inducible fork-block systems. Aside from the acentric/dicentric chromosome formation, Rad52p and Mus81 would be required for processing chromosomal ends during BFB cycles or initiating RCR (Figure 5B).
Another line of research genetically analyzed chromosomal rearrangements between endogenous long terminal repeat (LTR) sequences, presenting a replication-based model, faulty template switching (4,19). The LTR-mediated rearrangements were enhanced in rad9, and rad51, and rad52 mutants, whereas the frequency of amplification in this study was reduced in the rad9 mutant (Figure 5A and Supplementary Figure S4C) or not affected by rad51/52 mutations (Figure 5A). The frequency of LTR-mediated rearrangements moderately increased with mus81 mutation, which dramatically enhanced the colony formation (∼70-fold) in our system. The differences in genetic response suggest a diversity of molecular mechanism in mediating the chromosome rearrangements.
We here proposed a fork regression model for the chromosomal rearrangements (Figure 5D). Indications of replication stress, such as fork slowdown or stalling, could accumulate positive supercoil ahead of the forks, inducing fork regression (42). Oncogene-induced replication stress can cause replication fork slowdown and induce fork reversal, which accumulates by MUS81 depletion (43). A point to be solved is the RAD51 dependency of the process. Recently, whereas RAD51 was shown to mediate fork regression in sub-lethal genotoxic drug treatments, it was shown that RAD51 depletion did not reduce fork regression under an untreated condition (53,54). Consistently, in our genetic analysis, the rad51 mutant did not affect the frequency of colony formation. These findings indicate that RAD51 dependency of natural fork regression in untreated cells still has to be determined. We also presented an alternative model associated with single-strand DNA (ssDNA) gaps behind a replication forks (Supplementary Figure S4B, bottom left). This process at uncoupled forks may support the independence from Rad51 (55). Alternatively, random stochastic DNA replication errors may generate the initial recombinogenic lesion. The genomic region that the complex repeats were inserted into is reported to be a very late-replicating region, prone to generate chromosomal breaks. If the breaks can be repaired by a RAD51-independent pathway, the orientation of repeats would affect recombination outcomes, leading to BFB cycles in the case of the inverted repeats.
Mus81 is a structure-specific endonuclease (56) with diverse functions, including replication fork restart (32,57,58) and the resolution of recombination intermediates (59,60). Mus81 plays important roles in processing stalled or slowed forks, and MUS81 depletion slow down replication forks and accumulate regressed forks in human cells (43,44). Mus81 can cleave 3′-flap structures formed by invading DNA strands of D-loop structure (61). We propose that Mus81 resolves regressed or uncoupled forks to facilitate fork restarts free from ectopic DNA-strand invasions (Figure 5D and Supplementary Figure S4B, red arrowheads). Mus81 complex physically and functionally interacts with Rad27 (yeast FEN1); mutations in MUS81 and RAD27 are synthetically lethal (62) and these two nucleases mutually stimulate each other (35). FEN1 is a structure-specific endonuclease that cleaves 5′-flap structures, which could be produced during DNA strand invasion (Figure 5D and Supplementary Figure S4B, red arrowheads). FEN1 participates not only in Okazaki fragment processing during lagging strand DNA replication (63) but also has gap endonuclease activity (64), which may act similarly to Mus81. These endonucleases would prevent complex inverted structures from causing regressed forks or ectopic template switching.
Surprisingly, mutation of Rad9, a DNA damaging checkpoint protein, reduced the colony formation (Supplementary Figure S4C). Previously, Rad9 is shown to suppress activities of some exonucleases, including Exo1, particular at telomere (65). Thus, Rad9 may suppress a pathway (e.g. Exo1) that faithfully resumes regressed or uncoupled forks (Supplementary Figure S4B), and instead, such unrepaired forks may be processed harmfully by Rad9-independent pathways. This hypothesis may explain the negative impact of Rad9 on the chromosome rearrangements presented here.
In this study, the array CGH data indicated the palindromic duplications were followed by BFB cycles. Previously we demonstrated that the fusion step of BFB is mediated by hairpin-capped ends, which are key intermediates for palindromic chromosomal rearrangements. The centromeric boundary of higher copy number gain in Figure 2 contains short palindromic sequences that potentially form hairpin-capped ends under near-physiological conditions (Figure 2B). BFB cycles provide an ideal structural feature for RCR initiation (Figure 6). The RCR process can be induced by repetitive elements, including long/short interspersed nuclear element (LINE/SINE), highly abundant in the human genome (66). By a combination of repeats and genetic selection, RCR can create a variety of amplicons, as shown in Figure 4. Repetitive elements further could mediate amplicon evolution through the associated rearrangements. Of the resulting genetic variation of amplicons, some may confer much higher resistance to chemotherapy or the other may acquire enhanced the ability of metastasis in cancer cells. Thus, the RCR process can be a central process that links between BFB cycles and complex end products and diversifies amplification products for cancer evolution.
Supplementary Material
ACKNOWLEDGEMENTS
We thank D. Butler (Montana State University-Billings) for providing a yeast strain, NIBB Core Research Facilities for technical support, and Dr Horiuchi's lab members for critical discussions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Japan Society for the Promotion of Science (JSPS), Grant-in-Aid for Young Scientists [21770194 to T.W.]; National Cancer Institute [R01CA149385 to H.T.]; Grant for Research Projects from Hayama Center for Advanced Studies (to T.H. and T.W.); Kanzawa Medical Research Foundation (to T.W.). Cedars-Sinai Medical Center (to H.T.); Funding for open access charge: Tokai University.
Conflict of interest statement. None declared.
REFERENCES
- 1. Debatisse M., Malfoy B.. Gene amplification mechanisms. Adv. Exp. Med. Biol. 2005; 570:343–361. [DOI] [PubMed] [Google Scholar]
- 2. Narayanan V., Mieczkowski P.A., Kim H.M., Petes T.D., Lobachev K.S.. The pattern of gene amplification is determined by the chromosomal location of hairpin-capped breaks. Cell. 2006; 125:1283–1296. [DOI] [PubMed] [Google Scholar]
- 3. Mizuno K., Lambert S., Baldacci G., Murray J.M., Carr A.M.. Nearby inverted repeats fuse to generate acentric and dicentric palindromic chromosomes by a replication template exchange mechanism. Genes Dev. 2009; 23:2876–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Paek A.L., Kaochar S., Jones H., Elezaby A., Shanks L., Weinert T.. Fusion of nearby inverted repeats by a replication-based mechanism leads to formation of dicentric and acentric chromosomes that cause genome instability in budding yeast. Genes Dev. 2009; 23:2861–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mizuno K., Miyabe I., Schalbetter S.A., Carr A.M., Murray J.M.. Recombination-restarted replication makes inverted chromosome fusions at inverted repeats. Nature. 2013; 493:246–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Deng S.K., Yin Y., Petes T.D., Symington L.S.. Mre11-Sae2 and RPA collaborate to prevent palindromic gene amplification. Mol. Cell. 2015; 60:500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tanaka H., Tapscott S.J., Trask B.J., Yao M.C.. Short inverted repeats initiate gene amplification through the formation of a large DNA palindrome in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:8772–8777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McClintock B. The stability of broken ends of chromosomes in Zea Mays. Genetics. 1941; 26:234–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Marotta M., Chen X., Watanabe T., Faber P.W., Diede S.J., Tapscott S., Tubbs R., Kondratova A., Stephens R., Tanaka H.. Homology-mediated end-capping as a primary step of sister chromatid fusion in the breakage-fusion-bridge cycles. Nucleic Acids Res. 2013; 41:9732–9740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marotta M., Onodera T., Johnson J., Budd G.T., Watanabe T., Cui X., Giuliano A.E., Niida A., Tanaka H.. Palindromic amplification of the ERBB2 oncogene in primary HER2-positive breast tumors. Sci. Rep. 2017; 7:41921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Watanabe T., Horiuchi T.. A novel gene amplification system in yeast based on double rolling-circle replication. EMBO J. 2005; 24:190–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Watanabe T., Tanabe H., Horiuchi T.. Gene amplification system based on double rolling-circle replication as a model for oncogene-type amplification. Nucleic Acids Res. 2011; 39:e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Okamoto H., Watanabe T.A., Horiuchi T.. Double rolling circle replication (DRCR) is recombinogenic. Genes Cells. 2011; 16:503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Butler D.K., Yasuda L.E., Yao M.C.. Induction of large DNA palindrome formation in yeast: implications for gene amplification and genome stability in eukaryotes. Cell. 1996; 87:1115–1122. [DOI] [PubMed] [Google Scholar]
- 15. Friedman K.L., Brewer B.J.. Analysis of replication intermediates by two-dimensional agarose gel electrophoresis. Methods Enzymol. 1995; 262:613–627. [DOI] [PubMed] [Google Scholar]
- 16. Erhart E., Hollenberg C.P.. The presence of a defective LEU2 gene on 2 mu DNA recombinant plasmids of Saccharomyces cerevisiae is responsible for curing and high copy number. J Bacteriol. 1983; 156:625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kawagoe H., Kandilci A., Kranenburg T.A., Grosveld G.C.. Overexpression of N-Myc rapidly causes acute myeloid leukemia in mice. Cancer Res. 2007; 67:10677–10685. [DOI] [PubMed] [Google Scholar]
- 18. Lambert S., Mizuno K., Blaisonneau J., Martineau S., Chanet R., Fréon K., Murray J.M., Carr A.M., Baldacci G.. Homologous recombination restarts blocked replication forks at the expense of genome rearrangements by template exchange. Mol. Cell. 2010; 39:346–359. [DOI] [PubMed] [Google Scholar]
- 19. Kaochar S., Shanks L., Weinert T.. Checkpoint genes and Exo1 regulate nearby inverted repeat fusions that form dicentric chromosomes in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:21605–21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Paek A.L., Jones H., Kaochar S., Weinert T.. The role of replication bypass pathways in dicentric chromosome formation in budding yeast. Genetics. 2010; 186:1161–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thomas B.J., Rothstein R.. The genetic control of direct-repeat recombination in Saccharomyces: the effect of rad52 and rad1 on mitotic recombination at GAL10, a transcriptionally regulated gene. Genetics. 1989; 123:725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bai Y., Symington L.S.. A Rad52 homolog is required for RAD51-independent mitotic recombination in Saccharomyces cerevisiae. Genes Dev. 1996; 10:2025–2037. [DOI] [PubMed] [Google Scholar]
- 23. Fasullo M., Giallanza P., Dong Z., Cera C., Bennett T.. Saccharomyces cerevisiae rad51 mutants are defective in DNA damage-associated sister chromatid exchanges but exhibit increased rates of homology-directed translocations. Genetics. 2001; 158:959–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Voineagu I., Narayanan V., Lobachev K.S., Mirkin S.M.. Replication stalling at unstable inverted repeats: interplay between DNA hairpins and fork stabilizing proteins. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:9936–9941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Y., Saini N., Sheng Z., Lobachev K.S.. Genome-wide screen reveals replication pathway for quasi-palindrome fragility dependent on homologous recombination. PLoS Genet. 2013; 9:e1003979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Viterbo D., Michoud G., Mosbach V., Dujon B., Richard G.F.. Replication stalling and heteroduplex formation within CAG/CTG trinucleotide repeats by mismatch repair. DNA Repair (Amst.). 2016; 42:94–106. [DOI] [PubMed] [Google Scholar]
- 27. Shishkin A.A., Voineagu I., Matera R., Cherng N., Chernet B.T., Krasilnikova M.M., Narayanan V., Lobachev K.S., Mirkin S.M.. Large-scale expansions of Friedreich's ataxia GAA repeats in yeast. Mol. Cell. 2009; 35:82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gerhardt J., Tomishima M.J., Zaninovic N., Colak D., Yan Z., Zhan Q., Rosenwaks Z., Jaffrey S.R., Schildkraut C.L.. The DNA replication program is altered at the FMR1 locus in fragile X embryonic stem cells. Mol. Cell. 2014; 53:19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Voineagu I., Surka C.F., Shishkin A.A., Krasilnikova M.M., Mirkin S.M.. Replisome stalling and stabilization at CGG repeats, which are responsible for chromosomal fragility. Nat. Struct. Mol. Biol. 2009; 16:226–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Siow C.C., Nieduszynska S.R., Muller C.A., Nieduszynski C.A.. OriDB, the DNA replication origin database updated and extended. Nucleic Acids Res. 2012; 40:D682–D686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lambert S., Carr A.M.. Impediments to replication fork movement: stabilisation, reactivation and genome instability. Chromosoma. 2013; 122:33–45. [DOI] [PubMed] [Google Scholar]
- 32. Hanada K., Budzowska M., Davies S.L., van Drunen E., Onizawa H., Beverloo H.B., Maas A., Essers J., Hickson I.D., Kanaar R.. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat. Struct. Mol. Biol. 2007; 14:1096–1104. [DOI] [PubMed] [Google Scholar]
- 33. Rass U. Resolving branched DNA intermediates with structure-specific nucleases during replication in eukaryotes. Chromosoma. 2013; 122:499–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Osman F., Whitby M.C.. Exploring the roles of Mus81-Eme1/Mms4 at perturbed replication forks. DNA Repair (Amst.). 2007; 6:1004–1017. [DOI] [PubMed] [Google Scholar]
- 35. Kang M.J., Lee C.H., Kang Y.H., Cho I.T., Nguyen T.A., Seo Y.S.. Genetic and functional interactions between Mus81-Mms4 and Rad27. Nucleic Acids Res. 2010; 38:7611–7625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thu H.P., Nguyen T.A., Munashingha P.R., Kwon B., Dao Van Q., Seo Y.S.. A physiological significance of the functional interaction between Mus81 and Rad27 in homologous recombination repair. Nucleic Acids Res. 2015; 43:1684–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wyatt H.D., Sarbajna S., Matos J., West S.C.. Coordinated actions of SLX1-SLX4 and MUS81-EME1 for Holliday junction resolution in human cells. Mol. Cell. 2013; 52:234–247. [DOI] [PubMed] [Google Scholar]
- 38. Kannouche P.L., Wing J., Lehmann A.R.. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell. 2004; 14:491–500. [DOI] [PubMed] [Google Scholar]
- 39. Branzei D., Szakal B.. DNA damage tolerance by recombination: Molecular pathways and DNA structures. DNA Repair (Amst.). 2016; 44:68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Watanabe K., Tateishi S., Kawasuji M., Tsurimoto T., Inoue H., Yamaizumi M.. Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 2004; 23:3886–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Branzei D., Vanoli F., Foiani M.. SUMOylation regulates Rad18-mediated template switch. Nature. 2008; 456:915–920. [DOI] [PubMed] [Google Scholar]
- 42. Olavarrieta L., Martínez-Robles M.L., Sogo J.M., Stasiak A., Hernández P., Krimer D.B., Schvartzman J.B.. Supercoiling, knotting and replication fork reversal in partially replicated plasmids. Nucleic Acids Res. 2002; 30:656–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Neelsen K.J., Zanini I.M., Herrador R., Lopes M.. Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. J. Cell Biol. 2013; 200:699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fu H., Martin M.M., Regairaz M., Huang L., You Y., Lin C.M., Ryan M., Kim R., Shimura T., Pommier Y. et al. The DNA repair endonuclease Mus81 facilitates fast DNA replication in the absence of exogenous damage. Nat. Commun. 2015; 6:6746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Saharia A., Teasley D.C., Duxin J.P., Dao B., Chiappinelli K.B., Stewart S.A.. FEN1 ensures telomere stability by facilitating replication fork re-initiation. J. Biol. Chem. 2010; 285:27057–27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ruiz J.F., Gomez-Gonzalez B., Aguilera A.. Chromosomal translocations caused by either pol32-dependent or pol32-independent triparental break-induced replication. Mol. Cell. Biol. 2009; 29:5441–5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Smith C.E., Lam A.F., Symington L.S.. Aberrant double-strand break repair resulting in half crossovers in mutants defective for Rad51 or the DNA polymerase delta complex. Mol. Cell. Biol. 2009; 29:1432–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pardo B., Aguilera A.. Complex chromosomal rearrangements mediated by break-induced replication involve structure-selective endonucleases. PLoS Genet. 2012; 8:e1002979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Burrell R.A., McGranahan N., Bartek J., Swanton C.. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013; 501:338–345. [DOI] [PubMed] [Google Scholar]
- 50. Yates L.R., Campbell P.J.. Evolution of the cancer genome. Nat. Rev. Genet. 2012; 13:795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Harada S., Sekiguchi N., Shimizu N.. Amplification of a plasmid bearing a mammalian replication initiation region in chromosomal and extrachromosomal contexts. Nucleic Acids Res. 2011; 39:958–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Willis N.A., Chandramouly G., Huang B., Kwok A., Follonier C., Deng C., Scully R.. BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. Nature. 2014; 510:556–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Neelsen K.J., Lopes M.. Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 2015; 16:207–220. [DOI] [PubMed] [Google Scholar]
- 54. Zellweger R., Dalcher D., Mutreja K., Berti M., Schmid J.A., Herrador R., Vindigni A., Lopes M.. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 2015; 208:563–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mott C., Symington L.S.. RAD51-independent inverted-repeat recombination by a strand-annealing mechanism. DNA Repair (Amst.). 2011; 10:408–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ciccia A., McDonald N., West S.C.. Structural and functional relationships of the XPF/MUS81 family of proteins. Annu. Rev. Biochem. 2008; 77:259–287. [DOI] [PubMed] [Google Scholar]
- 57. Fugger K., Chu W.K., Haahr P., Kousholt A.N., Beck H., Payne M.J., Hanada K., Hickson I.D., Sørensen C.S.. FBH1 co-operates with MUS81 in inducing DNA double-strand breaks and cell death following replication stress. Nat. Commun. 2013; 4:1423. [DOI] [PubMed] [Google Scholar]
- 58. Pepe A., West S.C.. MUS81-EME2 promotes replication fork restart. Cell Rep. 2014; 7:1048–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Matos J., Blanco M.G., Maslen S., Skehel J.M., West S.C.. Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell. 2011; 147:158–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Szakal B., Branzei D.. Premature Cdk1/Cdc5/Mus81 pathway activation induces aberrant replication and deleterious crossover. EMBO J. 2013; 32:1155–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pepe A., West S.C.. Substrate specificity of the MUS81-EME2 structure selective endonuclease. Nucleic Acids Res. 2014; 42:3833–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tong A.H., Evangelista M., Parsons A.B., Xu H., Bader G.D., Pagé N., Robinson M., Raghibizadeh S., Hogue C.W., Bussey H. et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001; 294:2364–2368. [DOI] [PubMed] [Google Scholar]
- 63. Balakrishnan L., Bambara R.A.. Flap endonuclease 1. Annu. Rev. Biochem. 2013; 82:119–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zheng L., Zhou M., Chai Q., Parrish J., Xue D., Patrick S.M., Turchi J.J., Yannone S.M., Chen D., Shen B.. Novel function of the flap endonuclease 1 complex in processing stalled DNA replication forks. EMBO Rep. 2005; 6:83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zubko M.K., Guillard S., Lydall D.. Exo1 and Rad24 differentially regulate generation of ssDNA at telomeres of Saccharomyces cerevisiae cdc13-1 mutants. Genetics. 2004; 168:103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cordaux R., Batzer M.A.. The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 2009; 10:691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.