


Abstract
Thymine DNA glycosylase (TDG) excises thymine from mutagenic G·T mispairs generated by deamination of 5-methylcytosine (mC) and it removes two mC derivatives, 5−formylcytosine (fC) and 5−carboxylcytosine (caC), in a multistep pathway for DNA demethylation. TDG is modified by small ubiquitin-like modifier (SUMO) proteins, but the impact of sumoylation on TDG activity is poorly defined and the functions of TDG sumoylation remain unclear. We determined the effect of TDG sumoylation, by SUMO-1 or SUMO-2, on substrate binding and catalytic parameters. Single turnover experiments reveal that sumoylation dramatically impairs TDG base-excision activity, such that G·T activity is reduced by ≥45-fold and fC and caC are excised slowly, with a reaction half-life of ≥9 min (37°C). Fluorescence anisotropy studies reveal that unmodified TDG binds tightly to G·fC and G·caC substrates, with dissociation constants in the low nanomolar range. While sumoylation of TDG weakens substrate binding, the residual affinity is substantial and is comparable to that of biochemically-characterized readers of fC and caC. Our findings raise the possibility that sumoylation enables TDG to function, at least transiently, as reader of fC and caC. Notably, sumoylation could potentially facilitate TDG recruitment of other proteins, including transcription factors or epigenetic regulators, to these sites in DNA.
INTRODUCTION
Thymine DNA glycosylase (TDG) initiates base excision repair (BER) by removing modified bases from DNA, including those resulting from deamination or oxidation of 5-methylcytosine (mC), and it has important functions in DNA repair and epigenetic regulation (1). TDG was discovered for its ability to excise thymine from G·T mispairs (2,3), as needed to preclude C→T transitions arising via mC deamination. It also functions in a multistep pathway for active DNA demethylation, which likely explains findings that depletion of TDG causes embryonic lethality in mice (4,5). The established DNA demethylation pathway begins with a ten-eleven translocation (TET) enzyme, followed by TDG and then BER (6). TET enzymes oxidize mC to give 5-hydroxymethylcytosine (hmC), 5-formylcytosine (fC) and 5-carboxylcytosine (caC); the latter two are excised by TDG and follow-on BER yields unmodified cytosine (7–13). Human TDG (410 residues) has a central catalytic domain (∼195 residues) flanked by two disordered regions that mediate various functions and are subject to post-translational modifications (PTMs) including acetylation, phosphorylation, ubiquitination, and SUMO (small ubiquitin-like modifier) conjugation (Figure 1A) (14–23). TDG also has a SUMO-interacting motif (SIM) that binds non-covalently to SUMO proteins, including conjugated (intramolecular) SUMO domains (Figure 1B), and SUMO–SIM binding likely mediates many of the effects of SUMO conjugation on TDG function (18,24–26). However, the impact of sumoylation on TDG activity is poorly defined and the functions of TDG sumoylation remain unclear. In the studies reported here, we investigated the effects of SUMO conjugation (sumoylation) on the biochemical activities of TDG.
Figure 1.

SUMO modification of TDG. (A) Primary structure of TDG including the catalytic domain and two flanking disordered regions that mediate various functions. Shown are post-translational modification (PTM) sites, the SUMO-interacting motif (SIM), and the PIP degron motif that enables TDG interaction with PCNA, ubiquitin modification, and degradation. The site(s) of ubiquitin modification is currently unknown. (B) Crystal structure (2.1 Å) of the TDG catalytic domain (residues 112–339) modified by SUMO-1 (green) (PDB ID: 1WYW); residues 301–331 of TDG (magenta) exhibit some secondary structure (β-strand, α-helix) for sumoylated TDG but are likely disordered in unmodified TDG. Because no structure is available for sumoylated TDG bound to DNA, the DNA shown here (orange) was positioned by aligning a structure (1.54 Å) of DNA-bound TDG (PDB ID: 5HF7, hidden) with that of TDG∼SUMO-1 (shown), using PyMol (RMSD = 0.573 Å for 136 Cα atoms). While this model cannot be accurate, because the SUMO-induced helix of TDG overlaps with the DNA, it is useful for illustrating the location of the SUMO–SIM interface relative to the DNA binding region and the active site.
It is informative to briefly review what is known about TDG sumoylation. In an initial study, Schär et al. showed (by western blots) that TDG is modified by SUMO proteins, in multiple types of human cells, and that recombinant TDG is modified similarly in cell extracts (18). Subsequent studies confirmed these findings and demonstrated sumoylation of TDG in vitro, using recombinant forms of the SUMO-activating (E1) and SUMO-conjugating (E2) enzymes and a SUMO protein, or by co-expression of these factors with TDG in bacterial cells (20,21,25,27–29). Sumoylation occurs exclusively at a single Lys residue in a consensus site (VKEE) (30) located in the disordered C-terminal region of TDG (Figure 1A). TDG is modified by SUMO-1, SUMO-2 or SUMO-3 (18,25,26); the latter two are virtually identical (referred to as SUMO-2/3) and they share ∼48% amino acid sequence identity with SUMO-1 (31). SUMO domains that are tethered to TDG can bind the SIM of the same TDG molecule via intramolecular SUMO·SIM binding, as shown by crystal structures of a sumoylated form of TDG that includes the catalytic domain and the SUMO modification site (residues 112–339) (Figure 1B) (25,26). Notably, the structures indicate that SUMO–SIM binding induces the formation of secondary structure (α-helix, β-strand) in a C-terminal region of TDG (D316-K330) that is otherwise likely to be disordered (19). While there are no reported structures of sumoylated TDG (TDG∼SUMO) bound to DNA, it has been proposed that the SUMO-induced α-helix could impair DNA binding (Figure 1B) (25,26). Although the crystal structures suggest a stable intramolecular SUMO–SIM complex, it is possible that SUMO–SIM binding is dynamic in aqueous solution, particularly for DNA-bound TDG∼SUMO and/or for sumoylated full-length TDG.
Initial studies reported that sumoylated TDG does not bind to DNA containing a G·T or G·U mispair, a G·C pair (nonspecific DNA), or even to a G·AP product site, suggesting that sumoylation abolishes detectible DNA binding (18,25,26). The effects on catalytic turnover (kcat) of TDG were also studied, with the finding that sumoylation completely abrogates detectible processing of G·T substrates while it enhances kcat for G·U substrates (18). Multiple groups had shown that TDG, like many DNA glycosylases, binds tightly to its AP-DNA product, severely impeding catalytic turnover in vitro (32–35). It was proposed that sumoylation regulates TDG product release and thereby enables efficient catalytic turnover (kcat). More specifically, the model holds that sumoylation occurs selectively for product-bound TDG and that SUMO is removed from TDG after product release to enable processing of other substrates, such that SUMO modification and deconjugation occurs for each catalytic cycle of TDG (18). While this paradigm seems to be broadly accepted (25,31,36–38), it is not directly supported by experimental evidence and remains to be substantiated. Moreover, the model has been challenged by recent findings (20,21), and many studies have shown that follow-on BER proteins, which act on glycosylase-generated AP sites, can dramatically enhance the catalytic turnover of TDG, as discussed below (21,39,40).
To understand the potential role(s) of TDG sumoylation it is important to define the effects of SUMO modification on discrete activities of TDG. While the general perception may be that much is known about how sumoylation impacts TDG activity, in fact, the opposite is true. For example, in previous studies the kinetics experiments were performed under multiple turnover conditions, often without a determination of Km (hence with unknown [S]/Km) (18,28,29). As such, the reported rate constants were influenced by many reaction steps, including enzyme-substrate association, product release, and product inhibition (Figure 2) and are therefore of limited value for understanding the effect of sumoylation on TDG activity. To date, no studies have examined the impact of sumoylation on TDG glycosylase activity using single turnover experiments, which yield the maximal rate of base excision when collected under saturating enzyme conditions (41). In addition, previous studies into the impact of TDG sumoylation on DNA binding were performed with qualitative methods (electrophoretic mobility shift assay or EMSA) and a truncated form of the enzyme (TDG112–339) that lacks the N-terminal region, which has been shown to enhance DNA binding (16,28,42,43) and might interact with the SUMO domain of modified TDG (28). No prior studies have used a quantitative method that yields a dissociation constant (Kd) or used full-length TDG that is uniformly modified with a single SUMO isoform (e.g., SUMO-1 only). Thus, the effect of sumoylation on two critical phases of the TDG reaction, substrate binding and base excision, remain unknown for G·T substrates. Moreover, the effect of sumoylation on TDG activity for the epigenetic substrates G·fC and G·caC has not been investigated. We addressed these problems in the studies described below.
Figure 2.

Minimal kinetic mechanism for the TDG reaction. Association of TDG (E) and DNA substrate (S) gives a collision complex (E·S), and the reversible nucleotide flipping step (Kflip), involving conformational changes in E and S, gives the reactive enzyme-substrate complex (E′·S′). Cleavage of the N-glycosyl bond and addition of the (water) nucleophile in the chemical step (kchem) gives the ternary product complex (E′·B·P′). Dissociation of E′·B·P′ likely involves rapid release of the excised base (B) and much slower release (kdis) of abasic DNA (P). TDG is severely inhibited by abasic DNA but does not bind to the nucleobases it removes from DNA (51). Solid lines denote reaction steps that contribute to enzymatic rate constants (kmax, kcat), and the dissociation constant (Kd) for the reactive (E′·S′) complex. Note that catalytic turnover (kcat) is influenced by multiple steps, including dissociation of the initial product complex (kdis) and subsequent product inhibition.
MATERIALS AND METHODS
Materials
Full length human TDG (410 residues) and the N140A variant (TDGN140A) (44) were expressed in Escherichia coli growing in Terrific Broth (TB) and purified as described (45). TDG (or TDGN140A) conjugated by SUMO-1 or by SUMO-2 was produced in E. coli that was co-transformed with two expression plasmids, one for TDG (or TDGN140A) and another for human SUMO-1 or SUMO-2 (mature form) together with the activating E1 (SAE1-SAE2) and conjugating E2 (Ubc9) enzymes that mediate sumoylation (46), as described (20). Cells were grown in TB and expression was induced with 0.4 mM IPTG at 22°C for ∼16 h; sumoylated TDG was purified as described for unmodified TDG (45), with the final chromatographic step performed using a Mono Q anion exchange column (GE Healthcare). Protein preparations were >99% pure as determined by SDS-PAGE. The SUMO-conjugated protein was free of detectible unmodified protein, as shown by Western blots (Supplementary Figure S1). Purified protein was flash frozen and stored at −80°C. Protein concentration was determined by absorbance at 280 nm (47), using extinction coefficients of ϵ280 = 31.5 mM−1cm−1 for TDG (48), ϵ280 = 37.8 mM−1cm−1 for TDG∼SUMO-1 and ϵ280 = 34.8 mM−1cm−1 for TDG∼SUMO-2; ϵ280 is unchanged by the N140A mutation.
Standard oligodeoxynucleotides (ODNs) were obtained from IDT. ODNs that contained fC, caC or fluorescein (FAM) were synthesized by the Keck Foundation Biotechnology Research Laboratory at Yale University. ODNs containing 2′-fluoroarabinodeoxythymidine (TF) were also synthesized at Yale using a phosphoramidite from Link Technologies (44). The subtle 2′-F substitution precludes TDG cleavage of dT, dU, 5-formyl-dC and 5-carboxyl-dC, due to transition-state destabilization (44,49–51). Crystal structures show that the 2′-F analogs of dU, 5-formyl-dC, and 5-carboxyl-dC flip completely into the TDG active site, forming key E·S interactions (43,50–52). ODNs modified with a 5′ sulforhodamine (Texas Red, or TR) were synthesized by Midland Certified Reagent Company (Midland, TX). ODNs were purified by reverse phase HPLC as described (53), and purity was confirmed by denaturing anion-exchange HPLC (54). Purified ODNs were exchanged into 0.02 M Tris–HCl pH 7.5, 0.04 M NaCl, and their concentration was determined by absorbance (45). Duplex DNA included a 28mer target strand, 5′-GTG TCA CCA CTG CTC AxG TAC AGA GCT G-3′, where x is the target base (fC, caC, T, U, TF), and a complementary strand, 5′-CAG CTC TGT ACG TGA GCA GTG GTG ACA C-3′, such that the target base (x) is paired with G and located in a CpG context (underlined). FAM and TR labels were attached to the complementary strand.
Glycosylase assays
Glycosylase (base excision) activity of sumoylated and unmodified TDG was determined using kinetic experiments performed using either single turnover conditions (saturating enzyme) to obtain the maximal rate of base excision (kmax), or multiple turnover experiments (saturating substrate) to obtain the maximal rate of catalytic turnover (kcat) (21,41,55). Assays were performed at 37°C in HEN.1 buffer (0.02 M HEPES pH 7.5, 0.1 M NaCl, 0.2 mM EDTA) with 0.1 mg/ml BSA. The 28 bp duplex DNA substrates contained a single G·fC, G·caC or G·T base pair. Single turnover assays included 0.5 μM DNA substrate and 1.5 μM enzyme, while multiple turnover assays used 1.0 μM DNA substrate and 0.05 μM enzyme. Kinetics reactions were initiated by adding concentrated enzyme to substrate in reaction buffer, then aliquots were removed at selected time points, rapidly quenched with 3× quench buffer (0.3 M NaOH, 0.03 M EDTA), and heated at 85°C for 3 min to quantitatively cleave the target DNA strand at TDG-generated abasic sites. The resulting DNA fragments were resolved by denaturing anion-exchange HPLC, using a DNAPac PA200 column (Thermo Fisher), and peak areas were used to determine fraction product (54). For single turnover experiments, progress curves (fraction product versus time) were fitted by non-linear regression to Equation (1) using Grafit5 (56).
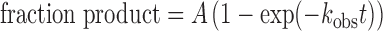 |
(1) |
where A is the amplitude, kobs is the rate constant, and t is reaction time. Experiments were performed with saturating enzyme ([E] >> Kd; [E] > [S]) such that the observed rate constant approximates the maximal rate of product formation (kobs ≈ kmax) and is not influenced by enzyme–substrate association or by product release or product inhibition (41). Saturating conditions were confirmed by observation of identical rate constants for experiments performed at other enzyme concentrations. For multiple turnover experiments, the linear (steady-state) portion of the progress curve was used to obtain the initial velocity (v0). Under these saturating substrate conditions ([S] ≫ Km), the Michaelis-Menten equation simplifies such that v0 approximates the maximal rate of catalytic turnover (kcat = v0/[E]).
Equilibrium binding monitored by fluorescence anisotropy
Fluorescence anisotropy experiments were used to monitor binding of enzyme (TDGN140A or sumoylated derivatives) to DNA containing a single G·fC, G·caC or G·TF pair and Texas Red (TR) at the 5′-end of the non-target strand (48,57). Individual samples (125 μl) contained DNA at a final concentration of 1 nM (G·fC, G·caC) or 10 nM (G·TF) and a varying enzyme concentration (0.01 nM to 10 μM) in binding buffer (20 mM HEPES pH 7.5, 0.1 M NaCl, 0.2 mM EDTA, 0.1 mg/ml BSA). Samples were equilibrated at 22°C for 1 h and then transferred to a quartz cuvette (Starna Cells). Anisotropy data were collected using a PTI Quantamaster 40 spectrofluorometer configured in T format, where one of the two PMTs is connected directly to the sample compartment, with no monochromator, and with wavelength selection provided by a 628-nm band pass filter (Semrock, Inc.) (48,57). The excitation wavelength was 590 nm (6-nm band pass), and the single-emission monochromator was set to 615 nm (8-nm band pass). Wavelength selection of the excitation and emission monochromators was enhanced by 586 nm and 624 nm band-pass filters, respectively (Semrock, Inc.). Dissociation constants (Kd) for enzyme-DNA complexes were determined by fitting the anisotropy data to binding models using DynaFit 4 (58,59); a representative DynaFit script is provided in the Supplementary Information. We previously described the rationale and benefits of using Dynafit for fitting anisotropy data in this system (TDG binding DNA) (48). The reported parameters were derived from global fitting of at least three independent experiments. The fitted parameters include the dissociation constant for binding of one enzyme subunit to one specific site of the DNA (Kd) and binding of a second TDG subunit to a nonspecific site (Kd2) at higher enzyme concentrations. Fitted parameters also include the anisotropy values for free DNA (rD) and for 1:1 and 2:1 complexes with enzyme (rED, rEED). As described in the figure legends, selected parameters were in some cases constrained to a fixed value if needed for proper fitting of the other parameters.
RESULTS
Effect of sumoylation on the rate of base excision by TDG
As noted above, previous studies of how sumoylation impacts the activity of TDG were limited to multiple turnover experiments, which give little insight into the effects on discrete elements of the enzymatic reaction. Moreover, the effect of sumoylation on TDG activity has been investigated for G·T and G·U mispairs but not for G·fC and G·caC substrates. Additionally, it is unknown whether conjugation by different SUMO isoforms might exert differential effects on the glycosylase (base-excision) activity of TDG and whether this might be substrate dependent. Unlike previous studies, we used single turnover kinetics under saturating enzyme conditions to obtain rate constants that approximate the maximal rate of base excision (kobs ≈ kmax) and are not influenced by enzyme-substrate association or steps after base excision (Figure 2). The experiments were performed using unmodified TDG or TDG modified uniformly by SUMO-1 or SUMO-2 (Figure 3, Table 1).
Figure 3.

Single turnover kinetics experiments (37°C) give the maximal glycosylase (base excision) activity of TDG∼SUMO-1 (squares) and TDG∼SUMO-2 (triangles). (A) Activity of sumoylated TDG (5 uM) and G·T substrate (1 uM); fitting to Equation (1) gives kmax = 0.020 ± 0.001 min−1 for TDG∼SUMO-1 and kmax = 0.019 ± 0.001 min −1 for TDG∼SUMO-2. (B) Activity of sumoylated TDG (1.5 uM) and G·fC substrate (0.5 uM); kmax = 0.079 ± 0.007 min−1 for TDG∼SUMO-1 and kmax= 0.078 ± 0.008 min−1 for TDG∼SUMO-2. (C) Sumoylated TDG (1.5 uM) acting on a G·caC substrate (0.5 uM); kmax= 0.069 ± 0.002 min−1 for TDG∼SUMO-1 and kmax = 0.074 ± 0.003 min−1 for TDG∼SUMO-2.
Table 1. Glycosylase activity for unmodified and sumoylated TDG.
| Enzyme | Substrate | k max (min−1) | Fold change in kmax | t 1/2 (min) |
|---|---|---|---|---|
| TDG | G·T | 0.89 ± 0.03 | - | 0.78 |
| TDG∼SUMO-1 | G·T | 0.020 ± 0.001 | 1/45 | 35 |
| TDG∼SUMO-2 | G·T | 0.019 ± 0.001 | 1/47 | 36 |
| TDG | G·fC | 1.69 ± 0.05 | - | 0.41 |
| TDG∼SUMO-1 | G·fC | 0.079 ± 0.007 | 1/21 | 8.7 |
| TDG∼SUMO-2 | G·fC | 0.078 ± 0.008 | 1/22 | 8.8 |
| TDG | G·caC | 0.50 ± 0.02 | - | 1.4 |
| TDG∼SUMO-1 | G·caC | 0.069 ± 0.002 | 1/7.2 | 10 |
| TDG∼SUMO-2 | G·caC | 0.074 ± 0.003 | 1/6.8 | 9.3 |
Fold change kmax gives the ratio of kmax values for unmodified and SUMO-modified TDG (kmaxTDG∼SUMO / kmaxTDG).
The largest effect of sumoylation on TDG base-excision activity is observed for G·T mispairs, where kmax is reduced by 45-fold and 47-fold upon modification by SUMO-1 or SUMO-2, respectively (Figure 3A). Turning to the epigenetic substrates, sumoylation also greatly impairs TDG glycosylase activity for G·fC pairs, where kmax is reduced 21- and 22-fold upon modification by SUMO-1 or SUMO-2, respectively (Figure 3B). Thus, sumoylation increases the reaction half-life from 25 s to 9 min. Sumoylation also exerts a large effect on TDG excision of caC, where modification with either SUMO-1 or SUMO-2 causes a 7-fold reduction in kmax (Figure 3C). Thus, the half-life for TDG excision of caC increases from 1.4 to 10 min. Notably, we find that sumoylated TDG exhibits nearly equivalent activity (kmax) for fC and caC substrates, while unmodified TDG has three-fold higher activity for fC relative to caC. It is also notable that for each substrate examined, the impact of sumoylation on TDG activity is nearly equivalent for the two different SUMO isoforms.
Sumoylation has little effect on TDG catalytic turnover for fC and caC substrates
The effect of sumoylation on the catalytic turnover (kcat) of TDG has been investigated for G·U and G·T (18,28,29) but not for G·fC and G·caC substrates. We performed multiple turnover experiments using a saturating (1.0 uM) concentration of G·fC or G·caC substrate and a limiting (0.05 uM) concentration of enzyme (TDG, TDG∼SUMO-1 or TDG∼SUMO-2) (Figure 4, Table 2). Under these conditions, the observed steady-state velocity (vo) approximates the maximal level (vo ≈ vmax) and yields the maximal catalytic turnover (kcat = vmax/[E]). For unmodified TDG, catalytic turnover is vastly lower than the maximal rate of base excision (kcat << kmax) for G·fC and G·caC substrates (Table 2), indicating that kcat is severely limited by steps after base excision, which could include product release and/or product inhibition. Previous studies reported similar results (kcat << kmax) for TDG acting on other substrates (including G·T, G·U) (21,33,35). Modification of TDG by SUMO-1 or SUMO-2 gives a 1.4-fold elevation in kcat for G·fC but has no impact for G·caC substrates. Previous studies reported that modification by SUMO-1 generated kcat enhancements of 2-fold and 4-fold for G·T and G·U substrates, respectively (29). As noted above, kcat reports on multiple steps of the TDG reaction, including nucleotide flipping, base excision, and product release and product inhibition (Figure 2). Observation that sumoylation greatly reduces kmax (Table 1) but has little effect on kcat seems likely to reflect offsetting effects on different steps of the reaction. For example, the impairment in base excision (kmax) could be offset by faster product release and/or diminished product inhibition, such that kcat is similar for sumoylated and unmodified TDG.
Figure 4.

Multiple turnover kinetics experiments for unmodified and sumoylated TDG (50 nM) acting on G·fC or G·caC substrates (1000 nM), performed at 37°C. (A) G·fC; linear fitting of data in the steady-state region gives velocities of v0 = 0.236 ± 0.011 nM·min−1 for TDG, v0 = 0.340 ± 0.036 nM·min−1 for TDG∼SUMO-1, and v0 = 0.315 ± 0.022 nM·min−1 for TDG∼SUMO-2. (B) G·caC; linear fitting of data in the steady-state region gives velocities of v0 = 0.363 ± 0.023 nM·min−1 for TDG, v0 = 0.356 ± 0.040 nM·min−1 for TDG∼SUMO-1, and v0 = 0.354 ± 0.037 nM·min−1 for TDG∼SUMO-2.
Table 2. Steady-state catalytic activity for unmodified and sumoylated TDG.
| Enzyme | Substrate | v o (nM·min−1) | k cat (min−1) | k max/kcat |
|---|---|---|---|---|
| TDG | G·fC | 0.236 ± 0.011 | (4.7 ± 0.3) × 10−3 | 359 |
| TDG∼SUMO-1 | G·fC | 0.340 ± 0.036 | (6.8 ± 0.7) × 10−3 | 12 |
| TDG∼SUMO-2 | G·fC | 0.315 ± 0.022 | (6.3 ± 0.5) × 10−3 | 12 |
| TDG | G·caC | 0.363 ± 0.023 | (7.3 ± 0.5) × 10−3 | 68 |
| TDG∼SUMO-1 | G·caC | 0.356 ± 0.040 | (7.1 ± 0.8) × 10−3 | 10 |
| TDG∼SUMO-2 | G·caC | 0.354 ± 0.037 | (7.1 ± 0.8) × 10−3 | 10 |
Effect of sumoylation on TDG substrate binding
While dissociation constants (Kd) were reported for unmodified TDG binding to G·T and G·U mispairs (48,57), binding to G·fC and G·caC has only been studied for the TDG catalytic domain (residues 111–308) (50), which lacks N-terminal residues that can greatly enhance substrate binding (28,43,50,60). Regarding sumoylated TDG, binding has been studied only qualitatively for G·T mispairs and not at all for G·fC or G·caC pairs. Accordingly, we used fluorescence anisotropy experiments to define the Kd for TDG and TDG∼SUMO binding to three different substrates (G·T, G·fC, G·caC) (48,57). The studies were performed using N140A-TDG (TDGN140A), a variant that binds substrates like wild-type TDG but has greatly reduced catalytic activity due likely to disrupted binding of the water nucleophile (44,50–52,61). Indeed, TDGN140A has no detectable activity on G·T substrates (over 48 h) (44) and has exceedingly slow activity for G·fC (kobs = 4 × 10−5 min−1; t1/2 = 329 h) (51) and G·caC substrates (kobs = 6 × 10−6 min−1; t1/2 = 1977 h) (Supplementary Figure S2). As such, enzyme-substrate binding experiments (<3 h) can readily be performed in the absence of base excision. Many previous studies show that TDG binds DNA with a stoichiometry of 1:1 or 2:1 (TDG:DNA), where a single TDG subunit binds a specific site of the DNA (e.g. a G·fC pair) and a second subunit binds to a non-specific site as the concentration of TDG increases (48,57,62). However, previous studies show that 1:1 binding is fully sufficient for catalysis and 1:1 complexes are observed in recent crystal structures, even though the DNA is long enough to accommodate two TDG subunits (48,51,53,63). Thus, our focus is on the affinity of TDG for binding the target site (G·T, G·fC, G·caC), that is, the Kd for 1:1 binding.
We first consider binding of unmodified TDGN140A to a G·TF mispair, where TF (2′-fluoroarabino-dT) is a dT analogue that is not cleaved by TDG and was used in our previous studies for unmodified TDG (48). We find that unmodified TDGN140A binds to a G·TF site with Kd = 16 ± 5 nM (Figure 5, Table 3). Notably, this is equivalent to the value determined previously for wild-type TDG binding to the same G·TF DNA (Kd = 18 ± 3 nM) (48), indicating that the N140A mutation does not substantially alter substrate binding, as previously reported (44). Regarding the impact of sumoylation, TDG affinity for G·T mispairs is weakened by 6- and 7-fold upon modification by SUMO-1 or SUMO-2, respectively (Figure 5, Supplementary Figure S3). While the impact of sumoylation on G·T binding is substantial, the impact on glycosylase activity (kmax) is much larger (≥45-fold; Table 1). We next consider the affinity of TDGN140A and its sumoylated derivatives for binding to DNA containing a single G·fC or G·caC pair. TDGN140A binds with high affinity to DNA containing a G·fC site (Kd = 4.0 ± 0.8 nM), and this binding is weakened by 8- or 9-fold upon modification of TDG by SUMO-1 or SUMO-2, respectively (Figure 6, Supplementary Figure S4). Unmodified TDGN140A binds with very high affinity to DNA containing a G·caC pair (Kd = 2.3 ± 0.6 nM), and sumoylation by either SUMO-1 or SUMO-2 weakens this binding by ∼6-fold (Figure 7, Supplementary Figure S5).
Figure 5.

Equilibrium binding of unmodified and sumoylated TDGN140A to G·TF DNA (10 nM) monitored by fluorescence anisotropy. (A) Data for TDGN140A binding to G·TF DNA, fitted to a two-site model (using Dynafit), gives Kd = 16 ± 5 nM and Kd2 = 1800 ± 2000 nM, and anisotropy values of rD = 0.184 ± 0.002, rED = 0.253 ± 0.007, rEED = 0.292 ± 0.014. (B) Binding of TDGN140A∼SUMO-1 to G·TF DNA yields Kd = 95 ± 21 nM, Kd2 = 186 ± 68 nM, rD = 0.183 ± 0.002, rED = 0.249 (fixed), and rEED = 0.281 ± 0.003. (C) Binding of TDGN140A∼SUMO-2 to G·TF DNA, fitted to a two-site model, yields Kd = 118 ± 22 nM, Kd2 = 135 ± 38 nM, rD = 0.190 ± 0.001, rD = 0.255 (fixed), and rEED = 0.285 ± 0.002. Fitting of the TDG∼SUMO-1 and TDG∼SUMO-2 data was performed using a fixed value for rED (anisotropy of 1:1 complex); fitting this parameter yielded unreasonably high values. The value used was determined using the relationship rED = rD + Δr1:1ave, where rD was fitted as noted above (B, C) and Δr1:1ave is the average anisotropy change associated with 1:1 binding (Δr1:1 = rED – rD) observed for TDG binding to three substrates (G·T, G·fC and G·caC).
Table 3. Dissociation constants for enzyme-substrate complexes.
| Enzyme | Substrate | K d (nM) | Fold change in Kd |
|---|---|---|---|
| TDG | G·TF | 16 ± 5 | - |
| TDG∼SUMO-1 | G·TF | 95 ± 21 | 5.9 |
| TDG∼SUMO-2 | G·TF | 118 ± 22 | 7.4 |
| TDG | G·fC | 4.0 ± 0.8 | - |
| TDG∼SUMO-1 | G·fC | 30 ± 4 | 7.5 |
| TDG∼SUMO-2 | G·fC | 36 ± 3 | 9.0 |
| TDG | G·caC | 2.3 ± 0.6 | - |
| TDG∼SUMO-1 | G·caC | 14 ± 2 | 6.1 |
| TDG∼SUMO-2 | G·caC | 13 ± 2 | 5.7 |
All of the equilibrium binding studies were performed using the N140A variant of TDG.
Figure 6.

Equilibrium binding of TDGN140A, unmodified and sumoylated, to G·fC DNA (1 nM) monitored by fluorescence anisotropy. (A) Data for TDGN140A binding to G·fC DNA, fitted to a two-site model gives Kd = 4.0 ± 0.8 nM and Kd2 = 1190 ± 370 nM, and anisotropy values of rD = 0.185 ± 0.002, rED = 0.248 ± 0.003, rEED = 0.293 ± 0.003. (B) Binding of TDGN140A∼SUMO-1 to G·fC DNA yields Kd = 30 ± 4 nM, Kd2 = 560 ± 130 nM, rD = 0.186 ± 0.002, rED = 0.252 (fixed), and rEED = 0.283 (fixed). (C) Binding of TDGN140A∼SUMO-2 to G·fC DNA, fitted to a two-site model, yields Kd = 36 ± 3 nM, Kd2 = 2370 ± 490 nM, rD = 0.187 ± 0.002, rED = 0.253 (fixed) and rEED = 0.284 (fixed). Fitting of TDG∼SUMO-1 and -2 data was performed using a fixed value for rED and rEED (anisotropy of 1:1 and 2:1 complexes, respectively), which were otherwise poorly fitted. The values used for rED were determined using the relationship rED = rD + Δr1:1ave, as described above (Figure 5). The rEED value was determined using the relationship rEED = rD + Δr2:1ave, where Δr2:1ave is the average total anisotropy change associated with forming 2:1 complex TDG∼SUMO-1 and -2 binding to G·T DNA.
Figure 7.

Equilibrium binding of TDGN140A, unmodified and sumoylated, to G·caC DNA (1 nM) monitored by fluorescence anisotropy. (A) Data for TDGN140A binding to G·caC DNA, fitted to a two-site model (using Dynafit), gives Kd = 2.3 ± 0.6 nM, Kd2 = 820 ± 500 nM, rD= 0.184 ± 0.0033, rED = 0.251 ± 0.004, and rEED = 0.284 ± 0.004. (B) Binding of TDGN140A∼SUMO-1 to G·caC DNA fitted to a two-site model (black line) gives Kd = 14 ± 2 nM, Kd2 = 24000 ± 16000 nM, rD= 0.189 ± 0.002, rED = 0.255 (fixed), and rEED = 0.285 (fixed). Fitting to a one site model (red line) yields essentially the same affinity, Kd = 16 ± 3 nM, with rD = 0.190 ± 0.002, and rED = 0.257 ± 0.002. (C) Binding of TDGN140A∼SUMO-2 to G·caC DNA fitted to a two-site model (black line) gives Kd = 13 ± 2 nM, Kd2 = 5000 ± 2000 nM, rD = 0.185 ± 0.003, rED = 0.251 (fixed), and rEED = 0.281 (fixed). Fitting to a one site model (red line) yields Kd = 20 ± 3 nM, with rD = 0.187 ± 0.002, and rED = 0.260 ± 0.002. Fitting for TDG∼SUMO-1 and -2 employed fixed values for rED and rEED, using the same approach described for fitting binding of these enzymes to G·fC DNA (Figure 6).
DISCUSSION
Unmodified TDG binds tightly to G·fC and G·caC pairs in DNA
While our studies focus largely on the effects of sumoylation on TDG activity, we also report that unmodified TDG possess high affinity for G·fC and G·caC pairs (in a CpG context), with Kd of 4.0 nM and 2.3 nM, respectively (Table 3). By comparison, TDG has greater affinity for G·U mispairs (Kd = 0.6 nM) but lower affinity for G·T mispairs (Kd = 16 nM), an unmodified CpG site (Kd = 63 nM), and nonspecific DNA (Kd = ∼300 nM) (48). Thus, TDG has high specificity for G·fC and G·caC pairs, binding these sites with ΔΔGbind of 2.5 kcal/mol and 2.9 kcal/mol, respectively, compared to nonspecific DNA (or ΔΔGbind of 1.6 kcal/mol and 2.0 kcal/mol relative to an unmodified CpG site). Because fC and caC impart only small, localized changes to DNA structure (64,65), the specificity of TDG for these sites is likely explained largely by specific interactions that it forms with the formyl and carboxyl groups, as observed in crystal structures (50,51). Although the affinity of TDG for G·hmC pairs has not been reported, evidence that TDG does not form specific interactions with the hydroxyl of hmC is provided by findings that the TDG catalytic domain (residues 111–308; TDG111–308) lacks detectable affinity for G·hmC (or G·mC) pairs while it binds specifically to G·fC (Kd 130 nM) and G·caC (Kd 70 nM) pairs (50). Importantly, our results reveal that the affinity of TDG for G·fC and G·caC pairs is much tighter than that reported in two previous studies. One study reported that TDG binds G·fC and G·caC with Kd values that are weaker by 11- and 41-fold, respectively, compared to those reported here (40). Another study, which used the catalytic domain (TDG111–308) (50), reported Kd values for G·fC and G·caC that are 33- and 30-fold weaker than those reported here for full-length TDG. This latter discrepancy could indicate that some portion of the disordered N-terminal region, which is absent in TDG111–308, enhances TDG binding to G·fC and G·caC pairs, as demonstrated for G·T mispairs (28,43).
Impact of TDG sumoylation on substrate binding and glycosylase activity
Our findings reveal the quantitative effect of TDG sumoylation on two key catalytic parameters, substrate binding (Kd) and the maximal rate of glycosylase activity (kmax). These are the first studies to define the effect of sumoylation on Kd and kmax for a G·T substrate, and the first studies to investigate the impact of sumoylation on TDG activity for G·fC and G·caC substrates. Moreover, this is the first investigation of whether modification by different SUMO isoforms causes differential effects on TDG substrate binding and catalysis. Remarkably, the observed parameters (kmax, kcat, Kd) varied by less than 10% for modification of TDG by SUMO-1 versus SUMO-2. This is significant, given that SUMO-1 and SUMO-2 share only 48% amino acid sequence identity, but perhaps not surprising given the high similarity of crystal structures of TDG modified with SUMO-1 or SUMO-3 (RMSD of 1.09 Å over 269 Cα atoms) (25,26). Notably, SUMO-2 and SUMO-3 are nearly identical (referred to as SUMO-2/3), thus the effects of TDG modification by SUMO-2 reported here are likely indicative of those that would be observed for SUMO-3.
Although sumoylation weakens TDG substrate binding by 6- to 9-fold, the modified enzyme retains substantial affinity for G·fC (Kd ≤ 36 nM) and G·caC (Kd ≤ 14 nM) pairs (Table 3). Indeed, the affinity of sumoylated TDG for G·caC pairs is greater than that of unmodified TDG for G·T substrates. Our studies also reveal that TDG∼SUMO exhibits weakened though readily measurable binding to G·T mispairs (Kd ≤ 118 nM). Similarly, we showed previously that sumoylated TDG retains substantial albeit weakened affinity for abasic sites in DNA (20). Thus, our results provide an important advance because in previous studies, substrate and product binding had not been observed for sumoylated TDG (18,25,26).
Importantly, our studies also reveal that sumoylation imparts large adverse effects on the base excision activity of TDG, with reductions in kmax of 46-, 22-, and 7-fold for G·T, G·fC, and G·caC substrates, respectively. Together, these results directly challenge the current paradigm that the predominant effect of sumoylation is on the binding affinity of TDG for DNA substrates and product, with minimal effect on catalytic activity (18,28). On the contrary, our results show that the adverse effects of sumoylation are greater for catalytic activity relative to those for substrate binding.
Our discovery that sumoylation dramatically impairs kmax reveals a corresponding decrease in the upper limit of catalytic turnover (kcat) for sumoylated versus unmodified TDG, even if kcat is enhanced by other factors (e.g., APE1), because kcat cannot exceed kmax (Figure 2). Thus, even if sumoylation potentiates the stimulatory effect of APE1 on TDG catalytic turnover, as reported for G·U substrates (18), the upper limit of APE1-stimulated kcat is far lower for sumoylated versus unmodified TDG. We note that kcat for sumoylated TDG is probably not particularly relevant anyway, because if sumoylation does serve to enhance TDG catalytic turnover, the mechanism seems likely to involve modification of product-bound TDG, with unmodified TDG handling the base excision step (18).
How might sumoylation impact substrate binding and glycosylase activity of TDG?
It is of interest to consider potential mechanisms by which sumoylation impacts substrate binding and base excision by TDG. Previous studies indicate that the adverse effects of sumoylation on DNA binding and glycosylase activity are likely mediated by non-covalent binding of the tethered SUMO domain to the SIM of TDG (Figures 1B and 8) (25,26). Crystal structures indicate that SUMO–SIM interactions lead to the formation of a β-strand and an α-helix in a C-terminal region of TDG (residues 307–331) (25,26), which is likely disordered in the unmodified enzyme (19,25). The nascent β-strand of TDG contacts a β-strand of SUMO to form an intermolecular β-sheet, while the α-helix forms few interactions with SUMO or other regions of TDG and protrudes away from the protein surface. The affinity of TDG for binding free SUMO proteins was shown to be reduced by mutation of TDG residues that mediate SUMO–SIM interactions, including R281, E310, Y313, and F315 (Figure 8), and these mutations also reverse (at least partially) the adverse effect of sumoylation on TDG binding to abasic DNA (25,26). Together, these previous findings suggest that non-covalent SUMO–SIM interactions mediate the adverse effects of sumoylation on TDG activity, but what is the molecular mechanism?
Figure 8.

Closeup view of the SUMO–SIM interface and the catalytic ‘insertion’ loop, using the same crystal structures and coloring scheme as described above for Figure 1B. Some of the residues that mediate SUMO–SIM binding, and residues of the catalytic ‘insertion’ loop are shown. Interactions between these residues could potentially link SUMO–SIM binding to impairment of productive nucleotide flipping. Because no structure is available for sumoylated TDG bound to DNA, the DNA shown (orange) was positioned as described for Figure 1B.
The current paradigm involves a model whereby the SUMO-induced α-helix of TDG perturbs DNA binding by clashing with the phosphodiester backbone (Figures 1B and 8) (25,26), which may seem consistent with previous reports that sumoylation fully abrogates detectable binding of TDG to DNA substrates and abasic DNA product (18,25,26,28). However, our studies reveal that the effects of sumoylation on TDG substrate binding are relatively modest (ΔΔGbind ≤ 1.3 kcal/mol), and sumoylated TDG retains substantial binding affinity for G·fC and G·caC pairs in DNA. Moreover, observation that sumoylated TDG retains some base excision activity requires that it can bind productively to DNA substrates. These results suggest that the SUMO–SIM-induced α-helix might be destabilized or displaced upon binding to DNA substrates or abasic product (20). Otherwise, binding would require substantial deformation of the DNA backbone (to avoid steric clash with the α-helix), which seems unlikely. Addressing the question of how the putative steric clash is resolved will require a structure of sumoylated TDG bound to DNA.
Our findings, together with structural observations, suggest an alternative mechanism for the effects of sumoylation on TDG substrate binding and base excision. Because our single turnover experiments were performed under saturating enzyme conditions, the rate constants (kmax) report on steps of the TDG reaction that occur after formation of the initial enzyme-substrate complex and up to formation of enzyme-bound product (Figure 2), which include the enzyme-substrate conformational changes associated with nucleotide flipping (E·S to E′·S′) and the chemical step(s) (E′·S′ to E′·B·P′). One plausible explanation for the dramatic SUMO-induced reductions in kmax is that sumoylation alters nucleotide flipping and thereby reduces the fraction of substrate-bound enzyme that forms a productive conformation (E′·S′). Notably, an adverse effect on nucleotide flipping could also account, at least in part, for the observed sumoylation-induced weakening of substrate binding (Kd) (Figure 2). The possibility that sumoylation perturbs nucleotide flipping is supported by structural observations. An important catalytic loop of TDG, termed the ‘insertion loop’, contains many conserved residues including Arg275, which penetrates the DNA helix to fill the void created by nucleotide flipping (Figure 8) (43,51,53). The Arg ‘plug’ is highly conserved in TDG and other G·T mismatch glycosylases and likely helps to stabilize nucleotide flipping (44,66). Residues of the insertion loop (Phe279, Arg281) interact with residues in the SIM or with SUMO, linking these regions of sumoylated TDG and suggesting a mechanism by which sumoylation could alter nucleotide flipping. Additional structural and biochemical studies will be needed to test this and other potential mechanisms for SUMO-induced reductions in substrate binding and catalysis.
What are the functions of TDG sumoylation?
It is of interest to consider the potential implications of our results regarding the role(s) of TDG sumoylation. The current paradigm is that sumoylation of TDG regulates product release and enhances its catalytic turnover (kcat) (18). TDG binds very tightly to its reaction product, abasic DNA, which severely impedes catalytic turnover in vitro (32–35). The current model holds that sumoylation occurs selectively for product-bound TDG, triggering product release, and that SUMO deconjugation reactivates TDG for processing additional substrates, such that each catalytic cycle of TDG requires sumoylation and desumoylation (18). While this model seems widely accepted (25,31,36–38) it is not directly supported by experimental evidence and has been challenged by recent studies. For example, in vitro studies showed that E2-mediated sumoylation of TDG is efficient but not specific for product-bound TDG (20). Moreover, sumoylation of TDG was shown to be dispensable for efficient removal of TET-generated caC from genomic DNA in human cells (21). In addition, catalytic turnover of TDG is strongly enhanced by follow-on BER enzymes, including AP endonuclease 1 (APE1), which process AP sites generated by DNA glycosylases. APE1 can enhance TDG catalytic turnover to a level that approaches the theoretical maximum (such that kcat approaches kmax) (21,39). The bifunctional enzymes NEIL1 and NEIL2 also stimulate TDG catalytic turnover in vitro, and they contribute to the efficient removal of fC and caC by TDG in cells (40). Additionally, the XPC repair complex (XPC, RAD23B, possibly CENT2) interacts with and stimulates catalytic turnover of TDG in vitro and in mammalian cells (67,68). Taken together, these observations indicate that other potential roles for TDG sumoylation should be considered.
Previous studies suggest sumoylation of TDG could serve to alter its subcellular localization and modulate its interactions with other proteins (15,27), which would be consistent with the roles of sumoylation that have been observed for most other proteins that undergo SUMO modification. Sumoylation might also help to suppress potentially deleterious activity of TDG in S phase of the cell cycle. TDG is depleted in S phase via ubiquitination and proteasome degradation (22,23,69), for reasons that remain unclear. One possibility is that degradation precludes TDG processing of G·T (or G·U) mismatches that arise from replication errors, which could potentially yield tight TDG-product complexes and disrupt DNA replication. Sumoylation might provide a safeguard in S phase, to suppress the glycosylase activity of TDG molecules that escape degradation. One study reported that the low residual level of TDG detected in S phase was predominantly sumoylated, suggesting that ubiquitin-mediated degradation is less efficient for sumoylated versus unmodified TDG (22).
Our findings raise the possibility of an intriguing new role for TDG sumoylation - it might enable TDG to function, at least transiently, as a reader rather than just an eraser of fC and caC. This idea stems from our findings that sumoylation dramatically reduces the glycosylase activity of TDG such that it excises fC and caC much less efficiently (t1/2 >9 min, 37°C). Meanwhile, sumoylated TDG retains relatively high affinity for these substrates, with Kd ≤36 nM for G·fC and Kd ≤14 nM for G·caC. Indeed, these affinities are similar to that of unmodified TDG for binding G·T mispairs in DNA. Thus, sumoylation yields a form of TDG that binds specifically to fC and caC but is far less likely excise them compared to the unmodified enzyme. TDG interacts with a broad range of proteins including transcription factors such as p300 (14), retinoic acid receptors (RAR, RXR) (70), and the estrogen receptor α (71), among others, and epigenetic regulators such as DNA methyltransferases (72) and the SIRT1 deacetylase (17). Many of these proteins are sumoylated and/or contain a SIM (20). Sumoylation could potentially afford TDG with an enhanced ability - and more time - to recruit other proteins to DNA sites containing fC and caC before it excises these bases. Notably, the SUMO domain of sumoylated TDG might participate in protein recruitment, depending on its accessibility for DNA-bound TDG∼SUMO and the presence of a SUMO-interacting motif in other proteins. Previous studies indicate that 20–50% of TDG is sumoylated in human cells, as determined from western blots of nuclear extract (15,18,27). However, the fraction of TDG that is sumoylated in vivo will likely depend on many factors, including the abundance and activity of enzymes that mediate SUMO conjugation (E1, E2, possibly E3) and deconjugation (SENPs) (21). Notably, TDG can be rapidly modified by the SUMO-conjugating enzyme (E2∼SUMO thioester), with maximal rate constant of kmax = 1.6 min−1 (t1/2 of 0.4 min) (20). It has been shown that SENP1 deconjugates modified TDG, but the rate constant has not been reported (21).
It is of interest to consider how the affinity of TDG∼SUMO for fC and caC compares to that of other potential readers of these bases. Mass spectrometry studies uncovered many putative readers of fC and caC (73,74) but only a few have been biochemically characterized. N-methylpurine DNA glycosylase (MPG, aka AAG, ANPG) binds (but does not excise) fC in DNA with a reported affinity of 13 nM (using an ELISA assay) (74). If accurate, this affinity is about twofold tighter than that of TDG∼SUMO for fC. MAX, a binding partner of the MYC transcription factor, binds enhancer-box (E-box) elements and has some specificity for caC; MAX binds with a Kd of ∼30 nM when the CpG site in these elements contains caC or unmodified C (75). Another putative caC reader is the CXXC domain of TET3, which binds unmodified CpG sites, and, with 3-fold higher affinity, CpG sites with a caC modification (76). Thus, our results reveal that sumoylated TDG binds fC and caC with similar affinity to previously-characterized readers of these bases in DNA.
Supplementary Material
ACKNOWLEDGEMENTS
The pGEX plasmids for co-expressing E1/E2/SUMO-1 and E1/E2/SUMO-2 were graciously provided by Professor M. Shirakawa (Kyoto University) and Professor Michael Matunis (Johns Hopkins University), respectively. The authors acknowledge Jake Dow for assistance with collection and analysis of some enzyme kinetics experiments and thank the reviewers for very helpful suggestions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R01-GM72711 to A.C.D.]; Support for procuring the imaging system (GE Typhoon FLA 9500) used in these studies was provided by the National Institutes of Health Grant [S10-OD011969]. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1. Bellacosa A., Drohat A.C.. Role of base excision repair in maintaining the genetic and epigenetic integrity of CpG sites. DNA Repair (Amst.). 2015; 32:33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Neddermann P., Jiricny J.. The purification of a mismatch-specific thymine-DNA glycosylase from HeLa cells. J. Biol. Chem. 1993; 268:21218–21224. [PubMed] [Google Scholar]
- 3. Neddermann P., Gallinari P., Lettieri T., Schmid D., Truong O., Hsuan J.J., Wiebauer K., Jiricny J.. Cloning and expression of human G/T mismatch-specific thymine-DNA glycosylase. J. Biol. Chem. 1996; 271:12767–12774. [DOI] [PubMed] [Google Scholar]
- 4. Cortazar D., Kunz C., Selfridge J., Lettieri T., Saito Y., Macdougall E., Wirz A., Schuermann D., Jacobs A.L., Siegrist F. et al. . Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature. 2011; 470:419–423. [DOI] [PubMed] [Google Scholar]
- 5. Cortellino S., Xu J., Sannai M., Moore R., Caretti E., Cigliano A., Le Coz M., Devarajan K., Wessels A., Soprano D. et al. . Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell. 2011; 146:67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Drohat A.C., Coey C.T.. Role of base excision “repair” enzymes in erasing epigenetic marks from DNA. Chem. Rev. 2016; 116:12711–12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maiti A., Drohat A.C.. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: Potential implications for active demethylation of CpG sites. J. Biol. Chem. 2011; 286:35334–35338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. He Y.F., Li B.Z., Li Z., Liu P., Wang Y., Tang Q., Ding J., Jia Y., Chen Z., Li L. et al. . Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011; 333:1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ito S., Shen L., Dai Q., Wu S.C., Collins L.B., Swenberg J.A., He C., Zhang Y.. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011; 333:1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pfaffeneder T., Hackner B., Truss M., Munzel M., Muller M., Deiml C.A., Hagemeier C., Carell T.. The discovery of 5-formylcytosine in embryonic stem cell DNA. Angew. Chem. Int. Ed. Engl. 2011; 50:7008–7012. [DOI] [PubMed] [Google Scholar]
- 11. Song C.X., Szulwach K.E., Dai Q., Fu Y., Mao S.Q., Lin L., Street C., Li Y., Poidevin M., Wu H. et al. . Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell. 2013; 153:678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shen L., Wu H., Diep D., Yamaguchi S., D’Alessio A.C., Fung H.L., Zhang K., Zhang Y.. Genome-wide analysis reveals TET- and TDG-dependent 5-methylcytosine oxidation dynamics. Cell. 2013; 153:692–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Weber A.R., Krawczyk C., Robertson A.B., Kusnierczyk A., Vagbo C.B., Schuermann D., Klungland A., Schar P.. Biochemical reconstitution of TET1-TDG-BER-dependent active DNA demethylation reveals a highly coordinated mechanism. Nat. Commun. 2016; 7:10806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tini M., Benecke A., Um S.J., Torchia J., Evans R.M., Chambon P.. Association of CBP/p300 acetylase and thymine DNA glycosylase links DNA repair and transcription. Mol. Cell. 2002; 9:265–277. [DOI] [PubMed] [Google Scholar]
- 15. Mohan R.D., Rao A., Gagliardi J., Tini M.. SUMO-1-dependent allosteric regulation of thymine DNA glycosylase alters subnuclear localization and CBP/p300 recruitment. Mol. Cell. Biol. 2007; 27:229–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guan X., Madabushi A., Chang D.Y., Fitzgerald M., Shi G., Drohat A.C., Lu A.L.. The Human Checkpoint Sensor Rad9-Rad1-Hus1 Interacts with and Stimulates DNA Repair Enzyme TDG Glycosylase. Nucleic Acids Res. 2007; 35:6207–6218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Madabushi A., Hwang B.J., Jin J., Lu A.L.. Histone deacetylase SIRT1 modulates and deacetylates DNA base excision repair enzyme thymine DNA glycosylase. Biochem. J. 2013; 456:89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hardeland U., Steinacher R., Jiricny J., Schar P.. Modification of the human thymine-DNA glycosylase by ubiquitin-like proteins facilitates enzymatic turnover. EMBO J. 2002; 21:1456–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Smet-Nocca C., Wieruszeski J.M., Chaar V., Leroy A., Benecke A.. The Thymine-DNA Glycosylase Regulatory Domain: Residual Structure and DNA Binding. Biochemistry. 2008; 47:6519–6530. [DOI] [PubMed] [Google Scholar]
- 20. Coey C.T., Fitzgerald M.E., Maiti A., Reiter K.H., Guzzo C.M., Matunis M.J., Drohat A.C.. E2-mediated small ubiquitin-like modifier (SUMO) modification of thymine DNA glycosylase is efficient but not selective for the enzyme-product complex. J. Biol. Chem. 2014; 289:15810–15819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McLaughlin D., Coey C.T., Yang W.C., Drohat A.C., Matunis M.J.. Characterizing requirements for SUMO modification and binding on base excision repair activity of thymine DNA glycosylase in vivo. J. Biol. Chem. 2016; 291:9014–9024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Slenn T.J., Morris B., Havens C.G., Freeman R.M. Jr, Takahashi T.S., Walter J.C.. Thymine DNA glycosylase is a CRL4Cdt2 substrate. J. Biol. Chem. 2014; 289:23043–23055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shibata E., Dar A., Dutta A.. CRL4Cdt2 E3 ubiquitin Ligase and PCNA cooperate to degrade thymine DNA glycosylase in S-phase. J. Biol. Chem. 2014; 289:23056–23064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Minty A., Dumont X., Kaghad M., Caput D.. Covalent modification of p73alpha by SUMO-1. Two-hybrid screening with p73 identifies novel SUMO-1-interacting proteins and a SUMO-1 interaction motif. J. Biol. Chem. 2000; 275:36316–36323. [DOI] [PubMed] [Google Scholar]
- 25. Baba D., Maita N., Jee J.-G., Uchimura Y., Saitoh H., Sugasawa K., Hanaoka F., Tochio H., Hiroaki H., Shirakawa M.. Crystal structure of thymine DNA glycosylase conjugated to SUMO-1. Nature. 2005; 435:979–982. [DOI] [PubMed] [Google Scholar]
- 26. Baba D., Maita N., Jee J.G., Uchimura Y., Saitoh H., Sugasawa K., Hanaoka F., Tochio H., Hiroaki H., Shirakawa M.. Crystal structure of SUMO-3-modified thymine-DNA glycosylase. J. Mol. Biol. 2006; 359:137–147. [DOI] [PubMed] [Google Scholar]
- 27. Takahashi H., Hatakeyama S., Saitoh H., Nakayama K.I.. Noncovalent SUMO-1 binding activity of thymine DNA glycosylase (TDG) is required for its SUMO-1 modification and colocalization with the promyelocytic leukemia protein. J. Biol. Chem. 2005; 280:5611–5621. [DOI] [PubMed] [Google Scholar]
- 28. Steinacher R., Schar P.. Functionality of human thymine DNA glycosylase requires SUMO-regulated changes in protein conformation. Curr. Biol. 2005; 15:616–623. [DOI] [PubMed] [Google Scholar]
- 29. Smet-Nocca C., Wieruszeski J.M., Leger H., Eilebrecht S., Benecke A.. SUMO-1 regulates the conformational dynamics of thymine-DNA Glycosylase regulatory domain and competes with its DNA binding activity. BMC biochem. 2011; 12:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sampson D.A., Wang M., Matunis M.J.. The small ubiquitin-like modifier-1 (SUMO-1) consensus sequence mediates Ubc9 binding and is essential for SUMO-1 modification. J. Biol. Chem. 2001; 276:21664–21669. [DOI] [PubMed] [Google Scholar]
- 31. Geiss-Friedlander R., Melchior F.. Concepts in sumoylation: a decade on. Nat. Rev. Mol. Cell. Biol. 2007; 8:947–956. [DOI] [PubMed] [Google Scholar]
- 32. Waters T.R., Swann P.F.. Kinetics of the action of thymine DNA glycosylase. J. Biol. Chem. 1998; 273:20007–20014. [DOI] [PubMed] [Google Scholar]
- 33. Waters T.R., Gallinari P., Jiricny J., Swann P.F.. Human thymine DNA glycosylase binds to apurinic sites in DNA but is displaced by human apurinic endonuclease 1. J. Biol. Chem. 1999; 274:67–74. [DOI] [PubMed] [Google Scholar]
- 34. Scharer O.D., Nash H.M., Jiricny J., Laval J., Verdine G.L.. Specific binding of a designed pyrrolidine abasic site analog to multiple DNA glycosylases. J. Biol. Chem. 1998; 273:8592–8597. [DOI] [PubMed] [Google Scholar]
- 35. Fitzgerald M.E., Drohat A.C.. Coordinating the initial steps of base excision repair. Apurinic/apyrimidinic endonuclease 1 actively stimulates thymine DNA glycosylase by disrupting the product complex. J. Biol. Chem. 2008; 283:32680–32690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wilkinson K.A., Henley J.M.. Mechanisms, regulation and consequences of protein SUMOylation. Biochem. J. 2010; 428:133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bergink S., Jentsch S.. Principles of ubiquitin and SUMO modifications in DNA repair. Nature. 2009; 458:461–467. [DOI] [PubMed] [Google Scholar]
- 38. Ulrich H.D. Two-way communications between ubiquitin-like modifiers and DNA. Nat. Struct. Mol. Biol. 2014; 21:317–324. [DOI] [PubMed] [Google Scholar]
- 39. Sassa A., Caglayan M., Dyrkheeva N.S., Beard W.A., Wilson S.H.. Base excision repair of tandem modifications in a methylated CpG dinucleotide. J. Biol. Chem. 2014; 289:13996–14008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schomacher L., Han D., Musheev M.U., Arab K., Kienhofer S., von Seggern A., Niehrs C.. Neil DNA glycosylases promote substrate turnover by Tdg during DNA demethylation. Nat. Struct. Mol. Biol. 2016; 23:116–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Coey C.T., Drohat A.C.. Kinetic methods for studying DNA glycosylases functioning in base excision repair. Methods Enzymol. 2017; 592:357–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gallinari P., Jiricny J.. A new class of uracil-DNA glycosylases related to human thymine-DNA glycosylase. Nature. 1996; 383:735–738. [DOI] [PubMed] [Google Scholar]
- 43. Coey C.T., Malik S.S., Pidugu L.S., Varney K.M., Pozharski E., Drohat A.C.. Structural basis of damage recognition by thymine DNA glycosylase: Key roles for N-terminal residues. Nucleic Acids Res. 2016; 44:10248–10258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Maiti A., Morgan M.T., Drohat A.C.. Role of two strictly conserved residues in nucleotide flipping and N-glycosylic bond cleavage by human thymine DNA glycosylase. J. Biol. Chem. 2009; 284:36680–36688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Morgan M.T., Bennett M.T., Drohat A.C.. Excision of 5-halogenated uracils by human thymine DNA glycosylase: Robust activity for DNA contexts other than CpG. J. Biol. Chem. 2007; 282:27578–27586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Uchimura Y., Nakamura M., Sugasawa K., Nakao M., Saitoh H.. Overproduction of eukaryotic SUMO-1- and SUMO-2-conjugated proteins in Escherichia coli. Anal. Biochem. 2004; 331:204–206. [DOI] [PubMed] [Google Scholar]
- 47. Gill S.C., von Hippel P.H.. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989; 182:319–326. [DOI] [PubMed] [Google Scholar]
- 48. Morgan M.T., Maiti A., Fitzgerald M.E., Drohat A.C.. Stoichiometry and affinity for thymine DNA glycosylase binding to specific and nonspecific DNA. Nucleic Acids Res. 2011; 39:2319–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Scharer O.D., Kawate T., Gallinari P., Jiricny J., Verdine G.L.. Investigation of the mechanisms of DNA binding of the human G/T glycosylase using designed inhibitors. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:4878–4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang L., Lu X., Lu J., Liang H., Dai Q., Xu G.L., Luo C., Jiang H., He C.. Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat. Chem. Biol. 2012; 8:328–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pidugu L.S., Flowers J.W., Coey C.T., Pozharski E., Greenberg M.M., Drohat A.C.. Structural basis for excision of 5-formylcytosine by thymine DNA glycosylase. Biochemistry. 2016; 55:6205–6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Maiti A., Noon M.S., Mackerell A.D. Jr., Pozharski E., Drohat A.C.. Lesion processing by a repair enzyme is severely curtailed by residues needed to prevent aberrant activity on undamaged DNA. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:8091–8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Malik S.S., Coey C.T., Varney K.M., Pozharski E., Drohat A.C.. Thymine DNA glycosylase exhibits negligible affinity for nucleobases that it removes from DNA. Nucleic Acids Res. 2015; 43:9541–9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bennett M.T., Rodgers M.T., Hebert A.S., Ruslander L.E., Eisele L., Drohat A.C.. Specificity of human thymine DNA glycosylase depends on N-glycosidic bond stability. J. Am. Chem. Soc. 2006; 128:12510–12519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Morgan M.T., Bennett M.T., Drohat A.C.. Excision of 5-halogenated uracils by human thymine DNA glycosylase. Robust activity for DNA contexts other than CpG. J. Biol. Chem. 2007; 282:27578–27586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Leatherbarrow R.J. 1998; Staines: Erithacus Software Ltd. [Google Scholar]
- 57. Maiti A., Drohat A.C.. Dependence of substrate binding and catalysis on pH, ionic strength, and temperature for thymine DNA glycosylase: Insights into recognition and processing of G.T mispairs. DNA Repair. 2011; 10:545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kuzmic P. Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal. Biochem. 1996; 237:260–273. [DOI] [PubMed] [Google Scholar]
- 59. Kuzmic P. DynaFit—a software package for enzymology. Methods Enzymol. 2009; 467:247–280. [DOI] [PubMed] [Google Scholar]
- 60. Coey C.T., Malik S.S., Pidugu L.S., Varney K.M., Pozharski E., Drohat A.C.. Structural basis of damage recognition by thymine DNA glycosylase: key roles for N-terminal residues. Nucleic Acids Res. 2016; 44:10248–10258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hardeland U., Bentele M., Jiricny J., Schar P.. Separating substrate recognition from base hydrolysis in human thymine DNA glycosylase by mutational analysis. J. Biol. Chem. 2000; 275:33449–33456. [DOI] [PubMed] [Google Scholar]
- 62. Maiti A., Morgan M.T., Pozharski E., Drohat A.C.. Crystal structure of human thymine DNA glycosylase bound to DNA elucidates sequence-specific mismatch recognition. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:8890–8895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Buechner C.N., Maiti A., Drohat A.C., Tessmer I.. Lesion search and recognition by thymine DNA glycosylase revealed by single molecule imaging. Nucleic Acids Res. 2015; 43:2716–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hardwick J.S., Ptchelkine D., El-Sagheer A.H., Tear I., Singleton D., Phillips S.E.V., Lane A.N., Brown T.. 5-Formylcytosine does not change the global structure of DNA. Nat. Struct. Mol. Biol. 2017; 24:544–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hardwick J.S., Lane A.N., Brown T.. Epigenetic modifications of cytosine: biophysical properties, regulation, and function in mammalian DNA. BioEssays. 2018; 40:doi:10.1002/bies.201700199. [DOI] [PubMed] [Google Scholar]
- 66. Manvilla B.A., Maiti A., Begley M.C., Toth E.A., Drohat A.C.. Crystal structure of human methyl-binding domain IV glycosylase bound to abasic DNA. J. Mol. Biol. 2012; 420:164–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shimizu Y., Iwai S., Hanaoka F., Sugasawa K.. Xeroderma pigmentosum group C protein interacts physically and functionally with thymine DNA glycosylase. EMBO J. 2003; 22:164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ho J.J., Cattoglio C., McSwiggen D.T., Tjian R., Fong Y.W.. Regulation of DNA demethylation by the XPC DNA repair complex in somatic and pluripotent stem cells. Genes Dev. 2017; 31:830–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hardeland U., Kunz C., Focke F., Szadkowski M., Schar P.. Cell cycle regulation as a mechanism for functional separation of the apparently redundant uracil DNA glycosylases TDG and UNG2. Nucleic Acids Res. 2007; 35:3859–3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Um S., Harbers M., Benecke A., Pierrat B., Losson R., Chambon P.. Retinoic acid receptors interact physically and functionally with the T:G mismatch-specific thymine-DNA glycosylase. J. Biol. Chem. 1998; 273:20728–20736. [DOI] [PubMed] [Google Scholar]
- 71. Chen D., Lucey M.J., Phoenix F., Lopez-Garcia J., Hart S.M., Losson R., Buluwela L., Coombes R.C., Chambon P., Schar P. et al. . T:G mismatch-specific thymine-DNA glycosylase potentiates transcription of estrogen-regulated genes through direct interaction with estrogen receptor {alpha}. J. Biol. Chem. 2003; 278:38586–38592. [DOI] [PubMed] [Google Scholar]
- 72. Li Y.Q., Zhou P.Z., Zheng X.D., Walsh C.P., Xu G.L.. Association of Dnmt3a and thymine DNA glycosylase links DNA methylation with base-excision repair. Nucleic Acids Res. 2007; 35:390–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Spruijt C.G., Gnerlich F., Smits A.H., Pfaffeneder T., Jansen P.W., Bauer C., Munzel M., Wagner M., Muller M., Khan F. et al. . Dynamic readers for 5-(hydroxy)Methylcytosine and its oxidized derivatives. Cell. 2013; 152:1146–1159. [DOI] [PubMed] [Google Scholar]
- 74. Iurlaro M., Ficz G., Oxley D., Raiber E.A., Bachman M., Booth M.J., Andrews S., Balasubramanian S., Reik W.. A screen for hydroxymethylcytosine and formylcytosine binding proteins suggests functions in transcription and chromatin regulation. Genome Biol. 2013; 14:R119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wang D., Hashimoto H., Zhang X., Barwick B.G., Lonial S., Boise L.H., Vertino P.M., Cheng X.. MAX is an epigenetic sensor of 5-carboxylcytosine and is altered in multiple myeloma. Nucleic Acids Res. 2017; 45:2396–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jin S.G., Zhang Z.M., Dunwell T.L., Harter M.R., Wu X., Johnson J., Li Z., Liu J., Szabo P.E., Lu Q. et al. . Tet3 reads 5-carboxylcytosine through its CXXC domain and is a potential guardian against neurodegeneration. Cell Rep. 2016; 14:493–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.