

ABSTRACT
Capillary rarefaction is broadly defined as a reduction in vascular density. Capillary rarefaction in the kidneys is thought to promote hypoxia, impair hemodynamic responses and predispose to chronic kidney disease (CKD) progression and hypertension development. Various mechanisms have been suggested to play a role in the development of capillary rarefaction, including inflammation, an altered endothelial-tubular epithelial cell crosstalk, a relative deficiency in angiogenic growth factors, loss of pericytes, increased activity of Transforming growth factor -β1 and thrombospondin-1, vitamin D deficiency, a link to lymphatic neoangiogenesis and INK4a/ARF (Cylin-dependent kinase inhibitor 2a; CDKN2A). In this review, we summarize the tools available to monitor capillary rarefaction noninvasively in the clinic, the contribution of capillary rarefaction to CKD and hypertension, the known mechanisms of capillary rarefaction, and potential future strategies to attenuate capillary rarefaction and reduce its negative impact. Therapeutic strategies to be explored in more detail include optimization of antihypertensive therapy, vitamin D receptor activators, sirtuin 1 activators, Hypoxia inducible factor prolyl hydroxylase inhibitors and stem cell therapy.
Keywords: capillary, chronic kidney disease, hypertension, hypoxia-inducible factor, pericyte, rarefaction
Introduction
Emerging evidence suggests that the kidney has considerable capacity to repair and regenerate. However, not all kidney cell types have the same regenerative capacity. Unlike proximal tubule cells, cells of the renal vasculature have a poor capacity for repair, which may lead to a persistent reduction in vascular density following an acute or chronic insult. The reduction in vascular density is broadly termed ‘capillary rarefaction’ and is thought to promote hypoxia, impair hemodynamic responses and potentially predispose to chronic kidney disease (CKD) progression and hypertension development [1]. In this review, we summarize the tools available to monitor capillary rarefaction noninvasively in the clinic, the role of capillary rarefaction in the progression of CKD and the development of hypertension, the known mechanisms of capillary rarefaction, and potential future strategies that attenuate capillary rarefaction and its negative impact on CKD and hypertension.
Noninvasive in vivo assessment of capillary rarefaction
In kidney biopsy specimens, capillary rarefaction is assessed histologically. However, kidney biopsy is an invasive procedure, not suitable for future clinical trials assessing the impact of therapeutic intervention on capillary rarefaction. Noninvasive quantitative analyses such as nailfold capillaroscopy a computed tomography (CT) are alternatives more suitable for repeated assessment in the clinic, the latter marred by radiation and contrast use, but allowing direct quantification of kidney vascular rarefaction.
Dermal capillaries represent an ‘open’ and representative window for the in vivo study of the human microcirculation that can be directly, repetitively and easily visualized by noninvasive techniques such as nailfold capillaroscopy to show capillary rarefaction [2, 3]. However, visual inspection of the capillaries is limited by the depth of penetrance of light photons and provides only a one-dimensional analysis of a three-dimensional problem.
Functional in vivo micro-CT imaging has allowed accurate assessment of vessel dysfunction in preclinical CKD [4]. Furthermore, small-caliber artery rarefaction (interlobular artery and more distal branches) can be followed separately from capillary rarefaction [4]. In humans, contrast-enhanced CT angiography has also been used to assess kidney vascular rarefaction by quantifying renal blood volume. Renal blood volume was lower in the cortex of CKD patients than in controls and closely mirrored capillary rarefaction in the corresponding nephrectomy specimens. In patients with follow-up CT angiography, reduction of renal function was paralleled by a decline in renal blood volume [5].
Capillary rarefaction and CKD progression
The major branches of the renal artery conduct more than 90% of renal blood flow directly to the glomerular capillary bed located in the kidney cortex [6]. Then, the efferent arteriole branches into peritubular capillaries, which initially supply oxygen and nutrients to the highly metabolically active proximal tubular cells. Less than 10% of the arterial blood flow is delivered to the medulla and then to more profound parts of the nephron. As a result, the cortex pO2 is between 30 and 50 mmHg, while in the medulla and medullary rays it is 10–20 mmHg, the lowest in the body [7, 8]. Thus, even under physiological circumstances, tubular cells, especially in some parts of the nephron, are relatively hypoxic. It is meaningful that erythropoietin-producing cells reside in the kidney, where they can sensitively detect hypoxia due to anemia. Under pathological circumstances, the hypoxic areas may extend even into cortex region [9]. In this regard, peritubular capillary rarefaction is a hallmark of CKD and of the acute kidney injury to CKD transition [10, 11]. Both acute and chronic kidney diseases result in capillary rarefaction in preclinical models and humans. Thus, unilateral ureteral obstruction [12], remnant kidney model [13], chronic allograft rejection [14] Col4a3-deficiency [15] and glomerulonephritis [16] are characterized by peritubular capillary loss associated with interstitial fibrosis and tubular atrophy. Although the sequence of events connecting peritubular capillary loss to fibrosis and tubular atrophy is still not completely characterized, hypoxia due to peritubular capillary rarefaction is thought to be a primary event in CKD and peritubular capillary rarefaction has been associated with reduced kidney regenerative capacity [17]. The functional micro-CT findings of capillary rarefaction were observed before the onset of overt fibrosis and may be the earliest diagnostic and prognostic sign for renal dysfunction. At histological level, CKD progression is associated with evidence of capillary injury, such as focal widening of the subendothelial space and higher numbers of endothelial vacuoles and caveolae, reduced numbers of endothelial fenestrations and increased thickness of the cell soma and lamina densa of the capillary basement membrane, and increased permeability [15].
Capillary rarefaction and hypertension
Capillary rarefaction has also been implicated in the pathogenesis of essential hypertension. The pathogenesis of capillary rarefaction in hypertension is unknown, but it may involve a low-grade inflammatory response [18, 19]. In spontaneously hypertensive rats, there is rarefaction of arterioles and capillaries in skeletal muscles [20]. Additionally, the number of nailfold capillaries is lower in patients with untreated essential hypertension than in controls [21]. In the non-renal population of hypertensive and normotensive individuals, capillary density significantly correlated with high-density lipoprotein/low-density lipoprotein ratio, but not with serum vascular endothelial growth factor (VEGF) or with high-sensitivity C-reactive protein. An inverse association was found with body mass index, insulin levels and homeostasis model assessment-insulin resistance [22].
In essential hypertension, capillary rarefaction was associated with cardiovascular reactivity and exercise-induced rheological abnormalities. In all, 61 men with essential hypertension and capillary rarefaction (<80 capillaries per field), and 20 age- and sex-matched controls underwent a strenuous cycle ergometer test to monitor, during exercise and recovery, the blood pressure profile, the hemorheological pattern and other parameters. Hypertensive men with <72 capillaries per field had an abnormal hemorheological profile before exercise. The physiological response to exercise was observed only in controls and in hypertensives with >73 capillaries per field. Abnormal responses to exercise worsened as capillaries were more rarefied [23].
Finally, hypertension is a frequent side effect of anti-angiogenesis therapy targeting VEGF receptor signaling in cancer patients, to the point that development of hypertension implies adequate VEGF inhibition and is associated with improved tumor responses [24]. In this regard, the receptor tyrosine kinase sunitinib promoted dermal capillary rarefaction and this could be one of the mechanisms for hypertension development in these patients [25].
Mechanisms of capillary rarefaction
Recent evidence has identified various mechanisms that contribute to the development of capillary rarefaction (Figure 1). These include inflammation, an altered endothelial-tubular epithelial cell crosstalk, a relative deficiency in angiogenic growth factors, loss of pericytes, increased activity of TGF-β1 and thrombospondin-1, vitamin D deficiency, a link to lymphatic neoangiogenesis and INK4a/ARF (Cylin-dependent kinase inhibitor 2a; CDKN2A).
Fig. 1.
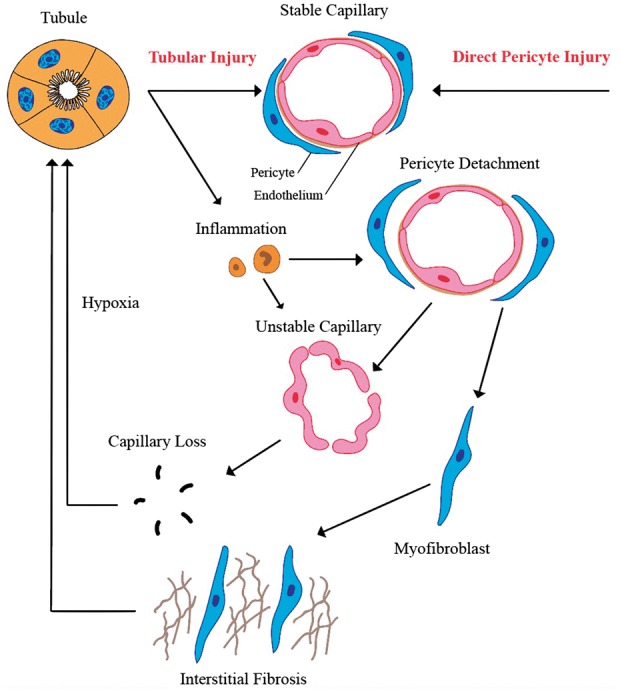
Factors playing a role in capillary rarefaction.
Inflammation and crosstalk between tubular epithelial cells and capillary endothelial cells
There is a bidirectional relationship between tubular epithelial cells and capillary endothelial cells. Primary tubular epithelial cell injury promotes capillary rarefaction [26] and capillary rarefaction further promotes hypoxic tubular cell injury, thus creating a vicious circle.
There are several examples in which tubular injury precedes capillary rarefaction. Chronic ureteral obstruction results in tubular atrophy, tubulointerstitial fibrosis and peritubular capillary rarefaction [12]. Exposure of proximal tubular cells to plasma proteins, as in proteinuric conditions, results in release of inflammatory cytokines from tubular cells, which may drive capillary rarefaction [27]. Genetically modified mice have been used in conjunction with diphtheria toxin-induced sublethal injury specific to proximal tubular cells, thus demonstrating that proximal tubular cell injury is sufficient to elicit a strong peritubular inflammatory response with secondary interstitial fibrosis and peritubular capillary rarefaction [28].
Additionally, capillary rarefaction decreases tubular blood and oxygen supply, promoting the loss of tubular cell viability and tubular atrophy and interstitial fibrosis. Hypoxia causes oxidative stress [29, 30] and increased expression of lethal inflammatory cytokines such as FasL, interleukin-1β and tumor necrosis factor α (TNF-α) [30]. Inflammatory factors and cells promote endothelial cell injury, including a pro-coagulant and pro-adhesive phenotype, leading to capillary occlusion by thrombosis, as well as to endothelial cell apoptosis. Impairment of blood flow decreases laminar shear stress on endothelial cells, resulting in further endothelial apoptosis and tubular hypoxia as a vicious circle [31]. This is especially striking in antibody-mediated rejection following kidney transplantation. During this type of rejection, endothelial cells become pro-thrombotic, causing platelet and leukocyte adhesion, which eventually leads to increased cell death [32].
VEGF
VEGF promotes peritubular capillary formation and proliferation [33, 34] and, as discussed above, anticancer drugs targeting VEGF signaling promote dermal capillary rarefaction [25].
Hypoxia resulting from peritubular capillary rarefaction promotes Hypoxia inducible factor (HIF) activation and the expression of HIF-dependent genes such as VEGF, potentially favoring new capillary formation and thus offsetting capillary rarefaction [27, 35]. Thus, VEGF release could be considered a compensatory response that enhances peritubular capillary density [36]. Indeed, kidney-derived mesenchymal stem cells (MSCs) reduce peritubular capillary rarefaction via secretion of VEGF [37] and cobalt-induced HIF activation mitigated renal injury in a CKD model [38]. However, in the remnant kidney model in adriamycin-induced CKD in mice, and in human CKD, a spontaneous increased HIF-1α expression was not associated with increased tubular cell VEGF, suggesting an HIF-VEGF blockade in chronically injured tubules [39, 40]. Indeed, loss of tubular VEGF resulted in substantial reduction of peritubular capillary density [7]. In this regard, a decreased renal expression of VEGF-A is associated with a reduction in peritubular capillary density in diabetic nephropathy [41]. The late stages of the remnant kidney model are also characterized by loss of VEGF expression and VEGF administration preserved peritubular capillaries and improved tubulointerstitial injury [13, 42]. In addition, as CKD progresses, shear stress in the peritubular capillary decreases, leading to lower nitric oxide and VEGF availability and facilitating Fas-FasL-mediated endothelial cell apoptosis [27]. However, the biology of VEGF is complex and tightly regulated, since excess VEGF may also be deleterious. Excessive and uncontrolled VEGF secretion may result in formation of leaky and nonfunctional vessels, favoring inflammation, macrophage recruitment and fibrosis [36, 43]. Furthermore, the VEGF120 and VEGF188 upregulated in preclinical CKD are dys-angiogenic isoforms [44]. Thus, the role of VEGF isoforms may have different impact on capillary rarefaction. There are various isoforms of VEGF such as VEGF121, VEGF165, VEGF189 or VEGF206 [45]. However, although these isoforms have been known for a long period, their specific impact on capillary rarefaction has not been studied widely. In one study it was shown that impaired adipose tissue angiogenesis is associated with overexpression of antiangiogenic isoform of VEGF-A165b [46]. As also suggested above, recent evidence showed that VEGF164 is proangiogenic, whereas VEGF120 and VEGF188 were dys-angiogenic [44]. Apparently, more studies are needed regarding VEGF isoforms and capillary rarefaction.
Loss of pericytes
Specific ablation of pericytes using a genetic model resulted in endothelial cell damage within 10 days and subsequent permanent peritubular capillary rarefaction [47]. An increase in the distance between pericytes and endothelial cells, heralding detachment of pericytes from capillaries, is an early feature of acute kidney injury [47]. Pericyte detachment and loss leads to structural instability of blood vessels and to capillary rarefaction [48–50]. Furthermore, detached pericytes are key precursors of myofibroblasts [51–53]. Pericytes-turned-myofibroblasts contribute to interstitial fibrosis that leads to further capillary rarefaction [43]. Additionally, pericytes serve as a local stem cell population that replenish differentiated interstitial and vascular cells lost during aging [54]. The loss of this reparative capacity in the toxic renal microenvironment after acute kidney injury or during CKD progression promotes cellular death of the unstable endothelium, with subsequent capillary rarefaction [54, 55].
A number of mediators are involved in the crosstalk between endothelial cells and pericytes via discontinuities in the capillary basement membrane that helps maintain the normal vessel structure and stability. platelet derived growth factor (PDGF)-β/PDGF receptor-beta (PDGFR-β) and angiopoietin-Tie2, appear to be crucial for pericyte differentiation, recruitment and expansion during angiogenesis. Pericytes produce angiopoietin-1, a growth factor that stabilizes the microvasculature by activating the endothelial Tie2 receptor. After renal injury endothelium-derived angiopoietin-2, an antagonist of angiopoietin-1, increases, favoring capillary leakiness and pericyte loss [56]. The endothelium and pericytes also communicate via ephrinB2, TIMPs/matrix metalloproteinases and others [57]. TGF-β, VEGF, Notch and Sphingosine-1-phosphate also regulate blood vessel stability [55]. In addition, pericyte detachment and myofibroblastic differentiation are associated with secretion of anti-angiogenic factors such as ADAMTS1 (a disintegrin and metalloproteinase with thrombospondin motifs-1), which further accelerate capillary regression induced by kallikrein [55].
Bearing these issues in mind one may think that replacement of stem cells and pericytes, in particular, may attenuate renal injury. However, it may not always be the case. For example, Kim et al. showed that administration of autologous MSCs resulted in rapid aggravation of preexisting renal insufficiency. Renal biopsy findings at dialysis showed severe interstitial fibrosis and inflammatory cell infiltration. This highlights the potential nephrotoxicity of autologous MSC therapy in CKD patients [58]. It was also concluded that regarding the results of the preliminary data about stem cell therapy, long-term follow-up data are not available and there is an absence of consensus between therapeutic protocols [59].
TGF-β1 and thrombospondin-1
During hypoxia, TGF-β1 stimulates angiogenesis indirectly by inducing VEGF-A expression [60]. However, TGF-β1 directly causes endothelial cell apoptosis and capillary pruning and this negative effect predominates during renal fibrosis [27].
Thrombospondin-1 could potentiate the fibrotic response by both activating TGF-β and exerting antiangiogenic actions, thus leading to capillary rarefaction [36]. Inhibition of thrombospondin expression suppressed tubulointerstitial fibrosis by promoting VEGF production and restoring peritubular capillary density [61].
Vitamin D deficiency
The role of vitamin D deficiency in tubulointerstitial damage and peritubular capillary rarefaction following acute kidney injury induced by ischemia-reperfusion was studied in rats fed vitamin D-free or standard diets for 35 days. On Day 28, rats were randomized into four groups: control, vitamin D deficient, bilateral kidney ischemia-reperfusion and a combination of both. Vitamin D deficiency alone led to reduced capillary density and it further exacerbated the capillary rarefaction induced by kidney ischemia-reperfusion [11].
Link to lymphatic neoangiogenesis
Peritubular capillary rarefaction may be associated with simultaneous proliferation of lymphatic vessels. Cortex and medulla microvascular density was lower in end-stage renal allografts than in controls, while new lymphatic vessels were observed in the graft tubulointerstitium, but not in controls [62, 63]. The drivers of the divergent response of peritubular capillaries (rarefaction) and lymphatic capillaries (neoangiogenesis) should be explored in further studies, but there is some evidence for a role of angiotensin II [63].
INK4a/ARF (CDKN2A)
Deletion of the INK4a/ARFlocus encoding p16 and p19 improved kidney regeneration and decreased capillary rarefaction after renal ischemia-reperfusion [64]. p16 and p19 play a role in tubular atrophy and interstitial fibrosis by promoting apoptosis and cell senescence.
Potential implications for therapy and future research
Since there is evidence that capillary rarefaction plays an important role in CKD progression, tubular atrophy and interstitial fibrosis and it contributes to the development of essential hypertension, prevention or treatment of capillary rarefaction may potentially halt the progression of CKD and hypertension. The potential for intervention includes the use of already available drugs (e.g. specific antihypertensive agents) or novel therapeutic approaches.
Antihypertensive medication
An unresolved issue is the distinct effect of different antihypertensive medications on capillary rarefaction. Angiotensin-converting enzyme inhibitors and angiotensin-1 receptor blockers may induce angiogenesis and reduce or even reverse microvascular rarefaction [65]. In rats with CKD, an angiotensin II antagonist for 10 weeks regenerated the kidney vasculature that had previously undergone rarefaction and this was associated with reduced apoptosis and increased endothelial cell proliferation [66]. Angiotensin-converting enzyme inhibitors also decreased both peritubular capillary rarefaction and lymphatic neoangiogenesisin a rat renal allograft model [63]. However, in observational cross-sectional human studies, dermal capillary density in treated hypertensive individuals has been reported to be lower than or higher than in control normotensive individuals [67, 68]. The reason for these seemingly contradictory findings is unclear and may depend on the specific antihypertensive agents, length of untreated or treated hypertension, or other factors. Only prospective studies are likely to provide significant insights.
Vitamin D receptor activators (VDRA)
The fact that vitamin D deficiency aggravates capillary rarefaction does not necessarily imply that pharmacological vitamin D doses of VDRA prevent kidney capillary rarefaction. We found no report addressing this. However, in a randomized clinical trial, the VDRA paricalcitol slowed the progressive endothelial dysfunction of moderate CKD, pointing to potential endothelial preservation capabilities [69]. Moreover, calcitriol prevented reduction of cardiac capillary density in rats with CKD [70].
Sirtuin 1 activators
A number of sirtuin 1 activators are known, most notably resveratrol, although the pharmacokinetic properties of resveratrol are suboptimal and additional sirtuin 1 (SIRT1) activators have been developed to delay aging and age-related diseases [71]. To our knowledge, these have not yet been tested for their preservation of kidney capillary density properties. However, sirtuin 1 may prevent capillary rarefaction. Sirtuin 1 is highly expressed in endothelial cells and regulates angiogenesis signaling pathways via its deacetylase activity [72]. In mice with inactive Sirtuin 1, angiogenesis is compromised [73, 74]. Endothelial Sirtuin 1 dysfunction causes activation of endothelial Notch1 signaling, which leads to enhanced apoptosis and senescence of peritubular capillary endothelial cells with impaired endothelial proliferation and expanded myofibroblast population, peritubular capillary rarefaction and fibrosis following kidney injury. Specifically, Sirtuin 1 mutant mice have more severe renal fibrosis and renal function impairment than wild-type mice following induction of folic acid nephropathy [75]. Compared with wild-type kidneys, SIRT1 mutant kidneys up-regulate Delta-like 4 (DLL4, a potent Notch1 ligand), Hey1 and Hes1 (Notch target genes) and Notch intracellular domain-1 (NICD1, active form of Notch1) in microvascular endothelial cells post-injury. SIRT1 mutant primary kidney microvascular endothelial cells display lower motility and vascular assembly, and faster senescence than wild-type cells [10].
VEGF
Since VEGF is the major survival factor for capillary endothelium, it may attenuate capillary rarefaction. Administration of recombinant VEGF-A121 decreased peritubular capillary rarefaction, improved renal function, lowered mortality and reduced fibrosis in a remnant kidney model [13]. However, VEGF is not yet in clinical use, in part due to the potential for harm due to excess VEGF activity, which may depend on the tissue microenvironment and on the existence of two VEGF receptors, VEGFR1, involved in the inflammatory responses, and VEGFR2, predominantly mediating angiogenesis [72]. In this regard, a VEGF mutant with specificity for VEGFR2 resulted in increased kidney damage, despite the supposed specificity for the VEGF receptor mediating the potentially beneficial effects [76]. A better way to enhance VEGF activity and keep it within physiological levels may involve a family of HIF activators, the HIF prolyl hydroxylase inhibitors, to which are undergoing clinical trials to treat anemia in patients with CKD [77]. These drugs increase hemoglobin levels without increasing blood pressure, an effect ascribed to increased VEGF secretion. These agents should be tested to prevent kidney capillary rarefaction.
Stem cell therapy
Kidney-derived MSCs have also been suggested to ameliorate capillary rarefaction through the release of proangiogenic factors, such as VEGF, or microparticles [78, 79]. Stem cell-derived microparticles decrease endothelial-to-mesenchymal transition, enhance endothelial cell proliferation and reduce apoptosis, resulting in decreased peritubular capillary rarefaction [78]. Microvesicles released from endothelial progenitor cells also protected against CKD progression by inhibiting capillary rarefaction [80]. Injection of kidney progenitor-like cells into animals with subtotal nephrectomy resulted in slower loss of renal function, and milder macrophage and myofibroblast recruitment, and vascular rarefaction [81]. Finally, adipose stromal cells accelerated recovery from renal ischemia-reperfusion, decreasing inflammation and tubular injury, and preserving peritubular capillaries [82]. The challenges for cell therapy are still enormous, but positive clinical trials have been reported in recent, large-scale, Phase 3 trials in other fields of medicine [83].
Conclusion
In conclusion, capillary rarefaction contributes to the development and progression of CKD and, potentially, to hypertension. Various mechanisms contribute to capillary rarefaction, which point to several potential therapeutic strategies. However, for some mechanisms, it is still unclear whether the improvement in capillary rarefaction is a primary event, or an event secondary to the improvement in other factors such as tubular cell injury or inflammation. Whether diminishing capillary rarefaction slows down the progression of CKD and the development of hypertension remains to be defined in clinical trials.
Funding
A.O. was supported by Intensificacion ISCIII and RETIC REDINREN RD 016/0009 FEDER funds.
Conflict of interest statement
None declared.
References
- 1. Basile DP, Friedrich JL, Spahic J. et al. Impaired endothelial proliferation and mesenchymal transition contribute to vascular rarefaction following acute kidney injury. Am J Physiol Renal Physiol 2011; 300: F721–F733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Triantafyllou A, Anyfanti P, Pyrpasopoulou A. et al. Capillary rarefaction as an index for the microvascular assessment of hypertensive patients. Curr Hypertens Rep 2015; 17: 33. [DOI] [PubMed] [Google Scholar]
- 3. Serne EH, Gans RO, ter Maaten JC. et al. Impaired skin capillary recruitment in essential hypertension is caused by both functional and structural capillary rarefaction. Hypertension 2001; 38: 238–242 [DOI] [PubMed] [Google Scholar]
- 4. Ehling J, Babikova J, Gremse F. et al. Quantitative micro-computed tomography imaging of vascular dysfunction in progressive kidney diseases. J Am Soc Nephrol 2016; 27: 520–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. von Stillfried S, Apitzsch JC, Ehling J. et al. Contrast-enhanced CT imaging in patients with chronic kidney disease. Angiogenesis 2016; 19: 525–535 [DOI] [PubMed] [Google Scholar]
- 6. Herzlinger D, Hurtado R.. Patterning the renal vascular bed. Semin Cell Dev Biol 2014; 36: 50–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dimke H, Sparks MA, Thomson BR. et al. Tubulovascular cross-talk by vascular endothelial growth factor A maintains peritubular microvasculature in kidney. J Am Soc Nephrol 2015; 26: 1027–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Safran M, Kim WY, O’Connell F. et al. Mouse model for noninvasive imaging of HIF prolyl hydroxylase activity: assessment of an oral agent that stimulates erythropoietin production. Proc Natl Acad Sci USA 2006; 103: 105–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Inoue T, Kozawa E, Okada H. et al. Noninvasive evaluation of kidney hypoxia and fibrosis using magnetic resonance imaging. J Am Soc Nephrol 2011; 22: 1429–1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kida Y, Zullo JA, Goligorsky MS.. Endothelial sirtuin 1 inactivation enhances capillary rarefaction and fibrosis following kidney injury through Notch activation. Biochem Biophys Res Commun 2016; 478: 1074–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de Braganca AC, Volpini RA, Mehrotra P. et al. Vitamin D deficiency contributes to vascular damage in sustained ischemic acute kidney injury. Physiol Rep 2016; 4; pii: 12829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ohashi R, Shimizu A, Masuda Y. et al. Peritubular capillary regression during the progression of experimental obstructive nephropathy. J Am Soc Nephrol 2002; 13: 1795–1805 [DOI] [PubMed] [Google Scholar]
- 13. Kang DH, Joly AH, Oh SW. et al. Impaired angiogenesis in the remnant kidney model: I. Potential role of vascular endothelial growth factor and thrombospondin-1. J Am Soc Nephrol 2001; 12: 1434–1447 [DOI] [PubMed] [Google Scholar]
- 14. Ishii Y, Sawada T, Kubota K. et al. Injury and progressive loss of peritubular capillaries in the development of chronic allograft nephropathy. Kidney Int 2005; 67: 321–332 [DOI] [PubMed] [Google Scholar]
- 15. Bábíčková J, Klinkhammer BM, Buhl EM. et al. Regardless of etiology, progressive renal disease causes ultrastructural and functional alterations of peritubular capillaries. Kidney Int 2017; 91: 70–85 [DOI] [PubMed] [Google Scholar]
- 16. Ohashi R, Kitamura H, Yamanaka N.. Peritubular capillary injury during the progression of experimental glomerulonephritis in rats. J Am Soc Nephrol 2000; 11: 47–56 [DOI] [PubMed] [Google Scholar]
- 17. Fine LG, Norman JT.. Chronic hypoxia as a mechanism of progression of chronic kidney diseases: from hypothesis to novel therapeutics. Kidney Int 2008; 74: 867–872 [DOI] [PubMed] [Google Scholar]
- 18. Pauletto P, Rattazzi M.. Inflammation and hypertension: the search for a link. Nephrol Dial Transplant 2006; 21: 850–853 [DOI] [PubMed] [Google Scholar]
- 19. Solak Y, Afsar B, Vaziri ND. et al. Hypertension as an autoimmune and inflammatory disease. Hypertens Res 2016; 39: 567–573 [DOI] [PubMed] [Google Scholar]
- 20. Chen II,, Prewitt RL,, Dowell RF.. Microvascular rarefaction in spontaneously hypertensive rat cremaster muscle. Am J Physiol 1981; 241: H306–H310 [DOI] [PubMed] [Google Scholar]
- 21. Gasser P, Bühler FR.. Nailfold microcirculation in normotensive and essential hypertensive subjects, as assessed by video-microscopy. J Hypertens 1992; 10: 83–86 [DOI] [PubMed] [Google Scholar]
- 22. Triantafyllou A, Anyfanti P, Triantafyllou G. et al. Impaired metabolic profile is a predictor of capillary rarefaction in a population of hypertensive and normotensive individuals. J Am Soc Hypertens 2016; 10: 640–646 [DOI] [PubMed] [Google Scholar]
- 23. Ciuffetti G, Schillaci G, Innocente S. et al. Capillary rarefaction and abnormal cardiovascular reactivity in hypertension. J Hypertens 2003; 21: 2297–2303 [DOI] [PubMed] [Google Scholar]
- 24. Abdel-Qadir H, Ethier JL, Lee DS. et al. Cardiovascular toxicity of angiogenesis inhibitors in treatment of malignancy: a systematic review and meta-analysis. Cancer Treat Rev 2017; 53: 120–127 [DOI] [PubMed] [Google Scholar]
- 25. van der Veldt AAM, de Boer MP, Boven E. et al. Reduction in skin microvascular density and changes in vessel morphology in patients treated with sunitinib. Anticancer Drugs 2010; 21: 439–446 [DOI] [PubMed] [Google Scholar]
- 26. Bonventre JV. Can we target tubular damage to prevent renal function decline in diabetes? Semin Nephrol 2012; 32: 452–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ballermann BJ, Obeidat M.. Tipping the balance from angiogenesis to fibrosis in CKD. Kidney Int Suppl 2014; 4: 45–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Grgic I, Campanholle G, Bijol V. et al. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int 2012; 82: 172–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Choi YJ, Chakraborty S, Nguyen V.. Peritubular capillary loss is associated with chronic tubulointerstitial injury in human kidney: altered expression of vascular endothelial growth factor. Hum Pathol 2000; 31: 1491–1497 [DOI] [PubMed] [Google Scholar]
- 30. Kida Y, Tchao BN, Yamaguchi I.. Peritubular capillary rarefaction: a new therapeutic target in chronic kidney disease. Pediatr Nephrol 2014; 29: 333–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dimmeler S, Assmus B, Hermann C. et al. Fluid shear stress stimulates phosphorylation of Akt in human endothelial cells: involvement in suppression of apoptosis. Circ Res 1998; 83: 334–341 [DOI] [PubMed] [Google Scholar]
- 32. Pober JS, Min W, Bradley JR.. Mechanisms of endothelial dysfunction, injury, and death. Annu Rev Pathol 2009; 4: 71–95 [DOI] [PubMed] [Google Scholar]
- 33. Hakroush S, Moeller MJ, Theilig F. et al. Effects of increased renal tubular vascular endothelial growth factor (VEGF) on fibrosis, cyst formation, and glomerular disease. Am J Pathol 2009; 175: 1883–1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Venkatachalam MA, Weinberg JM, Kriz W. et al. Failed tubule recovery, AKI-CKD transition, and kidney disease progression. J Am Soc Nephrol 2015; 26: 1765–1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci 2012; 33: 207–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gewin L, Zent R, Pozzi A.. Progression of chronic kidney disease: too much cellular talk causes damage. Kidney Int 2017; 91: 552–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ninichuk V, Gross O, Segerer S. et al. Multipotent mesenchymal stem cells reduce interstitial fibrosis but do not delay progression of chronic kidney disease in collagen4A3-deficient mice. Kidney Int 2006; 70: 121–129 [DOI] [PubMed] [Google Scholar]
- 38. Tanaka T, Kojima I, Ohse T. et al. Cobalt promotes angiogenesis via hypoxia-inducible factor and protects tubulointerstitium in the remnant kidney model. Lab Invest 2005; 85: 1292–1307 [DOI] [PubMed] [Google Scholar]
- 39. Kairaitis LK, Wang Y, Gassmann M. et al. HIF-1alpha expression follows microvascular loss in advanced murine adriamycin nephrosis. Am J Physiol Renal Physiol 2005; 288: F198–F206 [DOI] [PubMed] [Google Scholar]
- 40. Grone HJ, Simon M, Grone EF.. Expression of vascular endothelial growth factor in renal vascular disease and renal allografts. J Pathol 1995; 177: 259–267 [DOI] [PubMed] [Google Scholar]
- 41. Lindenmeyer MT, Kretzler M, Boucherot A. et al. Interstitial vascular rarefaction and reduced VEGF-A expression in human diabetic nephropathy. J Am Soc Nephrol 2007; 18: 1765–1776 [DOI] [PubMed] [Google Scholar]
- 42. Schrijvers BF, Flyvbjerg A, Tilton RG. et al. Pathophysiological role of vascular endothelial growth factor in the remnant kidney. Nephron Exp Nephrol 2005; 101: e9–e15 [DOI] [PubMed] [Google Scholar]
- 43. Kawakami T, Mimura I, Shoji K. et al. Hypoxia and fibrosis in chronic kidney disease: crossing at pericytes. Kidney Int Suppl 2014; 4: 107–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lin S-L, Chang F-C, Schrimpf C. et al. Targeting endothelium-pericyte cross talk by inhibiting VEGF receptor signaling attenuates kidney microvascular rarefaction and fibrosis. Am J Pathol 2011; 178: 911–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Park JE, Keller GA, Ferrara N.. The vascular endothelial growth factor (VEGF) isoforms: differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol Biol Cell 1993; 4: 1317–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ngo DT, Farb MG, Kikuchi R. et al. Antiangiogenic actions of vascular endothelial growth factor-A165b, an inhibitory isoform of vascular endothelial growth factor-A, in human obesity. Circulation 2014; 130: 1072–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kramann R, Wongboonsin J, Chang-Panesso M. et al. Gli1+ pericyte loss induces capillary rarefaction and proximal tubular injury. J Am Soc Nephrol 2017; 28: 776–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Armulik A, Abramsson A, Betsholtz C.. Endothelial/pericyte interactions. Circ Res 2005; 97: 512–523 [DOI] [PubMed] [Google Scholar]
- 49. Franco M, Roswall P, Cortez E. et al. Pericytes promote endothelial cell survival through induction of autocrine VEGF-A signaling and Bcl-w expression. Blood 2011; 118: 2906–2917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tanaka T. A mechanistic link between renal ischemia and fibrosis. Med Mol Morphol 2017; 50: 1–8 [DOI] [PubMed] [Google Scholar]
- 51. Ninichuk V, Anders HJ.. Bone marrow-derived progenitor cells and renal fibrosis. Front Biosci 2008; 13: 5163–5173 [DOI] [PubMed] [Google Scholar]
- 52. He J, Xu Y, Koya D. et al. Role of the endothelial-to-mesenchymal transition in renal fibrosis of chronic kidney disease. Clin Exp Nephrol 2013; 17: 488–497 [DOI] [PubMed] [Google Scholar]
- 53. Duffield JS, Humphreys BD.. Origin of new cells in the adult kidney: results from genetic labeling techniques. Kidney Int 2011; 79: 494–501 [DOI] [PubMed] [Google Scholar]
- 54. Kramann R, Humphreys BD.. Kidney pericytes: roles in regeneration and fibrosis. Semin Nephrol 2014; 34: 374–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fligny C, Duffield JS.. Activation of pericytes: recent insights into kidney fibrosis and microvascular rarefaction. Curr Opin Rheumatol 2013; 25: 78–86 [DOI] [PubMed] [Google Scholar]
- 56. Tsai Y-C, Chiu Y-W, Tsai J-C. et al. Association of angiopoietin-2 with renal outcome in chronic kidney disease. PLoS One 2014; 9: e108862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schrimpf C, Teebken OE, Wilhelmi M. et al. The role of pericyte detachment in vascular rarefaction. J Vasc Res 2014; 51: 247–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kim JS, Lee JH, Kwon O. et al. Rapid deterioration of preexisting renal insufficiency after autologous mesenchymal stem cell therapy. Kidney Res Clin Pract 2017; 36: 200–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Peired AJ, Sisti A, Romagnani P.. Mesenchymal stem cell-based therapy for kidney disease: a review of clinical evidence. Stem Cells Int 2016; 2016: 4798639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pertovaara L, Kaipainen A, Mustonen T.. Vascular endothelial growth factor is induced in response to transforming growth factor-beta in fibroblastic and epithelial cells. J Biol Chem 1994; 269: 6271–6274 [PubMed] [Google Scholar]
- 61. Sun D, Ma Y, Han H. et al. Thrombospondin-1 short hairpin RNA suppresses tubulointerstitial fibrosis in the kidney of ureteral obstruction by ameliorating peritubular capillary injury. Kidney Blood Press Res 2012; 35: 35–47 [DOI] [PubMed] [Google Scholar]
- 62. Adair A, Mitchell DR, Kipari T. et al. Peritubular capillary rarefaction and lymphangiogenesis in chronic allograft failure. Transplantation 2007; 83: 1542–1550 [DOI] [PubMed] [Google Scholar]
- 63. Hamar P, Kerjaschki D.. Blood capillary rarefaction and lymphatic capillary neoangiogenesis are key contributors to renal allograft fibrosis in an ACE inhibition rat model. Am J Physiol Heart Circ Physiol 2016; 311: H981–H990 [DOI] [PubMed] [Google Scholar]
- 64. Lee DH, Wolstein JM, Pudasaini B. et al. INK4a deletion results in improved kidney regeneration and decreased capillary rarefaction after ischemia-reperfusion injury. Am J Physiol Renal Physiol 2012; 302: F183–F191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Battegay EJ, de Miguel LS, Petrimpol M. et al. Effects of anti-hypertensive drugs on vessel rarefaction. Curr Opin Pharmacol 2007; 7: 151–157 [DOI] [PubMed] [Google Scholar]
- 66. Remuzzi A, Sangalli F, Macconi D. et al. Regression of renal disease by angiotensin II antagonism is caused by regeneration of kidney vasculature. J Am Soc Nephrol 2016; 27: 699–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Penna GLdeA, Garbero R, de F, Neves MF. et al. Treatment of essential hypertension does not normalize capillary rarefaction. Clinics 2008; 63: 613–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Debbabi H, Uzan L, Mourad JJ. et al. Increased skin capillary density in treated essential hypertensive patients. Am J Hypertens 2006; 19: 477–483 [DOI] [PubMed] [Google Scholar]
- 69. Lundwall K, Jörneskog G, Jacobson SH. et al. Paricalcitol, microvascular and endothelial function in non-diabetic chronic kidney disease: a randomized trial. Am J Nephrol 2015; 42: 265–273 [DOI] [PubMed] [Google Scholar]
- 70. Koleganova N, Piecha G, Ritz E. et al. Calcitriol ameliorates capillary deficit and fibrosis of the heart in subtotally nephrectomized rats. Nephrol Dial Transplant 2009; 24: 778–787 [DOI] [PubMed] [Google Scholar]
- 71. Hubbard BP, Sinclair DA.. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol Sci 2014; 35: 146–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kida Y, Goligorsky MS.. Sirtuins, cell senescence, and vascular aging. Can J Cardiol 2016; 32: 634–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Potente M, Ghaeni L, Baldessari D. et al. SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev 2007; 21: 2644–2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Guarani V, Deflorian G, Franco CA. et al. Acetylation-dependent regulation of endothelial Notch signalling by the SIRT1 deacetylase. Nature 2011; 473: 234–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Vasko R, Xavier S, Chen J. et al. Endothelial sirtuin 1 deficiency perpetrates nephrosclerosis through downregulation of matrix metalloproteinase-14: relevance to fibrosis of vascular senescence. J Am Soc Nephrol 2014; 25: 276–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sato W, Tanabe K, Kosugi T. et al. Selective stimulation of VEGFR2 accelerates progressive renal disease. Am J Pathol 2011; 179: 155–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gupta N, Wish JB.. Hypoxia-inducible factor prolyl hydroxylase inhibitors: a potential new treatment for anemia in patients with CKD. Am J Kidney Dis 2017; 69: 815–826 [DOI] [PubMed] [Google Scholar]
- 78. Choi HY, Lee HG, Kim BS. et al. Mesenchymal stem cell-derived microparticles ameliorate peritubular capillary rarefaction via inhibition of endothelial-mesenchymal transition and decrease tubulointerstitial fibrosis in unilateral ureteral obstruction. Stem Cell Res Ther 2015; 6: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Gatti S, Bruno S, Deregibus MC. et al. Microvesicles derived from human adult mesenchymal stem cells protect against ischaemia-reperfusion-induced acute and chronic kidney injury. Nephrol Dial Transplant 2011; 26: 1474–1483 [DOI] [PubMed] [Google Scholar]
- 80. Cantaluppi V, Gatti S, Medica D. et al. Microvesicles derived from endothelial progenitor cells protect the kidney from ischemia-reperfusion injury by microRNA-dependent reprogramming of resident renal cells. Kidney Int 2012; 82: 412–427 [DOI] [PubMed] [Google Scholar]
- 81. Chen CL, Chou KJ, Fang HC. et al. Progenitor-like cells derived from mouse kidney protect against renal fibrosis in a remnant kidney model via decreased endothelial mesenchymal transition. Stem Cell Res Ther 2015; 6: 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Collett JA, Traktuev DO, Mehrotra P. et al. Human adipose stromal cell therapy improves survival and reduces renal inflammation and capillary rarefaction in acute kidney injury. J Cell Mol Med 2017; 21: 1420–1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Panés J, García-Olmo D, Van Assche G. et al. Expanded allogeneic adipose-derived mesenchymal stem cells (Cx601) for complex perianal fistulas in Crohn's disease: a phase 3 randomised, double-blind controlled trial. Lancet 2016; 388: 1281–1290 [DOI] [PubMed] [Google Scholar]