

Abstract
A 14-year-old girl with a history of mid-line defects, basal encephalocele and morning glory disc anomaly presented with untreated growth hormone deficiency, pubertal delay and hypothyroidism. She was found to have a large craniopharyngeal canal based on MRI scan. Craniopharyngeal canal is an uncommon condition that has not been well described in the pediatric population. Consideration of craniopharyngeal canal in the differential diagnosis for basal encephaloceles and understanding its presentation can impact medical decision making and follow-up for patients.
INTRODUCTION
In 1886, Romiti et al. published the first described craniopharyngeal canal. Shortly thereafter in 1888, the presence of ectopic pituitary tissue in the nasopharynx was described. In 1950, Arey et al. reviewed autopsy data from 11 past studies, likely yielding the most accurate incidence estimation of craniopharyngeal canal to date. There was an aggregate of 8338 mostly adult subjects with an estimated incidence of 0.42% [1]. There has been limited data on the incidence craniopharyngeal canal in the pediatric population, although one would expect a similar incidence to adults as the etiology is likely congenital. The largest study was performed by Rizzo et al. in 1901 which included a subset of 44 pediatric autopsies. In that group, only one patient, a 15-year-old boy, was identified with having a craniopharyngeal canal [2]. Case reports of craniopharyngeal canal have been published but are sparse. The natural history and underlying disease associations, especially in the pediatric population, still remain unclear.
We present a case of a 14-year-old girl with a history of mid-line defects, basal encephalocele and morning glory disc anomaly that presented with untreated hypopituitarism and was found to have a craniopharyngeal canal.
CASE REPORT
A 14-year-old African–American female presented as a referral for evaluation of short stature. She has a history of basal meningoencephalocele [repaired during infancy], left-eye blindness due to morning glory disc anomaly, and cleft lip and palate [repaired during infancy]. Growth curves showed all of her growth parameters below −5 SD. Her estimated growth velocity was ~1 cm per year, annualized over 6 months. The mother reported normal growth during early infancy that slowed as she neared the age of 1 year. She was referred to another endocrinology group for growth failure at the age of 2 years.
On physical exam she was noted to have Tanner stage 1 breast development with lipomastia. No pubic hair or body odour was noted. She reported being premenarchal. She appeared small for age but exhibited a proportional habitus. Height 118.9 cm [−6.5 SD], weight 27.0 kg [−5 SD], BMI 19.1 kg/m2 [−0.2 SD], arm span 118.2 cm, upper segment 61 cm. Healed facial burns were noted from a scald injury during early childhood. Cardiac, respiratory, abdominal and genitourinary exam were unremarkable.
She denied symptoms and did not exhibit any manifestations of hypothyroidism except for dry skin. She also denied symptoms of adrenal insufficiency including hypoglycemic, hypotension and syncopal events. Mother noted that the patient had hypoglycemic during early childhood but this is now resolved. She denied any symptoms of diabetes insipidus including polyuria and polydipsia.
The patient was previously evaluated by endocrinology at 2 years of age but had been lost to follow-up at around the age of 3 years. During this time, she was diagnosed with growth hormone deficiency and hypothyroidism. Treatments were stopped as she was lost to follow-up. She had a non-contrast MRI of the brain at the age of 4 years, performed at our institution, that showed ‘broad depression of the sphenoid planum with meningoencephalocele’. The pituitary could not be visualized in full.
She has a history of left-eye blindness diagnosed at the age of 2 years. The mother stated that this was diagnosed by an optometrist. Records from her primary care provider confirm that she has a morning glory disc anomaly of the left eye.
She was the product of a full-term birth, with an uncomplicated pregnancy with good prenatal care. Antenatal course was complicated by cleft lip and cleft palate. Birth weight was 6 pounds and 14 ounces. Mother did not recall her birth length. The patient was in the NICU for feeding issues surrounding cleft lip and cleft palate. She also underwent cleft lip and cleft palate repair with simultaneous repair of her basal meningoencephalocele.
Family history includes a reported maternal height of 5 feet and 6 inches and reported paternal height 6 feet and 2 inches. Maternal onset of menarche was at 15 years old. Timing for paternal onset of puberty is unknown. Mother denied issues with infertility or consanguinity. No previous family history of genetic syndromes, short stature, endocrine disorders or autoimmune conditions.
The patient had multiple risk factors for having hypopituitarism that prompted endocrine testing, this included documented growth failure, previous diagnosis of growth hormone deficiency, previous diagnosis of hypothyroidism, coarse facial features, mid-line defects [cleft-lip and cleft palate] and pubertal delay.
Endocrine testing was obtained to screen for secondary/tertiary endocrinopathies. Cortisol level [8am]: 3.31 µg/dL [<10], 250 µg cosyntropin stimulation test peak cortisol level: 19.50 µg/dL [>18], ACTH level: 39 pg/mL [6–48], TSH: 2.110 mcIU/mL [0.5–4.5], free T4: 0.48 ng/dL [0.8–2.0], LH: 0.091 mIU/mL [0.4–11.7], FSH: 0.609 mIU/mL [1.0–9.2], insulin-like growth factor I [IGF-I]: 24 ng/mL [211–547], IGF binding protein 3 [IGFBP-3]: 0.7 mg/L [2.2–5.9]. Microarray showed normal female genotype, 46XX. Bone age according to standards of Greulich and Pyle is 8 years 10 months and 10 years for a chronologic age of 14 years 3 months [−5 SD].
As the patient was found to have hypopituitarism, contrast and non-contrast MRI of the brain and pituitary were obtained that revealed a craniopharyngeal canal (Fig. 1). Extension of CSF, meninges and the sellar contents into the craniopharyngeal canal was visualized although no intrasellar adenohypophyseal or neurohypophyseal tissue was identified. The transverse dimension of the defect was 6 mm and the oblique craniocaudal dimension was 7 mm. Her previously diagnosed morning glory disc anomaly was also seen. She was also noted to have a smaller than expected caliber of the orbital segment of the left optic nerve. Irregular appearance and caudal position of the cribriform plate, as well as the right fovea ethmoidalis, was also noted. Caudal protrusion of the right gyrus rectus and medial orbital gyrus was noted. The olfactory bulb could not be visualized. The patient has an intact sense of smell.
Figure 1:
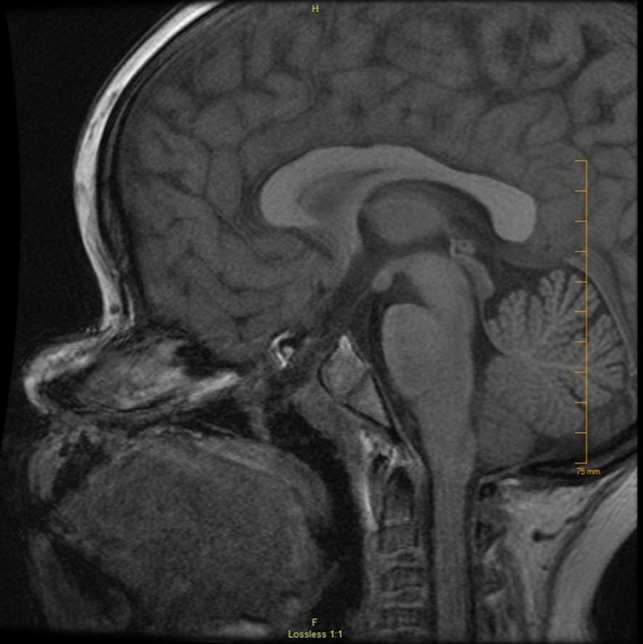
Sagittal MRI of the case demonstrating craniopharyngeal canal with a transverse measurement of 6 mm.
An MR angiogram was also obtained to help exclude Moyamoya disease, which has been associated with morning glory disc anomaly. Her MR angiogram was normal.
Unfortunately, the patient has been lost to follow-up despite multiple attempts to reach the family after her diagnosis. Therefore, we were unable to initiate therapy for any of her endocrinopthies.
DISCUSSION
Structurally, the craniopharyngeal canal is a mid-line corticated channel extending from the floor of the sella turcica to the nasopharynx. It is speculated that craniopharyngeal canal occurs as a result of the persistence of the embryologic stalk remnant of Rathkes pouch. Unlike Rathkes cleft, at 6 weeks of age, the pouch lengthens to form a narrow stalk. By the seventh week of age, the sphenoid base develops, obliterating the stalk through the transition from cartilage to cortication of the sphenoid body [3]. Persistence of the stalk occurs when there is abnormal fusion of the pre-sphenoidal and post-sphenoidal cartilages.
It has been difficult to classify craniopharyngeal canals due to the rarity of the condition and the differing clinical manifestations that can occur. The spectrum of pathology can range from a non-clinically significant defect to ectopic pituitary adenomas. In the presented case, the patient had an encephalocele and hypopituitarism, most likely due to ectopic adenohypophysis that may have been iatrogenically removed. Abele et al. proposed the first classification scheme for craniopharyngeal canal (Table 1). The classification was based on the presence of soft tissue within the defect such as adenohypophyseal tissue, a cephalocele, a tumor, or both. They found a significant difference in midpoint anteroposterior diameter between different types. Furthermore, there was an association between large Type 3 craniopharyngeal canals and ectopic adenohypophyseal tissue [4].
Table 1:
Classification of craniopharyngeal canal as proposed by Abele et al.
| Type | Description |
|---|---|
| Type 1 | Incidental canal |
| Type 2 | Canal with ectopic adenohypophysis |
| Type 3a | Canal containing cephalocele |
| Type 3b | Canal containing tumor |
| Type 3c | Canal containing both cephalocele and tumor |
Ectopy of pituitary tissue has been well established in the past in the context of basal encephaloceles. Encephaloceles have been classified into 2 subtypes based on the skull defect they originate from: transethmoidal and transsphenoidal [5]. Transsphenoidal encephaloceles are thought to originate from the craniopharyngeal canal and comprise of approximately 5% of basal encephaloceles. However, it is estimated that 50–60% of basal encephaloceles are associated hypopituitarism, prior to repair. The incidence of hypopituitarism in transsphenoidal basal encephaloceles was estimated to be ~60–78% [6, 7]. In the series published by Abele, all 4 patients with Type 3 A [encephaloceles] had hypopituitarism, prior to any corrective procedures [4].
Based on the proposed classification scheme by Abele et al., our case has a large Type 3 A with a midpoint anteroposterior diameter of 6 mm. Subsequently, it is possible that the patient had ectopic adenohypophyseal tissue that was iatrogenically removed during repair of her basal encephalocele during infancy. This is a consideration as the patient currently has tissue present in the canal that may represent ectopic adenohypophyseal tissue. This may also explain why the patient still has appropriate adrenal function. It is also possible that the patient has ectopic adenohypophyseal tissues that remains to be detected by MRI. Another etiology would be a hypoplastic or dysplastic adenohypophysis. Persons with reduction deformities of the brain, such as those with holoprosencephaly, have been shown to have abnormal formation of the sphenoid bone, which is the hypothesized mechanism for persistent craniopharyngeal canal [8]. There is also an association with hypopituitarism due to dysplastic pituitary formation in patients with reduction deformities of the brain [6]. Subsequently, one may postulate that persistent craniopharyngeal canal may be the lower-end of a spectrum of reduction deformities of the brain.
The presented case also had morning glory disc anomaly. Associations between encephaloceles and ocular manifestations, such as morning glory disc anomaly, optic nerve hypoplasia and hypertelorism, and have been described; interestingly, many of those patients also had associated brain reduction deformities such as agenesis of the corpus callosum [9]. There have also been case reports of morning glory disc anomaly in the context of craniopharyngeal canal as well [10]. In those cases, hypopituitarism ensued with progressive loss of hormonal function [7]. The natural history of these patients with ocular anomalies and craniopharyngeal canal are still unclear.
ACKNOWLEDGEMENTS
There are no acknowledgements for this study.
CONFLICT OF INTEREST STATEMENT
All authors have no conflicts of interest to disclose.
FUNDING
No funding was secured for this study.
ETHICAL APPROVAL
No ethical approval was needed.
CONSENT
Consent was obtained from patient’s guardian.
GUARANTOR
Nader Kasim is the assigned guarantor for this study.
REFERENCES
- 1. Arey LB. The craniopharyngeal canal reviewed and reinterpreted. Anat Rec 1950;106:1–16. [DOI] [PubMed] [Google Scholar]
- 2. Cave AJ. The craniopharyngeal canal in man and anthropoids. J Anat 1931;65:363–7. [PMC free article] [PubMed] [Google Scholar]
- 3. Cho KH, Chang H, Yamamoto M, Abe H, Rodriguez-Vazquez JF, Murakami G, et al. Rathke’s pouch remnant and its regression process in the prenatal period. Childs Nerv Syst 2013;29:761–9. [DOI] [PubMed] [Google Scholar]
- 4. Abele TA, Salzman KL, Harnsberger HR, Glastonbury CM. Craniopharyngeal canal and its spectrum of pathology. AJNR Am J Neuroradiol 2014;35:772–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Macfarlane R, Rutka JT, Armstrong D, Phillips J, Posnick J, Forte V, et al. Encephaloceles of the anterior cranial fossa. Pediatr Neurosurg 1995;23:148–58. [DOI] [PubMed] [Google Scholar]
- 6. Smith DE, Murphy MJ, Hitchon PW, Babin RW, Abu-Yousef MM. Transsphenoidal encephaloceles. Surg Neurol 1983;20:471–80. [DOI] [PubMed] [Google Scholar]
- 7. Morioka M, Marubayashi T, Masumitsu T, Miura M, Ushio Y. Basal encephaloceles with morning glory syndrome, and progressive hormonal and visual disturbances: case report and review of the literature. Brain Dev 1995;17:196–201. [DOI] [PubMed] [Google Scholar]
- 8. Kjaer I, Fischer-Hansen B. Human fetal pituitary gland in holoprosencephaly and anencephaly. J Craniofac Genet Dev Biol 1995;15:222–9. [PubMed] [Google Scholar]
- 9. Yokota A, Matsukado Y, Fuwa I, Moroki K, Nagahiro S. Anterior basal encephalocele of the neonatal and infantile period. Neurosurgery 1986;19:468–78. [DOI] [PubMed] [Google Scholar]
- 10. Ellika S, Robson CD, Heidary G, Paldino MJ. Morning glory disc anomaly: characteristic MR imaging findings. AJNR Am J Neuroradiol 2013;34:2010–4. [DOI] [PMC free article] [PubMed] [Google Scholar]