

Abstract
RNA-binding motif protein 20 (RBM20) is a cardiac splice regulator that adapts cardiac filling via its diverse substrates—including the sarcomeric protein titin. The molecular basis and regulation of RBM20-dependent exon exclusion are largely unknown. In tissue culture experiments, we show that the combination of RNA recognition motif (RRM) and C-terminus is necessary and sufficient for RBM20 activity, indicating an important function of the ZnF2 domain in splicing repression. Using splice reporter and in vitro binding assays targeting titin exons 241–243, we identified a minimal genomic segment that is necessary for RBM20-mediated splicing repression of the alternative exon. Here, RBM20 binds the cluster containing most RBM20 binding motifs through its RRM domain and represses the upstream and downstream introns. For subsequent exon exclusion, specific regions upstream, downstream and within the alternative exon 242 are required. Regulation of exon exclusion involves PTB4 as a novel titin splice regulator, which counteracts RBM20 repressor activity in HEK293 cells. Together, these mechanistic insights into the regulation and action of RBM20 and PTB4 provide a basis for the future development of RBM20 modulators that adapt titin elasticity in cardiac disease.
INTRODUCTION
Alternative splicing plays a major role in regulating gene expression by generating protein isoforms with distinct biological properties (1). The diversity of the resulting proteome relates to altered protein–protein interactions, subcellular localization or catalytic activity. Disrupted fine tuning of the alternative splicing process can cause disease or affect disease progression and severity (2,3).
Splicing is performed by a complex macromolecular machine—the spliceosome, which is composed of five uridine-rich small ribonucleoprotein particles (snRNPs) and several non-snRNP proteins (4). Major functions of the spliceosome are to integrate regulatory signals and to catalyze the splicing reaction. Multiple cis-acting RNA elements within the pre-mRNA act as splicing signals. They define exon/intron borders, which are recognized by the splicing machinery (the 5′ and the 3′ splice sites (ss), branch point and the polypyrimidine tract). 5′ss recognition occurs through RNA–RNA base pairing when the 5′ss sequence is bound by the 5′ end of U1 snRNA. On the other side of the intron the 3′ss is defined by non-snRNP factors such as SF1/mBBP interacting with the branch point and U2AF associating with the polypyrimidine tract and the 3′ss. The characterization of spliceosome assembly intermediates in vitro suggests a carefully orchestrated process (5). Spliceosome assembly starts with the recognition of the 5′ss by the U1 snRNP, proceeds with the recruitment of the U2 snRNP to the 3′ss and juxtaposition of splice sites, and continues with the recruitment of the U4/U5.U6 tri-snRNP, rearrangement of RNA–RNA and RNA–protein interactions and finally splicing catalysis (6).
Splicing regulation is frequently executed at the early events in splice site recognition, by positively or negatively affecting U1 snRNP/U2 snRNP recruitment to 5′ and 3′ss, respectively (7). Exon definition by binding of U1 and U2 snRNP to 5′and 3′ splice sites and subsequently pairing commits the pre-mRNA to the splicing pathway (8). Recognition of splicing signals and exon and intron definition are regulated by cis-acting RNA elements known as splicing regulatory elements. Exonic/intronic splicing enhancer/silencer sequences are bound by SR proteins (serine/arginine-rich) and the hnRNPs (heterogeneous nuclear riboproteins), which canonically promote and repress splicing, respectively (9).
The SR protein RBM20 (RNA binding motif protein 20) regulates isoform expression of the sarcomeric protein titin (TTN) and several other cardiac proteins (10). It is predominantly expressed in striated muscle with highest levels in the perinatal heart and was identified in a naturally occurring mutant rat deficient in titin splicing (10). Here, RBM20 deficiency leads to predominant expression of the higher molecular weight giant titin isoform N2BA-G as well as the N2BA isoform (10). The former is expressed in embryonic development, while the adult N2B and N2BA titin isoforms are expressed at ratios that vary between species and in healthy versus diseased hearts (11). These isoforms mainly differ in the I-band region, which determines the elastic properties of titin. The RBM20-deficient rats mirror the pathological features of patients deficient in RBM20 with left ventricular dilation, progressive subendocardial fibrosis and sudden death (10,12–13).
In addition to adapting titin isoform expression and thus cardiac filling in diastole, RBM20 affects a set of >30 genes, which have been implied in diastolic function, sarcomere assembly and ion transport. It regulates different types of alternative splicing, namely exon repression, mutually exclusive exon selection, exon inclusion, intron retention and exon shuffling (10,14–15). High-throughput analysis has identified UCUU as the RBM20 binding motif as well as RBM20 binding proteins that relate to early spliceosome formation (15), but how RBM20 leads to exon exclusion is largely unknown.
The splice factor PTB4 (alias PTBP1 or hnRNPI) is a member of the hnRNPs and can promote exon inclusion (16). It is expressed in the heart, skeletal muscle and the brain and is important for early embryonic development as the knockout is lethal (17).
In this study we characterize the molecular basis of RBM20-dependent alternative exon exclusion. We find RNA recognition motif (RRM) and C-terminus of RBM20 and a 994-nt minimal genomic region within the titin pre-mRNA essential to repress splicing. The minimal region contains exon 242 and its flanking introns and is necessary and sufficient for RBM20-mediated splicing regulation. In addition, RBM20 interferes with the removal of the upstream and downstream introns to prevent inclusion of titin exon 242. Mechanistically, the RRM of RBM20 binds to the downstream intronic region of the minimal genomic segment described above, where it competes with PTB4 to antagonistically affect exon inclusion.
MATERIALS AND METHODS
Molecular cloning
The titin expression construct (TTN241-3) was cloned by inserting human genomic titin fragment (GenBank NC_000002.12, chr2: 178622670–178625396), containing titin exons 241–243 together with intervening introns, into the pcDNA3.1zeo vector (Invitrogen). Exon 242 is part of I-band region of titin. Subsequent deletion mutants were created by polymerase chain reaction (PCR)-based gene assembly (please contact authors for primer sequences). The human hemoglobin beta expression construct was cloned by inserting a fragment containing exons 1–3 and intervening introns into the vector pcDNA3.1zeo (Invitrogen). Subsequent deletion mutants were created by PCR amplification and cloned into the same vector. Hybrid human titin/human beta globing constructs were created by subcloning PCR-amplified fragments into the pcDNA3.1zeo vector (Invitrogen). We cloned human RBM20 (the accession number in NCBI is NM_001134363) and its mutants as cDNA into pcDNA3.1(-)A-myc-his via NheI/KpnI sites. The RBM20-RRM (aa 511–601) was cloned into pGEX-6P-1 via BamHI/XhoI sites.
For all cloning amplifications we used the proofreading DNA polymerases (Phusion or Q5 from NEB) and final constructs were sequence-verified.
Cell culture and transfection
We cultured HEK293 cells in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (v/v) Fetal Bovine Serum (FBS), 100 U/ml penicillin and 100 ug/ml streptomycin. We used PEI40 (HEK293 cells) or Lipofectamine 2000 (Invitrogen) (C2C12, H9C2 cells) to transfect plasmids in DMEM without antibiotics (20 min complex formation, 48 h incubation). For cotransfection with RBM20 and PTBs we used equimolar ratios. DNA/PEI40 ratio of 1:3 at 1 µg DNA/per well was used for transfection of a 6-well dish. Total RNA was collected 48 h after transfection.
RNA isolation, RT- and qRT-PCR (SYBR Green)
To isolate total RNA from cells, we used Trizol (Invitrogen). Preparations of <2 μg of total RNA were treated with DNase I (Thermo Fisher Scientific), according manufacturer instructions. For first-strand cDNA synthesis, we used random octamers and oligo dT-16 primers and MuLV reverse transcriptase (High capacity RNA-to-cDNA kit, Thermo Fisher Scientific). PCR amplification was limited to 30 cycles with 60°C annealing temperature. We separated the PCR products on 3% agarose gel. PCR primer sequences are provided in the Supplementary Table S1. All experiments were repeated three times.
Quantitative reverse transcriptase-PCR (qRT-PCR) was performed using SYBR Green master mix (Applied Biosystems) in a 7900 HT cycler (Applied Biosystems). qRT-PCR primers are listed in Supplemental Table SII. The quantification of the gene expression was performed using the ΔΔCT method. Relative levels of splice isoforms are presented as a ratio of mRNAs, with exon 242 included, versus mRNAs, with exon 242 skipped. The fold change in inclusion/skipping ratio was obtained when compared to the control (transfection with RBM20 expressing plasmid). n = 3 for all samples, all data are expressed as mean ± SEM. Group comparisons were analyzed by one-way ANOVA and Bonferoni post test. P values were considered statistically significant as follows: *P < 0.05; **P < 0.01; ***P < 0.001.
In vitro transcription
Amplified PCR products were used as templates for in vitro transcription. Each template was generated using a forward primer containing a T7 polymerase promoter sequence [GGG to provide an efficient initiation site for transcription] and a sequence complementary to the beginning of the template. The reverse primer was complementary to the last nucleotides of a template. The PCR products were purified (MSB Spin PCR Rapace, Stratec Molecular) and used as templates for in vitro transcription. Uncapped, 32P-labeled transcripts (rUTP as the labeled nucleotide) were synthesized by in vitro run-off transcription from the purified PCR templates using T7 RNA polymerase (Promega).
Primer extension assays with splice junction oligonucleotides
Primer extension assays with splice junction oligonucleotides were carried out as described (18). Total RNA was extracted from for 48 h transfected HEK293 cells, enriched for mRNA using Promega PolyATract mRNA Isolation Systems III and IV kit (Cat Z5310) and ∼2 μg of mRNA used to carry out primer extension assay with indicated single 5′-labeled oligonucleotide (listed in Supplementary Table SIV).
Western blotting
We harvested transfected cells and lysed them in Radioimmunoprecipitation assay (RIPA) buffer. Protein concentration was measured using the BCA kit (Bicinchoninic Acid Assay - Thermo Scientific). We loaded 50 μg (or 100 μg) of total protein in a well. Proteins were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and subjected to immunoblotting. Membranes were probed with mouse antibodies against anti-c-Myc (13–2500, Invitrogen) and anti-α-Tubulin (DM1A, Calbiochem). The secondary antibody was HRP-conjugate anti-mouse IgG (H+L) (Calbiochem). For detection, we used an ECL kit (Thermo Scientific).
Recombinant proteins
Recombinant GST-tagged proteins were expressed in Escherichia coli Rosetta 2 strain (Novagen). Proteins were purified under native conditions by standard GST affinity chromatography, as described by the manufacturer (Sigma). Purified proteins were cleaved from the GST-tag with PreScission protease (GE Healthcare) and dialyzed against 20 mM HEPES (pH 7.9), 100 mM KCl, 20% glycerol, 0.2 mM ethylenediaminetetraacetic acid (EDTA), 0.5 mM Dithiothreitol (DTT) buffer and stored at −80°C. Quality of purification was assayed on an SDS-gel (protein marker PageRuler Prestained Plus Protein Ladder from Thermo Scientific was used) and the final protein concentration was determined using NanoDrop (Thermo Scientific).
Gel mobility shift assays
Each gel mobility shift reaction (10 μl) contained: 20 mM HEPES pH 7.9, 100 mM KCl, 3.2 mM MgCl2, 0.2 mM EDTA, 0.25 mM DTT, 15% glycerol, tRNA (1 mg/ml), heparin (0.5 mg/ml), the RNA probe (50 000 cpm, ∼10 fmol) and recombinant protein, where indicated recombinant human RBM20-RRM (50 nM–1μM). RNA probes were denatured at 80°C for 5 min and put on ice. All reaction components were mixed and incubated for 10 min at 30°C, then placed on ice and separated on an 8% native polyacrylamide gel. The apparent Kd was estimated as the concentration of protein where the RNA was half bound (19). Gel mobility shift assays using recombinant PTB4 were performed as described (19).
RESULTS
Identification of the genomic sequence necessary for RBM20-mediated titin exon 242 repression
We previously reported a role for RBM20 in regulating cardiac isoform expression, including conservation of target exons within titin (10). To study the molecular basis of RBM20-dependent titin exon exclusion, we based our analysis on human TTN exons 241–243, which is part of titin's I-band region—a main determinant of the elastic properties of the titin filament. The exon/intron cassette was transcribed under control of the CMV promoter (splicing reporter TTN241-3, Figure 1A). Transient transfection of TTN241-3 with or without human RBM20 expression resulted in a splicing pattern consistent with our predictions from the RNAseq analysis of rat and human cardiac transcriptomes (10) in all cell-lines tested (Figure 1B). RBM20 repression activity in HEK293 cells (human fibroblasts) appeared to be higher than in mouse or rat myoblasts (C2C12 or H9C2 cells) (Figure 1B). Accordingly, we selected HEK293 cells to study RBM20-mediated cassette exon repression and demonstrated that RBM20 is sufficient to regulate the alternative splicing of this pre-mRNA using quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR). In vivo, this region responds to RBM20, as TTN exons 241–243 are differentially regulated in the RBM20 deficient rat heart (Supplementary Figure S1E).
Figure 1.

Human titin-derived splicing reporters respond to RBM20-mediated splicing repression in cell culture. (A) The splicing reporter TTN241-3 contains the human titin (TTN) genomic sequence from exon 241 to 243. Exon 242 is excluded in the presence of RBM20. Alternative exons are represented as light boxes, constitutive exons as dark boxes and introns as lines (bp = base pairs). (B) Validation of RBM20-dependent alternative splicing by RT-PCR. Transient transfection of HEK293, C2C12 and H9C2 cells with the TTN241-3 and empty vector (−) or a plasmid expressing human RBM20 (+) produces the expected RT-PCR products—with and without the alternative exon 242 (top and bottom band, respectively). Quantification of three independent transfections are shown as percentages of mRNA excluding the alternative exon. (C) TTN241-3 deletion mutants, generated to identify the RBM20-responsive splicing repression element in the upstream intron. Dotted lines represent deleted sequences and the blue line a sequence replaced with the first intron of the human β-globin gene. (D) Analysis of RBM20-dependent alternative splicing by RT-PCR in HEK293 cells transiently transfected with each reporter construct, as indicated. The RT-PCR product at 268 bp represents mRNA species that does not contain exon 242. Ratio of exon inclusion/exclusion was reduced to 4% only in the X1 replacement construct, while deletion in construct Δ3 enhances the RBM20 effect (quantification of three independent transfections). (E) TTN241-3 deletion mutants generated to identify the RBM20-responsive splicing repression element in the downstream intron. Dotted lines indicate deleted sequences. (F) Analysis of RBM20-dependent alternative splicing of deletion constructs affecting the downstream intron of TTN241-3. Constructs where deletions extend into the mid intronic region (Δ4 and Δ5, indicated with a gray box) reduce their repressor activity to <10% and Δ7 to <30%. Presence of the mid intronic region (indicated with a black box, construct Δ6) restores repressor activity (quantification of three independent transfections). (G) TTN241-3 deletion mutants generated to integrate the information on splicing responsive elements and truncate the minigene for subsequent analysis. Thin (gray) lines indicate TTN sequences, dotted lines indicate deleted sequences. (H) Analysis of RBM20-dependent alternative splicing of deletion construct affecting both the upstream and the downstream introns of TTN241-3. RBM20 responsiveness is maintained in construct Δ10 that lacks >50% of the downstream intron sequence (quantification of three independent transfections).
To identify the minimal RBM20 responsive genomic segment within the TTN exons 241–243, we constructed a series of TTN241-3 deletion mutants. In the first set of mutants, depicted in Figure 1C, we sequentially deleted ∼100 bp fragments from the TTN241-3 upstream intron (Figure 1C, splicing reporter Δ1, Δ2 and Δ3). Although the repression was slightly increased, none of these deletions dramatically affected RBM20-mediated exon repression (Figure 1D). As indicated by quantification of the PCR products, alternative exon exclusion varies between 75 and 90% (Figure 1D, reporter TTN241-3). However, exchanging the last 146 nt of the upstream intron for the corresponding sequence of the human β-globin gene first intron resulted in a complete loss of RBM20 response (Figure 1C, reporter X1), underscoring the absolute requirement of this region for exon repression.
We applied the same approach to the downstream intron of the TTN241-3 reporter and found that a 910 nt long segment of the downstream intron, starting directly after the 5′ss, is required to retain RBM20 repressor activity (Figure 1E, reporter Δ6). Neither a 118 nt segment, nor a 466 nt were sufficient to mediate exon repression (Figure 1E, reporters Δ4 and Δ5). Subsequent deletions within the identified 910 nt long segment, retained repressor activity (Figure 1E, reporters Δ7 and Δ8). However, repression was not as efficient as with the splicing reporter, containing the full length fragment. Retaining sequences toward the middle of the downstream intron resulted in the activation of a cryptic 5′ss (Figure 1E, reporter Δ9; usage of the cryptic splice site was confirmed by sequencing).
Based on these results we maximally downsized upstream and downstream introns to create a TTN241-3 mini reporter (Figure 1G, Δ10). The response of the Δ10 reporter to RBM20 is slightly stronger than the full-length reporter (in Figure 1H; TTN241-3 versus Δ10). The band above the three joint exons-band (Figure 1H, lane 3) retains the intron sequence (verified by sequencing) and probably reflects removal of ISE sequences from this intron. Creation of an additional titin mini reporter, containing only 118 nt of the downstream intron (Supplementary Figure S1C), conveyed insufficient repressor activity of RBM20 (Supplementary Figure 1D compare TTN241-3 and Δ15). To identify the potential ISE region in the upstream intron we created the second set of mutants where we gradually re-inserted ∼150 bp long fragments from the original upstream intron (Supplementary Figure S1A, reporters Δ12, Δ13 and Δ14). Repression activity of the mutant splicing reporters was slightly increased compared to the wild-type (WT) reporter (Supplementary Figure S1B), suggesting that we did not eliminate sequences from the upstream intron that are essential for splicing repressor activity. Our deletion mutants within the upstream intron address splicing activation activity to the region between nt 278 and 440 (Supplementary Figure S1B, reporter Δ14 versus Δ11), as this reporter preferably spliced out the upstream intron sequence rather than included them (results confirmed by sequencing RT-PCR products).
Collectively these results indicate that a human titin genomic segment containing the regulated exon with the 102 and 910 nt of the upstream and downstream introns, respectively, is sufficient to mediate RBM20-dependent exon repression in cell culture.
A 994 nt titin genomic segment is sufficient to convert a human β-globin cassette into an RBM20 responsive splicing reporter
To confirm that the identified titin genomic segment is sufficient to mediate a RBM20 response, we inserted it into an RBM20 independent splicing reporter derived from the human β-globin gene, replacing the alternative exon and flanking intronic regions (Figure 2A, reporter X6). The human β-globin splicing reporter itself (Figure 2A, BG) or its derivative with a shortened second intron (Figure 2A, ΔBG) did not respond to RBM20 (Figure 2B). In the first set of mutants (Figure 2A), we tested the length requirement of the downstream intron segment for RBM20-mediated splicing repression. The length of the upstream intron segment was kept constant at 64 nt. Reduction of the downstream intron segment from 910 to 662 nt retained repressor activity of RBM20 (Figure 2B). Short 59 to 118 nt stretches of the downstream intron were not sufficient to efficiently support RBM20-mediated exon repression (Figure 2B, X2 and X3). Exon repression activity with the first 598 nt of the downstream intron included in the reporter was intermediate (Figure 2B, reporter X4).
Figure 2.

Transfer of RBM20 responsiveness from the titin to the β-globin splicing reporter by gradual exchange of the middle exon and the flanking intron regions. (A) Human titin/β-globin hybrid splicing reporters generated to analyze the minimal titin RBM20-responsive repressor element in the heterologous contents. Hybrids contain increasing amounts of TTN genomic segments (gray) inserted into the human β-globin minigene (exons 1–3 with interfering introns, blue; exon numbers on top). The gray bar indicates the RBM20 responsive region, the dispensable region in black. (B) RT-PCR validation of RBM20-mediated changes in splicing of human titin/β-globin hybrid splicing reporters X2–X6. The relative amount of mRNA excluding the alternative exon was determined for each mutant. Inclusion of intron segment 118–598 restored >20% of RBM20 response (X4). The additional inclusion of 598 to 663 restored >50% (X5) (quantification of three independent transfections). (C) Human titin/β-globin hybrid splicing reporters with TTN (gray) and β-globin sequences (blue) to evaluate the contribution of exon 242 and the upstream intron sequence to RBM20-mediated splicing repression. The black bar indicates the titin region not required for RBM20 responsiveness. (D) RT-PCR validation of RBM20 effects on alternative splicing of the human BG-TTN hybrid splicing reporters X7–X8. Constructs that contained the titin downstream intron or only the alternative exon with flanking exon definition sites did respond to RBM20 (quantification of three independent transfections). (E) Human titin/β-globin hybrid splicing reporters generated to evaluate the intron sequences proximal to the 5′splice site for splice repression activity. The 153 nt mid intron segment (illustrated in thin black line) was positioned at indicated distances from the 5′ss. (F) RT-PCR validation of RBM20-mediated changes in alternative splicing of the human titin/β-globin hybrid splicing reporters X9–X11. Adding the majority of the 3′ intron to the alternative exon with immediate flanking exon definition sites transfers RBM20 responsiveness to the β-globin splice reporter (quantification of three independent transfections). (G) Human titin/β-globin hybrid splicing reporters generated to evaluate the contribution of exon 242 and the upstream intron sequence to RBM20-mediated splicing repression. (H) RT-PCR validation of RBM20 effects on splicing of the human titin/β-globin hybrid splicing reporters X12–X15. Only the combination of complete exonic sequence, flanking region and 750 bp in the downstream intron restore RBM20 responsiveness (included/excluded >84%, quantification of three independent transfections). (I) Human titin/β-globin hybrid splicing reporters to evaluate the length of exon 242 required for RBM20-dependent splicing repression. The residual length of original exon sequence is indicated above the exon box. (J) RT-PCR validation of RBM20-mediated changes in alternative splicing of the human titin/β-globin hybrid splicing reporters X16–X19. None of the shortened constructs responded efficiently to RBM20 (quantification of three independent transfections). (K) Human titin/β-globin/lacZ hybrid splicing reporters generated to evaluate the sequence of exon 242 required in RBM20 mediated splicing repression. (L) RT-PCR validation of RBM20-mediated changes in alternative splicing of the human titin/β-globin/lacZ hybrid splicing reporters indicate that exonic sequence and size contribute to RBM20 responsiveness (quantification of three independent transfections).
After our demonstration that the flanking titin exons 241 and 243 are dispensable for RBM20-mediated exon repression, we evaluated if the upstream intronic region is sufficient to mediate RBM20-dependent exon repression. We created one mutant where only the downstream intron and exon 243 sequences in the TTN241-3 reporter were exchanged to β-globin (Figure 2C, reporter X7) and one where only exon 242 from titin was present in the β-globin splicing reporter (Figure 2C, reporter X8). None of these constructs responded to the addition of RBM20 (Figure 2D), demonstrating that exon 241, upstream intron and exon 242 sequences did not contain RBM20-responsive repressor elements. In addition, we moved a stretch of 3′ intronic titin sequence which contains several RBM20-responsive repressor elements closer to the 5′ss (Figure 2E). However, only the insertion of the complete 3′ titin intronic sequence up to nt 750 results in a reporter that efficiently responds to RBM20 (Figure 2F), suggesting that the downstream intron sequence architecture is important for RBM20-mediated exon repression.
To investigate the requirement of exon 242 and the upstream intron sequences for RBM20-mediated exon repression, we exchanged titin exon 242 with β-globin exon 2 sequence except for the last 13 or 75 nucleotides upstream of the 5′ss (Figure 2G, X12 or X13, respectively). We find exon repression dependent on the exon 242, as inclusion of only 13 or 75 nt from this exon completely eliminates RBM20-mediated exon repression (Figure 2H, X12 and 13 versus X11, 14 and 15). Inclusion of the full exon 242 sequence rescues RBM20-mediated exon repression, but is not sufficient to achieve the repression level of the identified minimal genomic segment (Figure 2H, X14 versus X11). At least 64 nt from the upstream intron sequence is required for RBM20-mediated exon repression (Figure 2H, TTN241-3 versus X15 and 11). qRT-PCR analysis was performed to confirm significant differences in repressor activity of X15 and X11 splicing reporters (Supplementary Figure S2A).
To investigate the requirement of exon 242 for RBM20-mediated repression of this exon, we gradually removed sequences from the middle of exon 242 (Figure 2I, X16–X19). This reduces exon 242 length from 267 to 200, 150, 100 and 41 nt in the respective mutants. In X16 the 41 nt exon was not recognized as flanking exons were joined together (Figure 2J). All the other shortened exons were recognized as being exons, but none of these splicing reporter mutants responded to RBM20 with the same exon repression efficiency as the reporter X5 containing the WT exon 242. To differentiate a size from a sequence effect, we exchanged exon 242 titin sequence with sequences derived from the bacterial lacZ gene (Figure 2K, X20–X22). Supplemented to its natural length of 267 nt, the hybrid titin-lacZ exons did not support efficient RBM20 dependent exon removal (Figure 2L, X20 and X21). As the exon was extended to more than 1000 nt in length, it was no longer recognized by the splicing machinery as an exon and resulted in the splicing pattern where the flanking exons were joined together (Figure 2L, X22).
In summary, the 994 nt-long human titin genomic segment is sufficient to convert the human β-globin splicing reporter into an RBM20-responding splicing reporter and suggests that both flanking introns and the alternative exon sequence are required for the repression activity in RBM20-mediated splicing regulation.
Both the RRM and the carboxy-terminal region of RBM20 are necessary for exon repression
The domain structure of RBM20 is conserved between species (10) and includes an amino-terminal domain of unknown structure and function, a central part with zinc finger 1 (ZnF1), RRM-type RNA binding domain (RRM) and an arginine-serine (RS) repeat-containing region and the carboxy-terminal domain with ZnF2 (depicted in Figure 3A) (10).
Figure 3.

The RBM20 RRM and C-terminus are sufficient for titin splicing repression. (A) Schematic representation of RBM20 and the deletion mutants. The two zinc finger (ZnF) domains are depicted in blue, the RRM green, the RS-like domain in orange. Unstructured regions are indicated as small gray boxes. The WT protein and all deletion mutants carry C-terminal myc- and his-epitope tags. RBM20 amino acid residues are indicated underneath the schemes and the corresponding molecular weight of proteins is depicted in brackets. (B) RT-PCR validation of RBM20-mediated changes in processing the human titin TTN241-3 splicing reporter in HEK293 cells. Gray boxes represent mRNA species with and without the alternative exon 242 (top and bottom band, respectively). Quantification of three independent transfections are shown as percentages of mRNA excluding the alternative exon. (C) qRT-PCR (SYBR Green) analysis of HEK293 cells transfected with the TTN241-3 splicing reporter and the RBM20 expression constructs with activity in panel B: n = 3; *P < 0.05; **P < 0.01; ***P < 0.001.
We used transient transfections to examine which RBM20 protein regions are required for splicing repressor activity. We created a set of RBM20 deletion mutants (Figure 3A) and transfected HEK293 cells with the TTN241-3 reporter (Figure 1A) and the RBM20 mutants. The effect on splicing repression examined by the RT-PCR (Figure 3B). All RBM20 deletion mutants localized to the nucleus of HEK293 cells (Supplementary Figure S3B) and had the expected molecular weight as determined by western blot (Supplementary Figure S3A). The alternative exon 242 in the TTN241-3 reporter was predominantly included when splicing reporter was co-transfected with vector alone or deletion mutants C-terminal of the RRM/RS domain. It was predominantly excluded with the N-terminal deletion construct (Figure 3B). Removal of the RRM or RS domain completely abolished the repressor activity (Figure 3B, RBM20Δ2, 3, 6, 8), indicating that the RRM and RS domains are necessary but not sufficient for splicing repression. Mutants with residual activity were evaluated using qRT-PCR (Figure 3C, RBM20Δ4, 5, 7) and apart from the essential RRM and RS domain, the ZnF2 domain had the strongest residual effect on the repressor activity of RBM20.
Taken together, the activity of RBM20 depends on the RRM and RS domains, necessary but not sufficient for exon exclusion. The addition of the C-terminus including the ZnF2 is sufficient for the repressor activity of RBM20.
RBM20-RRM binds to sequences within the minimal repression segment
The RNA binding motif of RBM20 contains a UCUU core (15). In a systematic approach we used gel-shift assays on serial truncations of the reporter construct to identify the minimal repression segment. The in vitro transcribed and uniformly radiolabeled RNA was exposed to increasing amounts of purified recombinant protein containing the RBM20-RRM domain (RBM20Δ2, residues 511–601; Supplementary Figure S3A) followed by separation on non-denaturing acrylamide gels. The 994 nt RBM20-responsive RNA segment was divided into the upstream intron transcript (A), the exon (B) and serial downstream intron transcripts (Figure 4A). RBM20Δ2 bound with highest affinity to transcripts C and D. RBM20Δ2 did not bind any other segments—independent of the number of consensus sites. A minor shift of transcript B when the highest RBM20Δ2 concentration was applied (Figure 4B) was reevaluated using shorter B-derived transcripts, but none of them bound RBM20Δ2 (Supplementary Figure S4B). C and D transcripts are situated immediately downstream the regulated exon and contain the most of the predicted RBM20 binding motifs (Figure 4A). Thus, RBM20 binds predominantly to an ∼300 nt RNA segment downstream of the regulated exon 242, which contains eight potential RBM20 binding motifs. The UCUU motif is sufficient for RBM20Δ2 binding specificity (Supplementary Figure S4C). To support our identification of the relevant motives, we introduced point mutations in TTN intron 242–243 eliminating the four UCUU motifs proximal to the 5′SS as potential RBM20 binding sites (Supplementary Figure S4D). Elimination of the first UCUU motif (mut1) had the strongest effect on RBM20-dependent alternative exon exclusion (Supplementary Figure S4E). However, this mutation was not sufficient to eliminate RBM20-mediatd exon exclusion (Supplementary Figure S4E). The remaining mutations (mut2, mut3 and mut4) had minor or negligible effects. RT-PCR results were confirmed by qRT-PCR (Supplementary Figure S4F).
Figure 4.
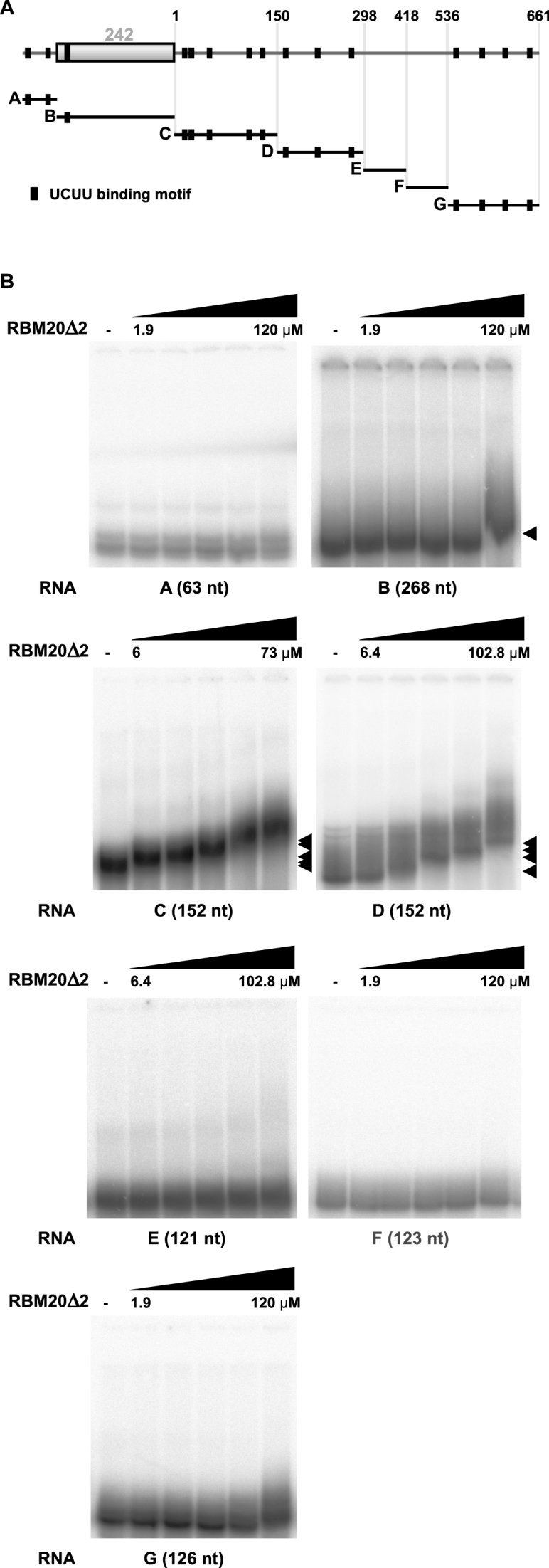
RBM20-RRM binds to the intron regions downstream of the alternative exon. (A) Schematic representation of the RNA transcripts used to study RBM20Δ2 binding. Titin transcripts were derived from the minimal region necessary for RBM20 function (labeled A–G. Black boxes indicate UCUU binding motifs—consensus sites for RBM20 binding. (B) RN–protein complex formation as determined by gel-shift assays. The same batch of recombinant RBM20Δ2 was used to allow direct comparisons of affinities. Black arrows indicate the RNA–protein complexes.
RBM20 prevents exon 242 inclusion by interfering with the removal of the upstream and downstream introns
So far, we demonstrated that exonic and flanking intronic sequences are necessary and addition of RBM20 is sufficient to cause exon 242 skipping. Based on thesis findings, we hypothesized that RBM20 simultaneously represses splicing of both individual flanking introns to exclude the alternative exon.
Building on established protocols (20), we used transient transfection of the TTN241-3 splicing reporter into the HEK293 cells followed by qRT-PCR to quantify splicing intermediates, where only one of two exon–exon junctions are generated (Figure 5A). Addition of RBM20 prevented splicing of both introns (Figure 5B and C). After addition of RBM20 the splicing intermediate lacking the down- or upstream intron were decreased 2- or 8-fold, respectively (Figure 5B and C). In contrast, addition of PTB4, which binds the same consensus motif as RBM20, differentially affected the splicing of the introns flanking titin exon 242: PTB4 increased the splicing intermediate lacking the downstream intron ∼6-fold (Figure 5D) with a minor decrease of the intermediate lacking the upstream intron that did not reach significance (Figure 5E). As the size of RBM20 and the minimal splicing reporter precluded their use in an in vitro splicing reaction (compare supplementary methods and Supplementary Figure S5A), we analyzed the accumulation of mRNA products, resulting from the cotransfection of TTN241-3 splicing reporter with RBM20, PTB4 or control into the HEK293 cells (Figure 5F and G). The mRNA was enriched, reverse transcribed and used for primer extension analysis (Figure 5F). The size of the resulting product was consistent with exclusion of the alternative exon 242 after addition of RBM20, but not after addition of PTB4 (Figure 5G). RBM20 dramatically reduced accumulation of the primer extension product over the downstream intron and slightly reduced upstream intron primer extension product accumulation, while PTB4 had no or the opposite effect. The accumulation of splicing intermediates and products, resulting from the cotransfection of TTN241-3 splicing reporter with RBM20 or PTB4 into the HEK293 cells was examined using the S1 nuclease assay. This analysis revealed that the exon 242–243 product is increased upon addition of PTB4 (Supplementary Figure S5C), while the exon 241–243 product is increased upon addition of RBM20 (Supplementary Figure S5D). These findings are consistent with a role of RBM20 in repressing the splicing of both upstream and downstream introns.
Figure 5.

Analysis of splicing intermediates of the RBM20-regulated TTN241-3 splicing reporter. (A) Schematic representation of intermediate splicing products and arrangement of primer pairs used to quantify splicing intermediates upon RBM20 addition on TTN241-3 splicing reporter. The pre-mRNA (depicted in the middle) was used for normalization (primers in red and blue). Primers specific for the intermediate RNAs were arranged at indicated exon–exon junctions (depicted in black). (B–E) RT-PCR (SYBR Green) validation of splicing intermediates upon transient transfection of TTN241-3 splicing reporter with/without RBM20/PTB4-expression plasmid into HEK293 cells. Splicing intermediates were normalized to the pre-mRNA. n = 3; ***P < 0.001. (F) Splice junction oligonucleotides to detect the alternative splicing products. (G) Alternative splicing with or without RBM20 and PTB was analyzed by primer extension. The expected products of primer extension are indicated. RBM20 suppresses and PTB4 enhances inclusion of titin exon 242.
Repressor activity of RBM20 is counteracted by PTB4
To investigate whether Polypyrimidine tract-binding protein (PTB) could regulate alternative splicing of titin-derived substrates, we first performed transient co-transfections of PTB and RBM20 together with a titin/β-globin mutant splicing reporter X11 (Figure 6A) in HEK293 cells and evaluated results by the RT-PCR (Figure 6B). We included RBM20-expressing plasmids in our transient transfection at 1:1 ratio with the reporter, to facilitate detection of the potential enhancing activity of the test protein. PTB exists in three alternatively spliced isoforms, PTB1, PTB2 and PTB4, which have distinct effects on alternative splicing of α-tropomyosin (TM) (21). As PTB2 had an intermediate effect in the repressive hierarchy (in the transfected smooth muscle cells), we restricted ourselves in cloning and testing only PTB1 and PTB4 isoforms. PTBP1 is expressed in HEK293 cells and in developing rat cardiomyocytes (Supplementary Figure S6I and J).
Figure 6.
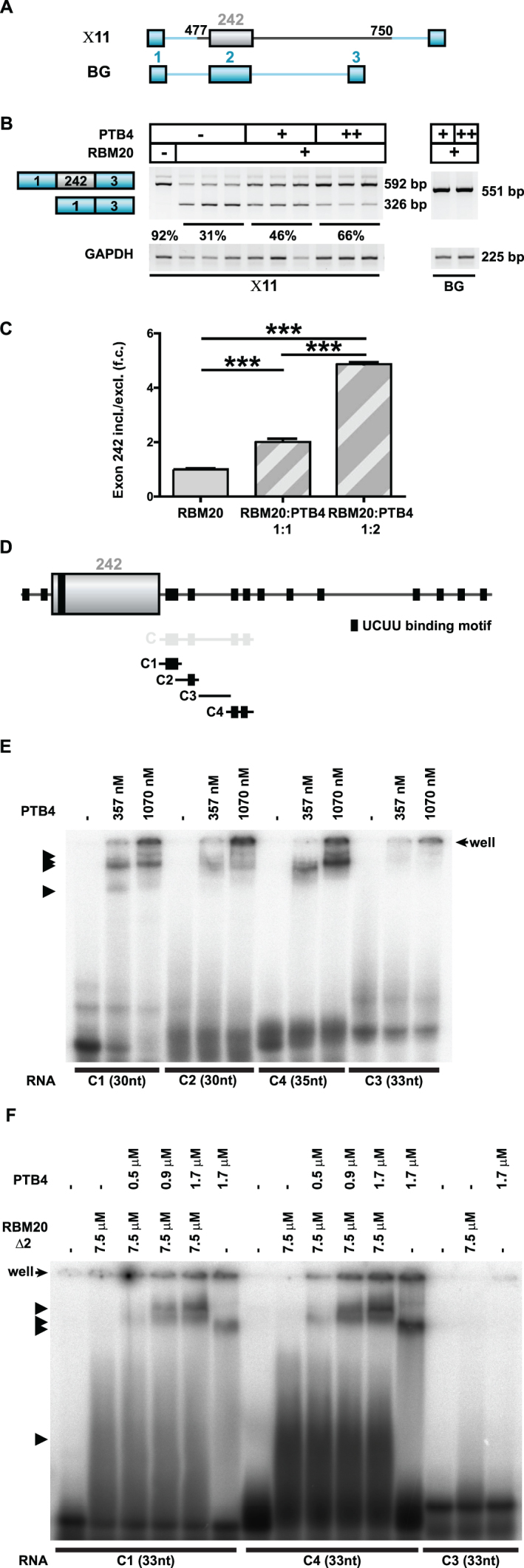
PTB4 enhancers TTN exon 242 inclusion dependent on the downstream intron. (A) Schematic representation of the human TTN (gray)/β-globin (blue) hybrid splicing reporters used to evaluate PTB4 effect on titin splicing regulation. (B) PTB4-mediated changes in the alternative splicing of the human titin/β-globin hybrid splicing reporter X11Changes in X11 alternative splicing were assayed in the background of RBM20. Increasing concentrations of PTB4 reverted the repressor activity of RBM20 on X11 splicing reporter. Titin-derived sequences missing splicing reporter (BG) was not responding to RBM20 or PTB4 (indicated on the right). (C) qRT-PCR (SYBR Green) quantification of PTB4-regulated alternative splicing events in the titin/β-globin mutant splicing reporter: n = 3; ***P < 0.001. (D) Schematic representation of the RNA transcripts used to study PTB4 binding. Black boxes indicate UCUU binding motifs. C1 transcript contains two UCUU binding motifs, organized in tandem, C2 contains one UCUU binding motif, C3 contains no UCUU binding motif and the C4 transcript contains two separated UCUU binding motifs. (E) RNA–protein complex formation as analyzed in a gel-shift assay. Black arrows indicate the observed formation of RNA–protein complexes. PTB4 binds all transcripts except for C3 that does not contain the consensus site. (F) RNA–protein complex formation analyzed by gel-shift assay using RBM20Δ2 and PTB4 protein. Black arrows indicate the formation of RNA–protein complexes. Increasing the concentration of PTB4 promotes the generation of additional RNA/protein complexes and leads to a reduction of RBM20Δ2/RNA complexes.
Increasing concentrations of transfected PTB4 significantly enhanced inclusion of the regulated middle exon in the X11 splicing reporter (Figure 6B and C; quantification in Supplementary Figure S6A and B). Similarly, co-transfection of equivalent or higher molar ratios of PTB1 did increase inclusion of the regulated exon 242 into the X11 splicing reporter (Supplementary Figure S6C–G). Toward understanding the molecular basis of PTB4 dependent titin splicing, we performed in vitro binding assays. Building on our RBM20Δ2 interaction data, we divided TTN 242–243 intron transcript C sequence into ∼30 nt RNAs with single, double or no binding motif UCUU (Figure 6D). Increasing amounts of recombinant PTB4 protein were incubated with those RNAs and formed mixtures were separated on a native gel (Figure 6E). PTB4/RNA complexes were formed only with RNAs containing UCUU motifs (C1, C2 and C4 versus C3). Adding increasing amounts of PTB4 to complexes of RBM20Δ2 and RNA containing two UCUU motives or only a single UCUU, resulted in complexes of higher molecular weight than complexes with RBM20 or PTB4 alone. This suggests simultaneous binding of PTB4 and RBM20 to transcripts (Figure 6F).
We conclude that both PTB4 and RBM20 bind the intron downstream of the alternative TTN exon 242 where PTB4 overcomes the repression by RBM20 as summarized in Figure 7.
Figure 7.

Model for the regulation of TTN241-3 splicing by RBM20 and PTB4. (A) In the absence of RBM20, spliceosomal complexes form over the exon and over the both introns (solid arrows) to include titin exon 242. Constitutive exons in dark gray, alternative exon 242 light gray and introns are indicated as thin lines. (B) Model of RBM20 mediated exclusion of exon 242: RBM20 binds downstream intron sequences and interferes with exon 242 5′ss recognition by the U1 snRNP resulting in reduced recognition of the downstream intron. This interaction destabilizes the upstream 3′ss recognition by the U2 snRNP and interferes with the exon definition complex and upstream intron recognition. As a result, the spliceosome recognizes only the external 5′ and 3′ splice sites and exon 242 is excluded. Transparent arrows indicate absent or incomplete communication between splice sites. (C) Addition of PTB4 overcomes the negative effect of RBM20 on exon retention as both factors bind the downstream intron, where they differentially interfere with U1 snRNP and 5′ss interactions, formation of the exon definition complex and subsequent inclusion of the alternative exon.
DISCUSSION
In this study, we investigated several aspects of alternative splicing regulation by the RBM20 protein and identified a 994-nt segment in the TTN pre-mRNA, which is necessary and sufficient to regulate exon 242 splicing by RBM20. The segment contains 64 nt from the upstream intron, the regulated exon 242 (267 nt) and 663 nt from the downstream intron. Additional truncation of this segment reduced or abolished the RBM20 repressor activity (Figures 1 and 2), indicating that the whole genomic segment contributes to splicing regulation. Interaction of the 994 nt segment with the RBM20-RRM domain involves a region immediately downstream of exon 242 which contains multiple UCUU binding motifs. A stretch of these motifs is required to mediate RBM20-mediated exon repression as demonstrated with the deletion analysis of the titin genomic segment (Figure 2A and B) and confirmed by mutagenesis gradually eliminating UCUU binding motifs in TTN241-3 downstream intron (Supplementary Figure S4E). Remarkably, the RBM20-RRM concentration required to achieve a gel-shift is lower for region C (immediately downstream of the alternative exon) versus region D (3′ of region C), which might indicate that sequences in the proximity to the 5′ss are bound with the higher affinity than more distal intronic sequences. This is consistent with the increased presence of UCUU motives and a cluster toward the 5′ss. Among the transcriptome-wide RBM20 binding sites in heart-specific transcripts identified using HITS-CLIP, intronic RBM20-binding positions flanking alternative exons correlate with splicing repression (15). In particular, RBM20 binding sites were significantly over-represented in a window spanning 400 bp upstream/downstream from the 3′/5′ splice sites of differentially spliced exons and RBM20 binding was found to peak 50 nt upstream and 100 nt downstream of repressed exons. Using RBM20-RRM in our binding assays we identified the region downstream of the repressed exon to be most strongly bound by RBM20 and no significant binding to the upstream intron of the repressed exon (Figure 4). This might relate to the averaging of consensus sites over the transcriptome versus the analysis of an individual exon: RBM20 might employ slightly different mechanisms for alternative exon repression versus mutually exclusive exons selection. Indeed, the presence of additional upstream RBM20 binding sites could thus relate to alternative splicing of mutually exclusive exons. An alternative explanation might be the differential binding of full length RBM20 versus the truncated isoform. Nevertheless, RNA-binding and repression are not directly connected, as exon 242 contains binding sites for RBM20 interacting proteins that can support the repressor activity of RBM20. An example is the RBM20-interacting protein FUS (15), which is predicted to bind titin exon 242 (RBPDB search (22)). A similar arrangement was recently described for splicing regulator RBfox that acts as part of a large complex of RNA-binding proteins (LASR) to stimulate splicing repression (23).
We found that the 5′ and 3′ splice sites of the flanking exons 241 and 243 did not contribute to the alternative splicing of exon 242, as their exchange for β-globin exons did not affect RBM20-dependent isoform expression (Figure 2). The minimal genomic segment required for RBM20-mediated exon repression includes the alternative exon and flanking intronic region. Together with the information on RBM20 binding, this data would be consistent with the molecular mechanism summarized in Figure 7. Binding of RBM20 to the downstream intron suggests that repression is achieved by interfering with intron definition. We find the 3′ss of exon 242 and the upstream intron sequences necessary for the repression, implying an even earlier role of RBM20—already at the level of spliceosome assembly during exon definition. This model suggests that inefficient recruiting of the spliceosome to the 5′ss of exon 242 would affect recognition of the upstream 3′ss needed to define the exon. Here, splicing repression would result from the poorly defined exon 242 3′ss and involve the inhibition of spliceosome assembly over the upstream intron (Figure 7B).
Diverse mechanisms of splicing repression have been described. Among them is binding of PTB at sites overlapping with splicing signals (polypyrimidine tract or branch point) and steric blockage of splicing factor access (24,25). Another scenario of PTB splicing repression requires binding to sequences located in both flanking introns and cooperative interaction between PTB molecules, with subsequent looping out of the intervening region or to its silencing by coating it with PTB molecules (19,26). Binding to an exonic splicing silencer and targeting molecular events that lead to exon definition (inhibiting the association of U2AF and U2 snRNP with the upstream 3′ss, without affecting recognition of the downstream 5′ss by the U1 snRNP) is used by PTB to exclude Fas exon 6 and generate mRNA encoding a soluble isoform of a receptor (18). Neuron-specific alternative splicing factors (Nova proteins) block U1 snRNP binding and inhibit exon inclusion by binding to an exonic YCAY cluster (20). Muscleblind-like 1 (MBNL1) protein binds to a structured intronic silencer to compete with U2AF65 binding and to repress cardiac troponin T exon 5 (22). hnRNP L together with hnRNP A1 repress spliceosome assembly and subsequently splicing of CD45 exon 4 by promoting aberrant U1 snRNA binding with exonic sequences upstream of the 5′ss (24). For RBM20 the details of the exon repression mechanism were unknown.
Here, we uncovered that RBM20 upon binding its functional response element in the downstream intron prevents removal of upstream and downstream introns and thus inhibits alternative exon inclusion. A similar mechanism has been described for activation of alternative exon inclusion by MBNL1 as binding of MBNL1 to its functional response element in the downstream intron and enhancing U2AF65 binding and splicing of the upstream intron leads to the inclusion of insulin receptor exon 11 (27). This mechanism relates to activation of exon inclusion, but has—to our knowledge—so far not been described for inhibition as described here for RBM20.
Toward understanding how splice factors concertedly shape cardiac isoform expression, we identified PTB4 as a novel titin splicing regulator that counteracts the splice repressor activity of RBM20. Interestingly, PTBP1 and RBM20 are both expressed from the late embryonic stages to the adulthood (Supplementary Figure S6J and (10)). In addition, we find that PTB4 and RBM20 bind the same motive on the 5′SS downstream of the alternative exon. Thus, we do not only show for the first time that PTB isoforms bind to titin-derived mRNA, but provide a possible mechanism of regulating titin isoform expression through additive binding of PTB and RBM20 to the downstream intron. Building on these findings, our future work will compare the titin splice regulators RBM20 and PTB4 as therapeutic targets to adapt cardiac isoform expression and improve diastolic function in patients with heart disease.
Supplementary Material
ACKNOWLEDGEMENTS
Author Contribution: V.D. designed and conducted experiments and analyzed the data. M.G. designed the project and experiments. V.D. and M.G. wrote the manuscript. We thank Beate Goldbrich-Hannig for excellent technical assistance. Michael Radke supported the initial cloning of the TTN241-3 splicing reporter.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Research Council [StG282078]; Deutsche Forschungsgemeinschaft, Bonn, Germany [Go865/11-1]; ‘Bundesministerium für Bildung und Forschung’ [CaRNAtion]; German Center for Cardiovascular Research (DZHK), Berlin. Funding for open access charge: Institutional funds.
Conflict of interest statement. None declared.
REFERENCES
- 1. Nilsen T.W., Graveley B.R.. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010; 463:457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chabot B., Shkreta L.. Defective control of pre-messenger RNA splicing in human disease. J. Cell Biol. 2016; 212:13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang G.S., Cooper T.A.. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat. Rev. Genet. 2007; 8:749–761. [DOI] [PubMed] [Google Scholar]
- 4. Hegele A., Kamburov A., Grossmann A., Sourlis C., Wowro S., Weimann M., Will C.L., Pena V., Lührmann R., Stelzl U.. Dynamic protein-protein interaction wiring of the human spliceosome. Mol. Cell. 2012; 45:567–580. [DOI] [PubMed] [Google Scholar]
- 5. Wahl M.C., Will C.L., Lührmann R.. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009; 136:701–718. [DOI] [PubMed] [Google Scholar]
- 6. Matera A.G., Wang Z.. A day in the life of the spliceosome. Nat. Rev. Mol. Cell Biol. 2014; 15:108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Black D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003; 72:291–336. [DOI] [PubMed] [Google Scholar]
- 8. Berget S.M. Exon recognition in vertebrate splicing. J. Biol. Chem. 1995; 270:2411–2414. [DOI] [PubMed] [Google Scholar]
- 9. Fu X.-D., Ares M.. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014; 15:689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guo W., Schafer S., Greaser M.L., Radke M.H., Liss M., Govindarajan T., Maatz H., Schulz H., Li S., Parrish A.M. et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 2012; 18:766–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Granzier H.L., Labeit S.. The giant protein titin: a major player in myocardial mechanics, signaling, and disease. Circ. Res. 2004; 94:284–295. [DOI] [PubMed] [Google Scholar]
- 12. Brauch K.M., Karst M.L., Herron K.J., de Andrade M., Pellikka P.A., Rodeheffer R.J., Michels V.V., Olson T.M.. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 2009; 54:930–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li D., Morales A., Gonzalez-Quintana J., Norton N., Siegfried J.D., Hofmeyer M., Hershberger R.E.. Identification of novel mutations in RBM20 in patients with dilated cardiomyopathy. Clin. Transl. Sci. 2010; 3:90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li S., Guo W., Dewey C.N., Greaser M.L.. Rbm20 regulates titin alternative splicing as a splicing repressor. Nucleic Acids Res. 2013; 41:2659–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maatz H., Jens M., Liss M., Schafer S., Heinig M., Kirchner M., Adami E., Rintisch C., Dauksaite V., Radke M.H. et al. RNA-binding protein RBM20 represses splicing to orchestrate cardiac pre-mRNA processing. J. Clin. Invest. 2014; 124:3419–3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kafasla P., Mickleburgh I., Llorian M., Coelho M., Gooding C., Cherny D., Joshi A., Kotik-Kogan O., Curry S., Eperon I.C. et al. Defining the roles and interactions of PTB. Biochem. Soc. Trans. 2012; 40:815–820. [DOI] [PubMed] [Google Scholar]
- 17. Suckale J., Wendling O., Masjkur J., Jäger M., Münster C., Anastassiadis K., Stewart A.F., Solimena M.. PTBP1 is required for embryonic development before gastrulation. PLoS One. 2011; 6:e16992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Izquierdo J.M., Majós N., Bonnal S., Martínez C., Castelo R., Guigó R., Bilbao D., Valcárcel J.. Regulation of Fas alternative splicing by antagonistic effects of TIA-1 and PTB on exon definition. Mol. Cell. 2005; 19:475–484. [DOI] [PubMed] [Google Scholar]
- 19. Amir-Ahmady B., Boutz P.L., Markovtsov V., Phillips M.L., Black D.L.. Exon repression by polypyrimidine tract binding protein. RNA. 2005; 11:699–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ule J., Stefani G., Mele A., Ruggiu M., Wang X., Taneri B., Gaasterland T., Blencowe B.J., Darnell R.B.. An RNA map predicting Nova-dependent splicing regulation. Nature. 2006; 444:580–586. [DOI] [PubMed] [Google Scholar]
- 21. Wollerton M.C., Gooding C., Robinson F., Brown E.C., Jackson R.J., Smith C.W.. Differential alternative splicing activity of isoforms of polypyrimidine tract binding protein (PTB). RNA. 2001; 7:819–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cook K.B., Kazan H., Zuberi K., Morris Q., Hughes T.R.. RBPDB: a database of RNA-binding specificities. Nucleic Acids Res. 2011; 39:D301–D308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Damianov A., Ying Y., Lin C.-H., Lee J.-A., Tran D., Vashisht A.A., Bahrami-Samani E., Xing Y., Martin K.C., Wohlschlegel J.A. et al. Rbfox proteins regulate splicing as part of a large multiprotein complex LASR. Cell. 2016; 165:606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ashiya M., Grabowski P.J.. A neuron-specific splicing switch mediated by an array of pre-mRNA repressor sites: evidence of a regulatory role for the polypyrimidine tract binding protein and a brain-specific PTB counterpart. RNA. 1997; 3:996–1015. [PMC free article] [PubMed] [Google Scholar]
- 25. Lin C.H., Patton J.G.. Regulation of alternative 3′ splice site selection by constitutive splicing factors. RNA. 1995; 1:234–245. [PMC free article] [PubMed] [Google Scholar]
- 26. Chou M.Y., Underwood J.G., Nikolic J., Luu M.H., Black D.L.. Multisite RNA binding and release of polypyrimidine tract binding protein during the regulation of c-src neural-specific splicing. Mol. Cell. 2000; 5:949–957. [DOI] [PubMed] [Google Scholar]
- 27. Echeverria G.V., Cooper T.A.. Muscleblind-like 1 activates insulin receptor exon 11 inclusion by enhancing U2AF65 binding and splicing of the upstream intron. Nucleic Acids Res. 2014; 42:1893–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.