

Abstract
Background
Models of epigenetic aging (epigenetic clocks) have been implicated as potentially useful markers for cancer risk and prognosis. Using 2 previously published methods for modeling epigenetic age, Horvath’s clock and epiTOC, we investigated epigenetic aging patterns related to World Health Organization grade and molecular subtype as well as associations of epigenetic aging with glioma survival and recurrence.
Methods
Epigenetic ages were calculated using Horvath’s clock and epiTOC on 516 lower-grade glioma and 141 glioblastoma cases along with 136 nontumor (normal) brain samples. Associations of tumor epigenetic age with patient chronological age at diagnosis were assessed with correlation and linear regression, and associations were validated in an independent cohort of 203 gliomas. Contribution of epigenetic age to survival prediction was assessed using Cox proportional hazards modeling. Sixty-three samples from 18 patients with primary-recurrent glioma pairs were also analyzed and epigenetic age difference and rate of epigenetic aging of primary-recurrent tumors were correlated to time to recurrence.
Results
Epigenetic ages of gliomas were near-universally accelerated using both Horvath’s clock and epiTOC compared with normal tissue. The 2 independent models of epigenetic aging were highly associated with each other and exhibited distinct aging patterns reflective of molecular subtype. EpiTOC was found to be a significant independent predictor of survival. Epigenetic aging of gliomas between primary and recurrent tumors was found to be highly variable and not significantly associated with time to recurrence.
Conclusions
We demonstrate that epigenetic aging reflects coherent modifications of the epigenome and can potentially provide additional prognostic power for gliomas.
Keywords: epigenetic clock, glioma, methylation
Importance of the study
Epigenetic age, a measure that is modeled using age-associated hyper- or hypomethylation of specific regions of the genome, has been suggested as a potentially useful marker in cancer prediction and prognosis. Not only is age the greatest single predictor of cancer risk, but studies have demonstrated epigenetic age acceleration in precancerous tissue and suggested that epigenetic aging in cancerous tissue reflects coherent epigenetic modifications. Here we investigate a focused application of 2 independent epigenetic aging models to high quality DNA methylation data obtained in gliomas, which comprise the majority of malignant brain tumor diagnoses but represent a highly heterogeneous class of tumors in terms of histology, molecular characterization, and prognosis. We demonstrate that this focused approach can yield insight into coherent modifications of the epigenome related to prognostic subtypes of glioma, and show that epigenetic aging of glioma tumor tissue can provide insight into survival and recurrence.
Research into mechanisms of cancer initiation and progression has traditionally focused on somatic mutation, but increasing evidence suggests that changes in the cancer epigenome can contribute a similarly important role in disease.1–4 One common modification that has been a major focus of epigenetic research is DNA methylation, which involves addition or removal of methyl groups on cytosines in cytosine-phosphate-guanine (CpG) dinucleotides and is involved in regulation of gene expression. Importantly, numerous studies have demonstrated that specific CpG sites are epigenetically modified in an age-dependent manner,5–8 being of specific interest to cancer researchers as age remains the single most significant predictor of incidence and survival in cancer.9–11
Multiple models of “biological” age based on DNA methylation have been developed5–7,12 that have shown potential for predicting risk of disease and survival in precancerous tissue,3,13,14 cancer,15–17 and a variety of other disease contexts.18 One such epigenetic age predictor is the epigenetic clock developed by Steve Horvath,7 which was trained on a wide range of tissue types to produce a highly accurate predictor of epigenetic age independent of tissue type or mitotic potential. As demonstrated by Horvath7 in his original article, however, the utility of epigenetic age predictors as applied to cancer tissue remains uncertain. Horvath observed highly heterogeneous changes in the epigenetic landscape across cancer types, and the functional relevance of those changes when applied to the dysregulated and aberrant machinery of cancer cells remains unknown. Horvath’s clock showed age accelerations ranging in magnitude and direction when applied to cancer with few discernible pan-cancer patterns,19 but other groups have since attempted to design epigenetic clocks capable of more transparently reflecting cancer biology. One such effort resulted in the epigenetic Timer Of Cancer (epiTOC)13 developed by Yang and colleagues, which was designed to reflect the number of mitotic divisions a cell has undergone.
Epigenetic changes are increasingly recognized as potential contributors to malignant transformation and progression of cancer, as specific gains and losses of DNA methylation throughout the cancer genome are predictive of treatment response and survival. This is particularly true in gliomas, where DNA methylation sites that predict treatment response have been defined, such as O6-methylguanine-DNA methyltransferase (MGMT) promoter methylation.20 Additionally, DNA methylation-based phenotypes of gliomas such as glioma CpG island methylator phenotype (G-CIMP),21 characterized by global hypermethylation of CpG islands, have been shown to reflect highly prognostic subgroupings of gliomas that are predictive of survival even after accounting for histopathological type and World Health Organization (WHO) grade.22 The G-CIMP methylation phenotype has in turn been shown to be associated with isocitrate dehydrogenase (IDH) mutation, and glioma molecular classification has been explored using a variety of approaches and features, including IDH-mutation status, 1p/19q codeletion status, telomerase reverse transcriptase mutation, as well as RNA and DNA methylation profiling.23–29 In this study, we investigated epigenetic aging in glioma using 2 independently designed epigenetic clocks, Horvath’s clock7 and epiTOC,13 and we assessed associations of these aging markers to glioma subtype, survival, and tumor recurrence. Through this in-depth analysis, we demonstrate the applications of epigenetic age as a marker in glioma and provide an example of the utility of epigenetic aging markers when focused on a specific cancer type.
Methods
DNA Methylation Data
Illumina HumanMethylation450 BeadChip DNA methylation data30 in normalized beta values format (Level 3) was downloaded from The Cancer Genome Atlas (TCGA) Legacy Archive (http://cancergenome.nih.gov/) for all available lower-grade glioma (LGG) and glioblastoma (GBM) cases. Five hundred sixteen LGG cases in TCGA had methylation data generated using the Illumina 450k platform, and 142 GBM cases had methylation data generated with the 450k platform and were included in this analysis. Relevant clinical data and case annotations (including molecular subtyping) on TCGA glioma cases were obtained from the most recent TCGA glioma study published by Ceccarelli et al22 (Table 1). One GBM case with 450k methylation data was not annotated and therefore was excluded from analysis, for a total of 657 DNA methylation profiles (516 LGG and 141 GBM). Normalized beta values generated on the Illumina HumanMethylation450 BeadChip on normal brain tissue were obtained on 136 samples (including glial, neural, and bulk samples) collected post mortem from 58 individuals as part of a previous study published by Guintivano et al.31 All normal brain methylation array data are publicly available under Gene Expression Omnibus (GEO) accession GSE41826.
Table 1.
Clinical characteristics of the sample set, arranged by study and by IDH and 1p/19q codeletion status
| By Study | By IDH and 1p/19q Codeletion Status | |||||
|---|---|---|---|---|---|---|
| LGG (n = 516) |
GBM (n = 141) | IDH-wt (n = 219) | IDH-mut-codel (n = 169) | IDH-mut-noncodel (n = 257) | Unknown (n = 12) | |
| Features | ||||||
| Histology (n) | ||||||
| Astrocytoma | 169 | 0 | 52 | 4 | 112 | 1 |
| Glioblastoma | 0 | 134 | 119 | 0 | 6 | 9 |
| Oligoastrocytoma | 114 | 0 | 15 | 30 | 69 | 0 |
| Oligodendroglioma | 174 | 0 | 19 | 117 | 37 | 1 |
| Unknown | 59 | 7 | 14 | 18 | 33 | 1 |
| WHO Grade (n) | ||||||
| II | 216 | 0 | 19 | 81 | 114 | 2 |
| III | 241 | 0 | 67 | 70 | 104 | 0 |
| VI | 0 | 134 | 117 | 0 | 6 | 9 |
| Unknown | 59 | 7 | 14 | 18 | 33 | 1 |
| Age | ||||||
| Median (LQ-UQ) | 41 (33–53) | 60 (52–69) | 59 (51–66) | 45 (35–54) | 36 (30–44) | 50 (43, 58) |
| Unknown (n) | 59 | 7 | 26 | 30 | 45 | 1 |
| Survival | ||||||
| Median (CI) | 87.4 (67.4–130.7) |
13.1 (11.3–16.7) |
14.6 (11.6–18.4) |
134.2 (78.2-Inf) |
79.9 (63.5-Inf) |
40 (17.6-Inf) |
| Unknown (n) | 59 | 7 | 26 | 30 | 45 | 1 |
| KPS (n) | ||||||
| <70 | 16 | 21 | 25 | 5 | 7 | 0 |
| 70–80 | 60 | 53 | 63 | 17 | 27 | 6 |
| 90 | 111 | 6 | 25 | 32 | 60 | 0 |
| 100 | 75 | 15 | 17 | 29 | 41 | 3 |
| Unknown | 254 | 46 | 89 | 86 | 122 | 3 |
| MGMT Promoter | ||||||
| Methylated | 425 | 60 | 84 | 168 | 227 | 6 |
| Unmethylated | 91 | 75 | 130 | 1 | 29 | 6 |
| Unknown | 0 | 6 | 5 | 0 | 1 | 0 |
| Epigenetic Clock | ||||||
| Horvath clock age, median (LQ-UQ) |
72.8 (55.4–95.3) |
76.3 (61.5–91.7) |
74.2 (55.6–86.4) |
96.7 (76.9–120.3) |
63.2 (50.8–76.0) |
70.6 (55.3–76.9) |
| epiTOC age, median(LQ-UQ) |
0.114 (0.092–0.147) |
0.142 (0.100–0.196) |
0.131 (0.091–0.184) |
0.147 (0.117–0.182) |
0.100 (0.087–0.116) |
0.118 (0.095–0.174) |
Abbreviations: LQ, lower quartile; UQ, upper quartile.
Survival: Unknown is number of cases where no survival time or status was available; not censored survival.
For the primary-recurrent analysis, raw IDAT methylation data generated on the Illumina HumanMethylation450 BeadChip on glioma recurrences were obtained from 63 samples from 18 patients from a study previously published by Mazor et al.32 For this dataset, normalized beta values were calculated from raw intensity files using the minfi package (v1.20.2) in R using quantile normalization preprocessing.33 These samples represent mostly primary–first recurrence pairs, with additional samples composed of multiple samples from the same tumor. If multiple samples were taken from the same tumor, the epigenetic age of that tumor was estimated as the average epiTOC and Horvath epigenetic age of all samples that were taken from the single tumor. In one case, patient 4, samples were taken from second and third recurrent tumors as well. One patient was dropped from analysis because records indicated that the second sample was residual disease rather than recurrence. All raw primary and recurrence methylation array intensity data are available under European Genome-Phenome Archive (EGA) accession EGAS00001001255.
Validation data for glioma subtyping associations were aggregated from 3 glioma DNA methylation studies.25–27 Data are available under GEO accessions GSE30339, GSE36278, and GSE61160. Pediatric gliomas were excluded, resulting in 203 glioma cases available for validation. Validation data for primary-recurrent analysis were obtained from Bai et al34 and are available under EGA accession EGAS00001001588, representing 24 individual primary-recurrent glioma pairs.
Calculations of epigenetic age
Epigenetic ages were calculated on all samples using R v3.3.2 according to previously published methods.7,13 Horvath clock age was calculated for LGG, GBM, normal, and primary-recurrent studies separately, with the normalization feature applied. Age acceleration for Horvath’s clock was defined as Horvath’s predicted age − chronological age at diagnosis/tumor resection. Age acceleration for epiTOC was defined as epiTOC value − predicted epiTOC value for a given age based on linear regression of epiTOC aging on normal brain samples. Performance of the linear regression model of epiTOC aging in normal brain was assessed and considered adequate for purposes of calculating epiTOC age acceleration (Supplementary Figure S8).
Statistics and Survival
All statistical analysis was performed in R v3.3.2. Multiple imputation of Karnofsky performance score (KPS; 300 cases), MGMT promoter methylation (6 cases), and pan-glioma DNA methylation cluster (6 cases) was done via the mice package in R35 using predictive mean matching. Multiple imputation was performed 100 times, with each imputation undergoing 10 iterations to generate pooled estimates for survival modeling. Assessment of imputation performance was done by plotting imputed distributions against complete data distributions without any signs of bias. Linear model diagnostics were performed and linear assumptions were not shown to be violated. Comparison of clock associations was performed using Pearson correlations and linear modeling, with hypothesis testing performed on Pearson correlations. Comparison of strength of correlations was performed by Williams test between 2 correlations sharing one variable. Hypothesis testing of the interaction between IDH-mutation and 1p/19q codeletion status and epiTOC/Horvath association was performed on the coefficients of the interaction terms using linear modeling. Reported R2 values were calculated using linear modeling on reported variables. Multivariable Cox regression analysis was performed on pooled multiple imputations utilizing survival. All variables were tested for violations of the proportional hazards assumption and none were found by visual inspection of Schoenfeld residuals or by the Schoenfeld test of each variable to have significant time-dependent coefficient estimates.
Results
Glioma Epigenetic Age
In order to investigate changes in epigenetic aging and its possible utility as a prognostic marker in glioma, we applied Horvath’s clock and epiTOC to 657 total glioma DNA methylation profiles classified as 141 GBM and 516 LGG cases in TCGA. We elected to use Horvath’s clock due to its universal applicability across tissue types,7 which makes it uniquely suited for estimating epigenetic age of tumors without bias as to tumor heterogeneity and composition. Horvath’s clock is not significantly attributable to any specific cellular functions, however, so we complemented our study with epiTOC, a DNA methylation-driven mitotic clock previously shown to be advanced in precancerous and cancerous tissues in association with expected mitotic age.13 It should be noted that epiTOC values are a unitless score reflecting cumulative DNA methylation that has been previously shown to be correlated to estimated number of cell divisions, but does not have a precise quantitative interpretation.
We first examined the clocks’ correlation with patient chronological age to investigate the extent of epigenetic age dysregulation. When applied to glioma tissue, both Horvath’s clock and epiTOC exhibited near-universal acceleration compared with normal brain (Fig. 1). Some small variations were observed across cellular components in normal brain (Supplementary Figure S1), but these differences were dwarfed by the changes in epigenetic age observed in comparison to glioma tissue. Acceleration of Horvath’s clock in brain tumors is consistent with Horvath’s previous results in smaller, non-TCGA GBM datasets.19 LGGs also followed a general pattern of age acceleration. Both clocks appeared to show age-dependent variance, with tumors diagnosed in older patients showing greater levels of acceleration from normal clock values. Despite the observed age acceleration, modest correlation between age at diagnosis and epiTOC (Pearson correlation coeff = 0.59, R2 = 0.35, P < 2.2E-16), as well as age at diagnosis and Horvath’s clock (Pearson coeff = 0.50, R2 = 0.25, P < 2.2E-16) was maintained in LGG (Fig. 1A, C, Supplementary Table S1). This correlation was notably weaker in GBM than in LGG (Pearson correlation coeff = 0.17, 0.32, R2 = 0.02, 0.09, P < 2.2E-16 and 2.40E-4 for epiTOC and Horvath, respectively), possibly due to the fact that GBMs are generally diagnosed in older patients and older patients appear to have greater clock variance at diagnosis compared with younger patients. Additionally, Horvath’s clock appeared to be less accelerated in GBMs compared with LGGs, a distinction that was not observed using epiTOC. Both Horvath’s clock and epiTOC recapitulated their reliability as aging markers in normal tissue, with Horvath’s clock being highly predictive of age and epiTOC showing modest but steady advancement in normal brain tissue with increasing age (Fig. 1, Supplementary Table S1).
Fig. 1.

(A) EpiTOC age versus chronological age at diagnosis, color coded by WHO grade. (B) EpiTOC age versus chronological age at diagnosis, color coded by IDH-mutation–1p/19q codeletion status. (C) Horvath clock age versus chronological age at diagnosis, color coded by WHO grade. (D) Horvath clock age versus chronological age at diagnosis, color coded by IDH-mutation–1p/19q codeletion status.
Horvath’s Clock and epiTOC Association
Horvath’s clock is independent of mitosis by design, whereas epiTOC is designed to be a mitotic clock, and the 2 clocks share only a single CpG probe in common. Hence, we next investigated whether the age measurements of these 2 clocks were associated with each other in glioma. We found a stronger association between the 2 epigenetic age measurements in glioma tumor tissue than either clock with chronological age (Fig. 2, Supplementary Table S2), which was found to be statistically significant (epiTOC:Horvath’s clock versus epiTOC:chronological age and Horvath’s clock:chronological age, P = 6.2E-22, 5.4E-10, respectively in GBM and P = 5.7E-22, 7.3E-11, respectively in LGG). This suggests that glioma DNA methylomes are still coherently modified even though the expected associations with chronological age in normal tissue have deteriorated. Of further note, the slope of the association between the 2 clocks is shallowest in normal tissue, indicating relatively slow advancement of the mitotic clock over epigenetic age as measured by Horvath’s clock (fitted linear model [LM] slope = 1.6E-4; Fig. 2A, Supplementary Table S2). In LGG, however, the slope of this relationship is markedly steeper compared with normal, and in GBM it is even steeper than in LGGs (fitted LM slopes = 1.25E-3 and 2.40E-3 for LGG and GBM, respectively), suggesting that although the clocks display a degree of coherent modification, the associations of these changes in methylation differ based upon tumor type. This effect, modeled as the interaction effect of LGG versus GBM on epiTOC age against Horvath’s clock age was statistically significant (coeff = −0.0010, P < 2E-16, LGG:Horvath age on epiTOC).
Fig. 2.

(A) All glioma samples with epiTOC age plotted against Horvath clock age, color coded by WHO grade. (B) All glioma samples with epiTOC age plotted against Horvath clock age, color coded by IDH-mutation–1p/19q codeletion status. (C) Overlap between methylation probe sets used to calculate Horvath’s clock and epiTOC and used in supervised subtyping of IDH-wt gliomas. (D) IDH-wt samples with epiTOC age plotted against Horvath clock age, color coded by methylation cluster subtype.
Epigenetic Clocks and Glioma Subtype
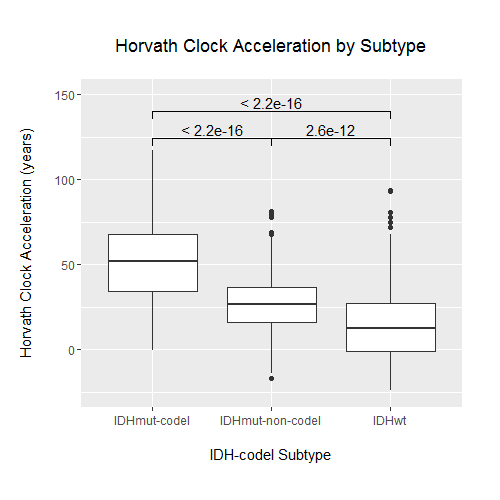
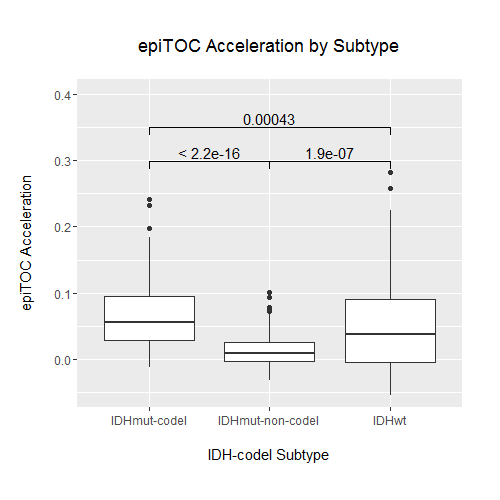
Studies have previously demonstrated that a small number of key molecular features are capable of categorizing gliomas into distinct groups with demonstrable differences in DNA methylation, RNA expression, and clinical outcome.22,29 We examined whether there were differences in epigenetic age across these known glioma molecular subtypes, in particular those defined by IDH1/2 mutation status and 1p/19q codeletion. Wildtype IDH (IDH-wt) gliomas make up the vast majority of GBMs, and histologically categorized LGGs that are IDH-wt have similar clinical outcomes to GBMs. When LGGs were categorized into known prognostic molecular subtypes, IDH-wt LGGs showed a similar epigenetic aging profile to GBM, underscoring a growing understanding of IDH-wt gliomas as members of a common glioma subgroup regardless of WHO grade22 (Fig. 2A, B). Because of this similarity, we reexamined the associations that were previously observed across WHO grade across IDH-mutation–1p/19q codeletion status instead of WHO grade. When broken down by IDH-mutation–1p/19q codeletion status, each group exhibited distinct epigenetic aging patterns (Fig. 1B, Supplementary Figure S2). Tumors with IDH mutation and 1p/19q codeletion (IDH-mut-codel) showed the highest levels of age acceleration as measured using Horvath’s clock, and IDH-wt gliomas showed the lowest levels of age acceleration. This finding was interesting not only because it reflects broad methylome changes that distinguish IDH-mut gliomas, which are known to be globally hypermethylated at CpG islands (ie, G-CIMP),21 but also because these levels of age acceleration reflect an overall negative association of age acceleration with survival in gliomas, as IDH-wt gliomas and IDH-mut-codel gliomas have the worst and best prognoses of the 3 subtypes, respectively. These differences in epigenetic age, while still statistically significant, were less clear using epiTOC and did not appear to reflect any apparent trend (Supplementary Figure S3). Notably, the association observed previously between epiTOC and Horvath’s clock appeared to also be dependent on IDH-mutation–1p/19q codeletion status, with IDH-wt showing the steepest epiTOC/Horvath clock slope followed by IDH-mut-codel and then IDH-mut-noncodel gliomas (Fig. 2B, Supplementary Table S2). This interaction between IDH-mutation–1p/19q codeletion status was shown to be statistically significant (coeff = 6.7E-4, 8.4E-4, P = 2.6E-9, 1.48E-14, for IDH-mut-noncodel and IDH-wt, respectively, interaction with Horvath’s age on epiTOC).
The IDH-wt subgroup was then further categorized into DNA methylation-based signature subgroupings previously identified by Ceccarelli et al in a recent TCGA pan-glioma study.22 Despite sharing only a relatively small proportion of probes between subtyping signature probes and clock probes (Fig. 2C), these subgroups based on methylation clustering exhibited distinct patterns of epigenetic aging (Fig. 3). The classic-like DNA methylation subtype demonstrated the highest level of epiTOC mitotic acceleration compared with other methylation subtypes; however, the IDH-mut-codel subtype had the highest level of age acceleration according to Horvath’s clock. No significant differences in epigenetic age were observed between the G-CIMP-high and G-CIMP-low IDH-mutant subtypes identified by Ceccarelli et al, which is interesting due to the expectation of global methylation differences between the 2 according to their previously published description. Strong reversals of epigenetic age were observed in the LGm6-GBM subtype and pilocytic astrocytoma (PA)–like subtype. Interestingly, these 2 subgroups were indistinguishable using the CpG probe sets utilized by Ceccarelli et al and were distinguished by histology. However, our epigenetic clock aging study suggests the PA-like group is additionally distinguishable from LGm6-GBM by a severely regressed epiTOC age compared with other IDH-wt gliomas and even compared with normal brain (Fig. 2D, Fig. 3). These findings as a whole contribute evidence that epigenetic age in gliomas may reflect coherent changes in the tumor methylome that are biologically and clinically relevant.
Fig. 3.

Epigenetic age acceleration (glioma tissue epigenetic age − patient age at diagnosis) for TCGA glioma samples plotted by supervised methylation subtype.
Validation of Pan-Glioma Epigenetic Clock Associations
To validate the associations we observed in gliomas across tumor epiTOC and Horvath’s clock age, patient age at diagnosis, and methylation subtype, we aggregated Illumina 450k DNA methylation data across several glioma studies, which to our knowledge represent all large-scale study 450k DNA methylation data currently available in gliomas25–27 (Supplementary Table S6). We ran similar tests compared with our analysis on the data from TCGA. Although 1p/19q codeletion information was not available for the entire validation set, we observed similar patterns of epigenetic clock associations with patient age at diagnosis, as well as epiTOC association with Horvath’s clock depending on IDH-mutation status (Supplementary Figure S4). Furthermore, we observed similar patterns of epigenetic age acceleration across previously identified methylation subtypes in our validation set (Supplementary Figure S5) compared with our initial TCGA dataset. As a whole, these findings suggest that the patterns of epigenetic aging in glioma are reproducible and represent coherent changes in epigenetic modification. Epigenetic aging patterns appear to be associated with IDH-mutation status as well as the methylation subtype classifications previously identified by Ceccarelli et al. Distinguishable patterns in epigenetic age of these different subtypes suggest that outside of signature molecular features such as IDH-mutation status and methylation subtype signature, these glioma subtypes are subject to coherent modifications of their epigenome that can be estimated by their epigenetic age.
Survival Modeling
To assess whether either epigenetic clock was associated with glioma survival, we performed Cox regression modeling on the pan-glioma TCGA dataset with known glioma survival predictors, including age at diagnosis, WHO grade, IDH-mutation–1p/19q codeletion status, MGMT promoter methylation,20 and KPS.36 A large proportion of glioma cases in TCGA were missing KPS information, therefore Cox regression was performed on both complete cases as well as an imputed dataset for comparison (using multiple imputation35). Cox modeling of survival on complete cases only affirmed the predictive power of age, IDH-mutation–1p/19q codeletion status, WHO grade, and KPS across all models tested (Supplementary Table S3). Inclusion of epiTOC with the base model in complete cases demonstrated a significant, negatively associated effect on survival (P = 0.016), but Horvath’s clock did not (P = 0.10). When both clocks were included in the model, however, epiTOC marginally lost its statistically significant association (P = 0.06), possibly due to collinearity with Horvath’s clock. Imputation of missing KPS allowed for inclusion of 236 additional cases with 55 additional recorded death events in the analysis (Supplementary Table S4). Pooled analysis again affirmed the importance of age, IDH-mutation–1p/19q codeletion status, and WHO grade across all models, but significant association of KPS with survival was lost. This larger analysis reaffirmed epiTOC as significantly associated with survival in addition to the base model (P = 0.025, Table 2). An additional, significant negative association of Horvath’s clock age with survival was detected (P = 0.0003, Table 2). Addition of both clocks to the base model showed no significant contribution of epiTOC to survival, but the negative association of Horvath’s clock to survival persisted (P = 0.003). The loss of significance of epiTOC in the combined epigenetic clocks model is likely due to collinearity. As previously mentioned, epiTOC and Horvath’s clock are highly correlated (Pearson’s coeff = 0.76, P < 2.2E-16 across all samples). Multivariable model results as well as detailed demographic information for both complete and imputed cases are included for completeness (Table 2, Supplementary Tables S3 and S4). Taken as a whole, these results suggest a possible negative association between epigenetic age of glioma tissue and patient survival in addition to known associated clinical variables and molecular features.
Table 2.
Cox regression results on TCGA glioma survival (pooled results, multiply imputed 100x)
| Base | |||||
|---|---|---|---|---|---|
| Variable | Beta Coeff | SE | 95% CI | P | |
| Age at diagnosis | 0.05 | 0.01 | [0.03, 0.06] | 4.08E-09 | * |
| Sex (male) | 0.35 | 0.18 | [−0.01, 0.7] | 5.50E-02 | |
| IDH-mut 1p/19q status (IDH-mut-non-codel) | 0.73 | 0.32 | [0.1, 1.35] | 2.31E-02 | * |
| IDH-mut 1p/19q status (IDH-wt) | 1.64 | 0.37 | [0.91, 2.37] | 9.57E-06 | * |
| KPS | −0.01 | 0.01 | [−0.03, 0.01] | 2.21E-01 | |
| MGMT promoter (unmethylated) | −0.01 | 0.21 | [−0.43, 0.41] | 9.75E-01 | |
| Grade II | −1.04 | 0.33 | [−1.69, −0.39] | 1.65E-03 | * |
| Grade III | −0.31 | 0.24 | [−0.77, 0.15] | 1.88E-01 | |
| Base + epiTOC | |||||
| Variable | Beta Coeff | SE | 95% CI | P | |
| Age at diagnosis | 0.05 | 0.01 | [0.04, 0.07] | 5.86E-10 | * |
| epiTOC | −3.85 | 1.72 | [−7.22, −0.49] | 2.48E-02 | * |
| Sex (male) | 0.32 | 0.18 | [−0.03, 0.67] | 7.67E-02 | |
| IDH-mut 1p/19q status (IDH-mut-non-codel) | 0.50 | 0.33 | [−0.15, 1.15] | 1.34E-01 | |
| IDH-mut 1p/19q status (IDH-wt) | 1.47 | 0.38 | [0.72, 2.21] | 1.24E-04 | * |
| KPS | −0.01 | 0.01 | [−0.03, 0.01] | 1.79E-01 | |
| MGMT promoter (unmethylated) | −0.08 | 0.22 | [−0.5, 0.35] | 7.23E-01 | |
| Grade II | −1.21 | 0.34 | [−1.88, −0.55] | 3.41E-04 | * |
| Grade III | −0.42 | 0.24 | [−0.89, 0.05] | 7.83E-02 | |
| Base + Horvath’s Clock | |||||
| Variable | Beta Coeff | SE | 95% CI | P | |
| Age at diagnosis | 0.06 | 0.01 | [0.05, 0.08] | 3.91E-12 | * |
| Sex (male) | 0.28 | 0.18 | [−0.08, 0.63] | 1.24E-01 | |
| Horvath age | −0.01 | 0.00 | [−0.02, −0.01] | 2.68E-04 | * |
| IDH-mut 1p/19q status (IDH-mut-non-codel) | 0.35 | 0.33 | [−0.31, 1] | 3.00E-01 | |
| IDH-mut 1p/19q status (IDH-wt) | 1.07 | 0.40 | [0.29, 1.85] | 7.20E-03 | * |
| KPS | −0.01 | 0.01 | [−0.03, 0.01] | 1.58E-01 | |
| MGMT promoter (unmethylated) | −0.04 | 0.21 | [−0.46, 0.38] | 8.42E-01 | |
| Grade II | −1.24 | 0.33 | [−1.89, −0.58] | 2.14E-04 | * |
| Grade III | −0.42 | 0.24 | [−0.89, 0.04] | 7.55E-02 | |
| Base + epiTOC + Horvath’s Clock | |||||
| Variable | Beta Coeff | SE | 95% CI | P | |
| Age at diagnosis | 0.07 | 0.01 | [0.05, 0.08] | 7.88E-12 | * |
| epiTOC | 0.87 | 2.33 | [−3.69, 5.43] | 7.08E-01 | |
| Sex (male) | 0.28 | 0.18 | [−0.08, 0.63] | 1.23E-01 | |
| Horvath age | −0.02 | 0.01 | [−0.03, −0.01] | 3.46E-03 | * |
| IDH-mut 1p/19q status (IDH-mut-non-codel) | 0.36 | 0.34 | [−0.3, 1.02] | 2.82E-01 | |
| IDH-mut 1p/19q status (IDH-wt) | 1.06 | 0.40 | [0.28, 1.84] | 8.04E-03 | * |
| KPS | −0.01 | 0.01 | [−0.03, 0.01] | 1.62E-01 | |
| MGMT promoter (unmethylated) | −0.03 | 0.22 | [−0.45, 0.39] | 8.87E-01 | |
| Grade II | −1.22 | 0.34 | [−1.88, −0.56] | 3.02E-04 | * |
| Grade III | −0.41 | 0.24 | [−0.88, 0.06] | 8.74E-02 | |
N = 580, deaths = 149.
Seventy-seven observations deleted due to missingness (12 missing IDH information, 66 missing survival information).
Reference groups: Sex (female), IDH-mut 1p/19q status (IDH-mut-codel), MGMT promoter (methylated), grade (IV).
*P-value < .05.
This survival association was unable to be independently validated in our validation dataset, however. With inclusion of age at diagnosis, IDH-mutation status, and WHO grade as predictive variables, epiTOC and Horvath clock age were not associated significantly with survival (P = 0.44 and 0.97 for epiTOC and Horvath’s clock age, respectively). However, it should be noted that our validation data were significantly smaller (n = 203) than even the complete, non-imputed dataset of TCGA. Furthermore, because our validation dataset was aggregated across multiple studies, molecular and clinical annotations were incomplete, forcing us to run a simplified model lacking 1p/19q codeletion status, KPS, and MGMT promoter methylation. While our findings in TCGA data suggest there may be an independent association with epigenetic age and survival in glioma, it remains to be seen whether this finding can be validated in similarly large DNA methylation studies.
Epigenetic aging of tumor recurrences
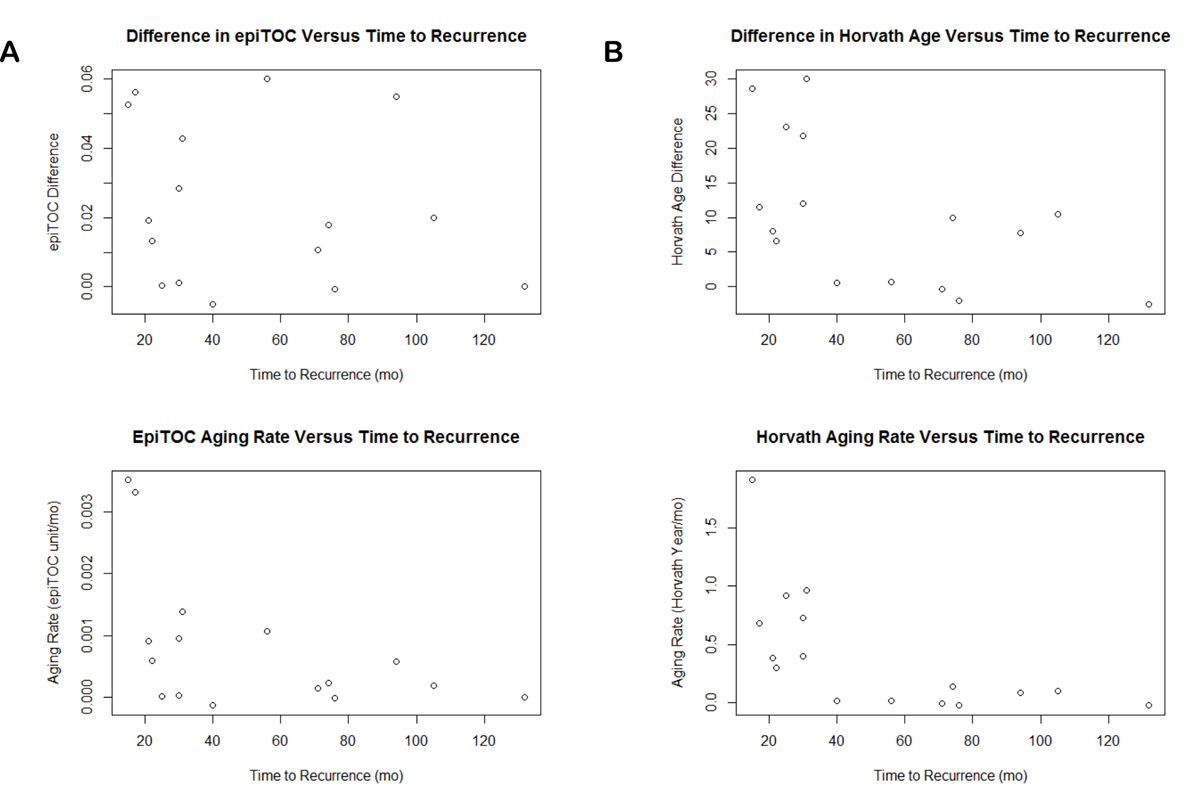
We further investigated changes in epigenetic age across low-grade primary gliomas and paired recurrences from 16 patients in a previous study published by Mazor et al.32 We found diverse changes in epigenetic age across glioma recurrences. Recurrent tumors showed a variety of aging changes, with some tumors having marginally regressed age, some having near-equivalent aging compared with normal tissue, and some demonstrating highly accelerated aging between their primary and recurrent tumors (Fig. 4). This diversity was observed across both epigenetic clocks. The rate of this aging was not associated with histopathological diagnosis of the primary or recurrent tumors or treatment type. The primary-recurrent analysis also yielded interesting insight into heterogeneity of epigenetic age within a tumor. We observed that even within samples obtained from the same tumor, epigenetic ages could vary significantly, as seen in the primary tumors of Patients 1, 4, 18, and 90, and the first recurrent tumor of Patient 1 (Fig. 4). Interestingly, comparing epigenetic age of different tumor portions with published predicted lineage32 using somatic mutation and methylation dynamics shows that in some cases the epigenetic age reflects similarity of the tumor to germline cells, which is the case for the “youngest” sample from Patient 90 (identified in original publication as Patient90 Initial C), but can also be counterintuitive, as the outlying primary tumor sample from Patient 18 (Patient18 Initial A) is predicted to be closely related in lineage to at least 2 of the other primary samples.
Fig. 4.

Plot of all primary and recurrent tumor sample epigenetic ages showing intratumoral heterogeneity and aging of tumor from primary to recurrence. Numbers in the center of points designate patient number from original publication.32
A negative correlation was observed between time to recurrence and epigenetic age difference between primary and recurrent tumors using Horvath’s clock (Pearson’s correlation −0.56, P = 0.025; Supplementary Figure S7); however, this finding could not be recapitulated in a validation dataset of LGG methylation34 (Pearson’s correlation 0.18, P = 0.39; Supplementary Figure S8). Although no additional significant associations were found between primary-recurrent epigenetic age differences and time to recurrence, it should be noted that this may still reflect an underlying relationship between epigenetic aging and recurrence. If primary-recurrent gliomas all aged at similar rates, we would have expected an overall increase in epigenetic age differences as time to recurrence increased; however, this was not observed in either test or validation dataset. To illustrate this point visually, we adjusted differences in epigenetic age by time to recurrence to obtain an estimate of epigenetic aging rate over the period of time between primary-recurrent resections (Supplementary Figures S6 and S7). The observation that epigenetic age differences are relatively unassociated with time to recurrence suggests that tumors that recur quickly might be aging more quickly, and that tumors that recur slowly are aging at a relatively slower rate. Neither clock age in primary or recurrent tumors was significantly associated with survival or time to recurrence when considered alone.
Discussion
By focusing our analysis scope on gliomas, we were able to identify patterns of epigenetic aging in tumor tissue that reflect known prognostic molecular subtypes. In addition, we were able to identify aging patterns that may contribute to glioma survival independent of molecular subtype and other known prognostic factors, and gained insight into epigenetic aging of tumors between primary and recurrence.
In our investigation of glioma epigenetic aging, we discovered that different subtypes of glioma demonstrated different epigenetic aging patterns. This observation not only contributes an independent line of verification of these subtypes as distinct biological classifications, but also showed that epigenetic age may be a useful measurement for further elucidating differences within molecular subtypes, such as between LGm6-GBM and PA-like gliomas, which were previously distinguished histologically rather than molecularly.
Within gliomas, we observed that overall lower epigenetic age is associated with poor survival. IDH-wt gliomas, which have generally poorer prognosis than IDH-mutant gliomas, had lower epigenetic age acceleration compared with IDH-mutant gliomas. These trends are contrary to most epigenetic aging studies, which have generally found that advanced epigenetic age is often associated with higher risks of disease and mortality in normal tissues. This opposite finding within glioma samples may be indicative of biological mechanisms in gliomas that oppose epigenetic aging and that lead to more aggressive or treatment-resistant disease. Another possible explanation for observed differences in epigenetic age is that age measurements may be reflective of variable tumor compositions, a factor that must be considered when studying invasive and highly heterogeneous tumors such as gliomas, particularly given the evidence that stemlike cells have younger epigenetic clocks.7 Currently, the biological drivers that determine epigenetic aging in gliomas remain unknown, and therefore the relationship of epigenetic age to biological mechanisms in glioma survival remains open for further investigation. There were several limitations to our analysis that bear mentioning. While missing KPS scores were imputed to provide as complete a dataset as possible and all efforts were made to prevent bias, there were differences in male/female ratio and between MGMT promoter methylation statuses between imputed cases and cases with complete data (Supplementary Tables S3 and S4). Furthermore, the survival associations with epigenetic clock age could not be validated in limited available data, emphasizing the need for additional large, high quality methylation data in glioma to investigate these types of associations. We also note that the strong correlation between the 2 epigenetic clocks is an interesting finding in itself, but likely contributes to collinearity in our survival models.
Inclusion of recurrent gliomas in our analysis allowed for study of epigenetic aging in gliomas over time. Although no reproducible association between primary-recurrent epigenetic age difference and time to recurrence was observed, the lack of this expected association intuitively reflects variable aging rates across gliomas. Given that the majority of primary-recurrent pairs move in the same positive direction measured by both epigenetic clocks, however, it seems unlikely that the epigenetic aging process is altogether dysregulated. This lack of association between epigenetic aging and time to recurrence can be explained by higher aging rate in faster recurrences and slower aging rate in delayed recurrences, but the evidence for this in our data is circumstantial. Our primary-recurrent analysis showed evidence of significant intratumoral heterogeneity with regard to epigenetic age, and the variety of treatments and responses of patients after resection of their primary glioma complicates any firm conclusions that can explain these variable aging rates. This observation is therefore not necessarily useful for predicting recurrence using epigenetic age, but is reported here solely to serve as a reference for further investigation into the drivers of epigenetic aging in gliomas.
Although epigenetic clocks appear broadly dysregulated in cancer without any clear pan-cancer utility, application of epigenetic clocks specifically to glioma demonstrated that epigenetic age can be a potentially useful biomarker in isolated cancer contexts. Furthermore, we identified the need for additional investigation of the mechanisms of epigenetic aging in glioma, where we observed associations between epigenetic age, glioma subtypes, and glioma survival and recurrence.
Supplementary Material
Supplementary material is available at Neuro-Oncology online.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Funding
This work was supported by the National Institutes of Health (T32 GM007250 and P30 5P30CA043703-27).
Acknowledgments
Thanks to Houtan Noushmehr and Thais Sabedot for assistance with validation methylation data.
Conflict of interest statement. None.
References
- 1. Baylin SB, Jones PA. A decade of exploring the cancer epigenome‒biological and translational implications. Nat Rev Cancer. 2011;11(10):726–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cruickshanks HA, McBryan T, Nelson DM, et al. . Senescent cells harbour features of the cancer epigenome. Nat Cell Biol. 2013;15(12):1495–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Klutstein M, Moss J, Kaplan T, Cedar H. Contribution of epigenetic mechanisms to variation in cancer risk among tissues. Proc Natl Acad Sci U S A. 2017;114(9):2230–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Teschendorff AE, Menon U, Gentry-Maharaj A, et al. . Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010;20(4):440–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Weidner CI, Lin Q, Koch CM, et al. . Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014;15(2):R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bocklandt S, Lin W, Sehl ME, et al. . Epigenetic predictor of age. PLoS One. 2011;6(6):e14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10):R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Christensen BC, Houseman EA, Marsit CJ, et al. . Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009;5(8):e1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Balducci L, Ershler WB. Cancer and ageing: a nexus at several levels. Nat Rev Cancer. 2005;5(8):655–662. [DOI] [PubMed] [Google Scholar]
- 10. Fraga MF, Agrelo R, Esteller M. Cross-talk between aging and cancer: the epigenetic language. Ann N Y Acad Sci. 2007;1100:60–74. [DOI] [PubMed] [Google Scholar]
- 11. Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hannum G, Guinney J, Zhao L, et al. . Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49(2):359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang Z, Wong A, Kuh D, et al. . Correlation of an epigenetic mitotic clock with cancer risk. Genome Biol. 2016;17(1):205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Curtius K, Wong CJ, Hazelton WD, et al. . A molecular clock infers heterogeneous tissue age among patients with Barrett’s esophagus. PLoS Comput Biol. 2016;12(5):e1004919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Agrawal S, Unterberg M, Koschmieder S, et al. . DNA methylation of tumor suppressor genes in clinical remission predicts the relapse risk in acute myeloid leukemia. Cancer Res. 2007;67(3):1370–1377. [DOI] [PubMed] [Google Scholar]
- 16. Bullinger L, Ehrich M, Döhner K, et al. . Quantitative DNA methylation predicts survival in adult acute myeloid leukemia. Blood. 2010;115(3):636–642. [DOI] [PubMed] [Google Scholar]
- 17. Xu Z, Taylor JA. Genome-wide age-related DNA methylation changes in blood and other tissues relate to histone modification, expression and cancer. Carcinogenesis. 2014;35(2):356–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marioni RE, Shah S, McRae AF, et al. . DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015;16:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Horvath S. Erratum to: DNA methylation age of human tissues and cell types. Genome Biol. 2015;16:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hegi ME, Diserens AC, Gorlia T, et al. . MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. [DOI] [PubMed] [Google Scholar]
- 21. Noushmehr H, Weisenberger DJ, Diefes K, et al. ; Cancer Genome Atlas Research Network Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17(5):510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ceccarelli M, Barthel FP, Malta TM, et al. ; TCGA Research Network Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. 2016;164(3):550–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Eckel-Passow JE, Lachance DH, Molinaro AM, et al. . Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N Engl J Med. 2015;372(26):2499–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yan H, Parsons DW, Jin G, et al. . IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Turcan S, Rohle D, Goenka A, et al. . IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483 (7390):479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mur P, Mollejo M, Ruano Y, et al. . Codeletion of 1p and 19q determines distinct gene methylation and expression profiles in IDH-mutated oligodendroglial tumors. Acta Neuropathol. 2013;126(2):277–289. [DOI] [PubMed] [Google Scholar]
- 27. Sturm D, Witt H, Hovestadt V, et al. . Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22(4):425–437. [DOI] [PubMed] [Google Scholar]
- 28. Brat DJ, Verhaak RG, Aldape KD, et al. . Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. 2015; 372(26):2481–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cohen AL, Holmen SL, Colman H. IDH1 and IDH2 mutations in gliomas. Curr Neurol Neurosci Rep. 2013;13(5):345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bibikova M, Barnes B, Tsan C, et al. . High density DNA methylation array with single CpG site resolution. Genomics. 2011;98(4):288–295. [DOI] [PubMed] [Google Scholar]
- 31. Guintivano J, Aryee MJ, Kaminsky ZA. A cell epigenotype specific model for the correction of brain cellular heterogeneity bias and its application to age, brain region and major depression. Epigenetics. 2013;8(3):290–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mazor T, Pankov A, Johnson BE, et al. . DNA methylation and somatic mutations converge on the cell cycle and define similar evolutionary histories in brain tumors. Cancer Cell. 2015;28(3):307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Aryee MJ, Jaffe AE, Corrada-Bravo H, et al. . Minfi: a flexible and comprehensive bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30(10):1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bai H, Harmancı AS, Erson-Omay EZ, et al. . Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat Genet. 2016;48(1):59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. van Buuren S, Groothuis-Oudshoorn CGM.. mice: Multivariate Imputation by Chained Equations in R. 2011; 45(3). [Google Scholar]
- 36. Walid MS. Prognostic factors for long-term survival after glioblastoma. Perm J. 2008;12(4):45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.