

Abstract
The merging of two divergent genomes in a hybrid is believed to trigger a “genomic shock”, disrupting gene regulation and transposable element (TE) silencing. Here, we tested this expectation by comparing the pattern of expression of transposable elements in their native and hybrid genomic context. For this, we sequenced the transcriptome of the Arabidopsis thaliana genotype Col-0, the A. lyrata genotype MN47 and their F1 hybrid. Contrary to expectations, we observe that the level of TE expression in the hybrid is strongly correlated to levels in the parental species. We detect that at most 1.1% of expressed transposable elements belonging to two specific subfamilies change their expression level upon hybridization. Most of these changes, however, are of small magnitude. We observe that the few hybrid-specific modifications in TE expression are more likely to occur when TE insertions are close to genes. In addition, changes in epigenetic histone marks H3K9me2 and H3K27me3 following hybridization do not coincide with TEs with changed expression. Finally, we further examined TE expression in parents and hybrids exposed to severe dehydration stress. Despite the major reorganization of gene and TE expression by stress, we observe that hybridization does not lead to increased disorganization of TE expression in the hybrid. Although our study did not examine TE transposition activity in hybrids, the examination of the transcriptome shows that TE expression is globally robust to hybridization. The term “genomic shock” is perhaps not appropriate to describe transcriptional modification in a viable hybrid merging divergent genomes.
Keywords: hybridization, genomic shock, transposable element, stress, Arabidopsis
Introduction
Interspecific hybridization is an important factor in plant evolution: While fertile allopolyploid hybrids may become the founders of new species (Soltis and Soltis 2009), the merging of two divergent genomes in a hybrid can also cause mild outbreeding depression or even result in complete incompatibility (Bomblies and Weigel 2007; Todesco et al. 2016). At the molecular level, hybridization can lead to a genome-wide misregulation of the transcriptome and epigenome (Lafon-Placette and Köhler 2015). The mobilization of up-regulated transposable elements (TE) can further result in chromosomal rearrangements. Such phenomena, first described by Barbara McClintock in maize (McClintock 1984) and termed “genomic shock”, are thought to provide a postzygotic barrier against gene flow between species.
Yet, a recent study of the homoploid hybrid of Arabidopsis thaliana and Arabidopsis lyrata did not report a major disruption of the transcriptome and epigenome. Moderate differences in expression of protein-coding genes were reported and the parental pattern of the histone mark H3K27me3 appeared to be maintained (Zhu et al. 2017). These species, however, differ in both genomic structure and TE content. A recent increase in TE number has been reported for the A. lyrata genome (Hu et al. 2011). Arabidopsis thaliana TEs are concentrated in pericentromeric regions, rarely venturing in gene rich chromosome arms. By contrast, in A. lyrata TEs account for most of the 40% larger genome and are broadly distributed, occurring in closer vicinity to expressed genes (Hu et al. 2011). The relatively low frequency of insertion polymorphisms within species revealed evolutionary tensions on insertions in gene rich regions, where permanent TE silencing can have deleterious consequences on the expression of neighboring genes (Hollister and Gaut 2009; Lockton and Gaut 2010; Hollister et al. 2011). Finally, several studies have observed that A. lyrata TEs tended to be more highly expressed than A. thaliana alleles in hybrids (He et al. 2012). In view of these differences in number, distribution, and regulation of transposable elements, we hypothesized that for hybrids between these species, a genomic shock is more likely to occur at the level of TE expression. To test this hypothesis, we quantified the impact of parental versus hybrid genomic background on TE regulation.
We studied transposable elements in the A. thaliana (Col-0) × A. lyrata (MN47) hybrid and its respective parental lines, using RNA-Seq expression data and ChIP-seq data of the histone marks H3K27me3 and H3K9me2. It is difficult to establish orthology relationships for TEs, because many of them duplicated after the species separation (Hu et al. 2011; de la Chaux et al. 2012). Thus, our analysis focuses on trans-acting effects experienced by TEs of each genome after hybridization. Contrary to our primary expectations, we find TE silencing to be largely unaffected in the hybrid. No systematic relationship could be found between a change of TE expression in the hybrid and the gain or loss of histone marks. We further exposed interspecific hybrids and their parents to severe dehydration stress and confirm that TE regulation remains robust to hybridization in stress conditions.
Materials and Methods
Plant Materials and Growth Conditions
Seeds of A. thaliana accession Col-0 were obtained from the Arabidopsis Biological Resource Center (ABRC, USA). Arabidopsis lyrata ssp. lyrata genotype MN47 was obtained from the lab of D. Weigel (Max Planck Institute for Development, Tübingen, Germany). Parental lines were crossed by pollinating emasculated A. thaliana flowers with A. lyrata pollen, as described in De Meaux et al. (2006). We obtained approximately 40 seeds per silique, 90% of which aborted. We did not use embryo rescue to generate more hybrids as in Zhu et al. (2017). Reciprocal crosses using A. thaliana as pollen donor were not successful.
RNA Sampling and Sequencing in Standard and Stress Conditions
Seeds were stratified for 3 days at 4°C, germinated on soil and grown for 4 weeks in a growth chamber at 20°C under 14 h light/16°C 10 h dark under dim light (100 mmol s−1 m−2). A dehydration treatment was applied following Seki et al. (2002). Plants were removed from the soil and dehydrated on paper for 0 and 3h, in the same growth chamber. The aerial part of the plant was sampled, flash-frozen in liquid nitrogen, and RNA was extracted as previously described (He et al. 2016). Four biological replicates were collected in 1-week intervals. Two micrograms of total RNA were used for library preparation following the TruSeq Illumina RNA Sample Preparation v2 Guide. This includes poly-A+ RNA selection and the use of random primers for reverse-transcription. Sequencing was performed on Illumina HiSeq2000 following the manufacturer’s protocols and paired-end 100-bp-long reads were obtained. In total, 15–20 million paired reads for the parents and 30–40 million reads for the hybrid were produced (supplementary table 1, Supplementary Material online). RNA-seq reads were filtered and trimmed for quality control as in He et al. (2016) and mapped on the hybrid genome, a concatenation of the A. thaliana Col-0 reference genome (TAIR10, www.arabidopsis.org; last accessed may 25, 2018) and the A. lyrata MN47 reference genome (Araly1, Nordberg et al. [2014]), using STAR with default parameters (Dobin et al. 2013).
TE and Gene Read Counting
The gene annotations TAIR10 (Col-0, Berardini et al. 2015), Alyrata_107_v1.0 (MN47, Hu et al. 2011) and TE annotations (e.g., position and TE family memberships) for A. lyrata and A. thaliana (Pietzenuk et al. 2016) were merged to form the genome annotation file used in this analysis. Aligned reads were filtered as follows: for the MN47 genome, only the main scaffolds 1–8 were considered. Read or fragment alignments shorter than 20 or longer than 700 base pairs (bp) were discarded. A minimum alignment score of −33 was required in order to accept an RNA-seq match. We allowed multiple RNA-seq read matches on TEs if they were entirely within a single (super)family, following Pietzenuk et al. (2016), and Jin et al. (2015). A read was assigned to a (super)family F on genome G if its primary match was in F on G, and any secondary matches on G were in F, too. In order to assign a match to a (super)family F, a minimum overlap of 50 bp to a member of F was required. Secondary matches on the alternative genome were allowed but not counted. Each read thus either contributed a single count to a (super)family or was discarded as multiply matching. If a read matched to TE1 . . TEn of family F, the read was defined to contribute 1/n counts to each of these TEs. Reads from a region of overlap a TE and a gene were not discarded, but counted with the TE. These summed fractional per-element counts were the final output from the counting procedure. They were either used directly as counts per individual TE or the counts of the members of (super)families were summed up to yield aggregated per-(super)family counts. We implemented the counting procedure in the R programming language. After the counting step, annotated TEs without any read count were excluded from the analysis. Genes were counted with the same procedure as the TEs.
Read Count Normalization
Read counts from genes and TEs were simultaneously normalized. Each count was first multiplied by a factor of 1,000/l, where l is the length in base pairs of the respective annotated element (gene, individual TE, or entire TE (super)family). This removes the dependence of the read count on element length or (super)family size. The total length of a TE (super)family was defined to equal the summed length of all (super)family members considered, discounting any regions of overlap with other annotated elements.
Differential Expression Analysis
The Generalized Linear Models (GLM) support of the R package edgeR (Robinson et al. 2010; McCarthy et al. 2012) was used to test the significance of differences in length-normalized counts between the four biological replicates of hybrid and parental transcriptomes. P values were adjusted with the FDR correction (Benjamini and Hochberg 1995). For visualization in scatterplots and for the stringent definition of differential TE expression, length-normalized counts were converted to Counts per Million (CPM) and scaled (Robinson and Oshlack 2010) using the calcNormFactors function of the R package edgeR. This transformation mirrors what edgeR does internally when computing differential expression. It removes global count biases due to library size differences, easing the interpretation of the scatterplots and allowing the set-up of a sample-independent cutoff on the absolute expression value at 0 and 10 CPM.
Analysis of Distance Distribution of TEs to Neighboring Genes
Distances of differential and nondifferential TEs to the nearest gene or gene 5′-end were binned into classes 0–1, 1–2, 2–3, and ≥ 3 kb. (TEs overlapping a gene were not considered.) A pseudocount of 1 was added to each bin count. A saturated log-linear model of the counts was computed using the R glm() function (family = “poisson”), using sum-to-zero contrasts. The P value for distance d in supplementary table 1, Supplementary Material online, is the Pr(>|z|) value of the interaction coefficient between the factors distance category and significance of hybrid-dependent TE expression.
Random Expectations of Superfamily Frequencies
To evaluate the enrichment of TEs changing their expression in hybrid background within each superfamily, we computed a random expectation. For this, we used the set of TEs showing differential expression in the hybrid background as a reference set and performed 10,000 random resamplings of the same number of TEs from all TEs with evidence of expression. To account for the skewed distribution of these TEs in the vicinity of non-TE genes, both the reference set and the set of all TEs were binned by distance to the closest gene 5′-end. For each bin of the reference sample, the same number of TEs as contained in the bin was randomly selected from the corresponding distance bin of all TEs. The union of the resampled bins then formed one resampled set. The width of the bins increased with increasing distance: up to a distance of 1 kb, the bin width was 200 base pairs (bp), between 1 and 5 kb it was 500 bp, and beyond 5 kb it was 1,000 bp, to avoid having bins with sparse numbers of TEs. A median, 5% and 95% quantile of each superfamily frequency was extracted from the 10,000 random samples and compared with the observed frequency of TEs with hybrid-dependent expression.
Chromatin Immune-Precipitation Experiments
Plants were germinated and grown on germination medium containing ½ Murashige and Skoog salts, 3% sucrose, and 0.8% agar. Seeds were stratified for 5 days at 4°C, and then transferred to a growth chamber under long-day conditions (14 h cool white light supplemented with red light at 20°C, and 10 h dark at 18°C). One week after germination, plants were transferred to soil and grown in the same chamber. Finally, leaves larger than 0.5 cm were sampled from 3-week-old Col-0, and 6-week-old MN47 and F1 hybrids for chromatin immunoprecipitation (ChIP). Leaves from 10 to 15 plants were pooled. Sampling was shifted in the hybrid and the A. lyrata individual, in order to sample material of similar development (∼3 cm rosette diameter).
ChIP experiments were performed with the MAGnify Chromatin Immunoprecipitation System (49-2024, Thermo Fisher Scientific) according to the manufacturer’s instructions, with the following modifications. Plant material was fixed in 20 ml Crosslinking Buffer (0.4 M Sucrose, 10 mM Tris–HCl, pH 8.0, 10 mM MgCl2, 1% formaldehyde) by applying vacuum twice for 15 min. Nuclei and chromatin were purified as in He et al. (2012). Chromatin was sonicated with a BioRuptor device (Diagenode, Liège, Belgium) for 30 times 30 s at high setting with 30 s intermittent cooling in ice-water. A DNA fragment size of 300–600 bp was controlled by running an aliquot of decrosslinked and purified DNA on 1.5% agarose gels. The following antibodies were used in immunoprecipitation: anti-rat IgG (R9255, Sigma; St. Louis, MO), as well as anti-H3K27me3, anti-H3K9me2 and anti-H3 (07-449, 07-441, and 07-690, respectively, Millipore; Temecula, CA). To reach enough precipitated DNA, the ChIP-DNA precipitation protocol was performed independently for eight aliquots of the collected leaf material, pooled and purified by Qiagen MinElute Reaction Cleanup Kit. Finally, we obtained ChIP-DNA samples from Col-0, MN47, and Col-0xMN47 with antibodies targeting H3K27me3 and H3K9me2 marks. In addition, we generated a ChIP-DNA sample targeting Histone 3 in a Col-0xMN47 sample to verify that both genomes could be efficiently immuno-precipitated and assess ChIP-seq quality (see supplementary Methods, Supplementary Material online).
ChIP-Seq Library Preparation and Sequencing
DNA was sheared with Ion Shear reagents and the Ion Xpress Fragment Library Kit (Thermo Fisher Scientific, Waltham, MA) was used for subsequent library preparation. [AQ10] After ligation of the respective Ion Xpress barcode adapters, samples were amplified by 17 or 23 PCR cycles. Amplified libraries were quantified on Bioanalyzer DNA High-Sensitivity Chips (Agilent Technologies, Santa Clara, CA) and diluted to 9 pM. Emulsion PCR was performed on the Ion OneTouch System, followed by enrichment for template positive Ion Sphere Particles. Sequencing sample was loaded on an Ion Proton chip and sequenced on the Ion proton sequencer (Thermo Fisher Scientific) according to the manufacturer’s instructions.
ChIP-Seq Read Processing, Mapping and Peak Detection
Raw data were preprocessed using the TORRENT SUITE version 4.0.1 (Thermo Fisher Scientific) to trim adapter sequences. Using the FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/; last accessed May 25, 2018), reads were then trimmed from the 3′ end to remove low quality bases (phred <15). Reads shorter than 30 bases and reads of poor overall quality (more than half of the bases with phred < 20) were discarded. The mapping of the reads was performed with bowtie2 (Langmead and Salzberg 2012), with the end-to-end parameter, without allowing mismatches in the seed (-N 0), and allowing for up to two alignments per read (-k 2); although then only the best alignment was kept. All samples were mapped to a hybrid reference genome obtained by concatenating the genomes of the parent as described above for RNAseq read mapping. Cross-hybridization between genomes was negligible (0.5–1% of mapped reads). A summary of ChIP seq read data are given in supplementary table 2, Supplementary Material online.
Quality assessment of the assays was performed using phantompeaktools (Kharchenko et al. 2008; Landt et al. 2012) to calculate NSC and RSC metrics (normalized and relative strand coefficients, respectively), and the bioconductor ChIPQC package (Carroll et al. 2014) to calculate the square sum of deviation. NSC was low but RSC metrics were well within the recommended range (Landt et al. 2012). Mapped reads were filtered with samtools (Li et al. 2009) for a mapping quality of 3 (-q 3), which discards multimapping reads with two equally good mappings but retains those with a single best mapping location. Duplicate reads from the amplification step during library preparation were removed with the MarkDuplicates program from Picard Tools (http://broadinstitute.github.io/picard; last accessed May 25, 2018). ChIP-seq read counts for TEs and non-TE genes were counted following the same pipeline as for RNAseq reads. A first analysis of ChIP-seq count distribution confirmed that the distribution of marks recovered known chromatin domains but revealed 64 genomic regions giving aberrant ChIP signals (see supplementary Methods, Supplementary Material online). These 64 regions were removed from all analyses presented in the manuscript.
Results
Only a Minor Fraction of All Expressed TEs Is Differentially Expressed in the Hybrid
Our annotation contains 10863 TEs on the A. thaliana Col-0 genome and 30,868 elements on the A. lyrata MN47 genome. After excluding genome-specific TE families, 10,805 and 29,150 elements remained on the Col-0 and MN47 genomes, respectively. Of these, 1,592 (15%) of the A. thaliana TEs and 7,045 (24%) of the A. lyrata TEs showed evidence of expression in at least one replicate. We quantified TE expression in normalized Counts Per Million (CPM, see Materials and Methods). TEs contributed but a minor fraction of all reads (on average, only 0.07% in A. thaliana samples and 0.24% in A. lyrata samples). This fraction remained comparable in the hybrid transcriptome (on average 0.06% and 0.30% of A. thaliana and A. lyrata reads, respectively in the hybrid). Only a small fraction (maximally 2%) of the expressed TEs of both genomes were significantly differentially expressed between parental and hybrid backgrounds, using different cutoffs to define significance (table 1). Moreover, this percentage dropped below 0.5% when the median expression was required to be above ten CPM in at least one of the two backgrounds (table 1). These observations show that TE transcription is not severely disturbed in the hybrid.
Table 1.
Numbers and Fractions of Differentially Expressed TEs at Two Different False-Discovery Rates (FDR ≤ 0.01 or ≤0.05) and Two Different Thresholds on the Normalized Counts Per Million (CPM) (all: ≥0 CPM; ≥10 CPM)
| FDR ≤ 0.01 |
FDR ≤ 0.05 |
|||
|---|---|---|---|---|
| All | ≥10 CPM | All | ≥10 CPM | |
| Arabidopsis thaliana | 3 + 2 (0.3%) | 1 + 1 (0.1%) | 5 + 5 (0.6%) | 1 + 2 (0.2%) |
| Arabidopsis lyrata | 57 + 2 (0.8%) | 14 + 0 (0.2%) | 73 + 6 (1.1%) | 18 + 1 (0.3%) |
Note.—Numbers are given as “number of up-regulations” + “number of down-regulations.” Fractions were computed relative to all TEs with evidence of expression in standard conditions, discounting members of genome-specific families.
Specific Rather than Global Effects Act on TE Expression in the Hybrid
In order to investigate the effect of hybridization at the levels of individual elements, families and superfamilies of TEs, we correlated expression of individual TEs and combined expression levels of TE families between parents and hybrids (fig. 1). Although TE transcription in the two genetic backgrounds correlated well at all levels, the correlation increased from the individual TEs to the family and superfamily levels. Spearman’s rank correlation of the median expression of replicates in the two backgrounds under control conditions was 0.73, 0.89, 0.97 on the three levels of individual TE, family, and superfamily, respectively in A. thaliana, and 0.65, 0.86, 0.91, respectively, in A. lyrata. Including four replicates in the RNA-seq analysis was necessary to establish these relationships with increased confidence. Using subsets of only two replicates, for example, reduced the parent–hybrid correlation coefficient of TE expression to 0.53, 0.77, 0.87 on the three levels for A. thaliana, and to 0.51, 0.84, 0.89, respectively, for A. lyrata.
Fig. 1.

—Correlation of TE expression levels, estimated as the median of normalized counts per million over the replicate plants grown in standard conditions, in parent and hybrid for individual TEs, TE families, and superfamilies. TEs or (super)families with significant expression change are marked in red.
The small subset of TEs with significantly different expression level between parent and hybrid was restricted to lower level units (individual TEs and families), indicating that the differences are case-specific rather than caused by a global disruption of TE silencing.
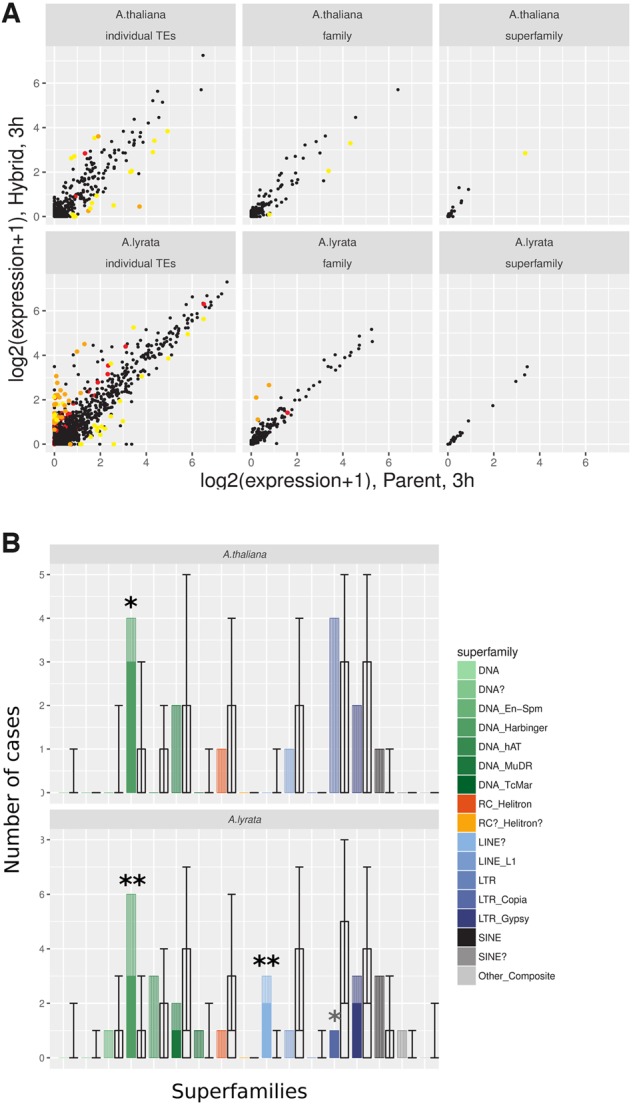
In A. lyrata, the 59 (0.8%) TEs that respond significantly to hybridization at FDR ≤ 0.01 were generally induced. In A. thaliana, the 5 (0.3%) affected elements were either induced or repressed (supplementary fig. S1, Supplementary Material online). SINEs dominated by Sadhu elements (Rangwala and Richards 2010) and Harbingers dominated by the ATIS112A family (Kapitonov and Jurka 2004) stood out as enriched among the responding A. lyrata TEs (fig. 2). These superfamilies were significantly overrepresented relative to the set of expressed TEs (≥1 RNA-seq count, permutation test, FDR < 0.05). The fact that only a small number of TE families showed enrichment for differential expressed elements indicates that hybridization does not have a global effect on TE expression.
Fig. 2.

—Observed and expected superfamily distribution of differentially expressed TEs (parent vs. hybrid). Colored bars show the observed numbers of TEs showing hybrid-dependent expression in each TE superfamily. Each color corresponds to one superfamily as indicated in the legend. Superfamily names are as given in the annotation of (Pietzenuk et al. 2016). Names with trailing question marks were annotated as deviant/uncertain members of the respective superfamily. Transparent bars indicate expected median count for random sampling within each superfamily. Error bars indicate the 5% and 95% quantile of the 10,000 random draws. Single black stars mark superfamilies whose observed count is significantly higher than expected from random expectations. Gray stars indicate depletion (maximally 5% of the universe have counts ≤ the observed count of the respective superfamily). Double stars indicate significant enrichment or depletion that is robust to FDR correction.
Rare TE Expression Changes Coincide with Alterations in Nearby Gene Expression
TEs can change expression of nearby genes by influencing local chromatin accessibility. We therefore asked whether hybrid-specific expression changes of TEs predict a corresponding change in neighboring genes.
Compared with TEs with expression not affected by hybridization, A. lyrata TEs expressed differentially in the hybrid were depleted in genome regions distant more than ≥ 3 kb from the closest gene 5′ ends (log-linear model, P ≤ 5 × 10−4, table 2). In addition, counts of hybridization-sensitive TEs were enriched in the bins 1–2 kb upstream of gene 5′-ends (log-linear model, P ≤ 0.01, table 2). This indicates a preferential location of hybridization-sensitive TEs in euchromatic, gene-rich regions in A. lyrata. In A. thaliana, there were too few differentially expressed TEs in the hybrid background to reliably compare the distance distributions.
Table 2.
Distance Distributions to Nearby Genes of Arabidopsis lyrata TEs with Differential Expression in Parent and Hybrid Under Control Conditions
| Distance to | Closest Gene |
Closest 5′ End of a Gene |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| <1 kb | 1–2 kb | 2–3 kb | ≥3 kb | <1 kb | 1–2 kb | 2–3 kb | ≥3 kb | ||
| Number of TEs | Diff. exp. PvsH | 15 | 18 | 13 | 13 | 7 | 24 | 12 | 16 |
| Not Diff. Expr. | 1,238 | 1,675 | 913 | 3,160 | 654 | 1,455 | 1,078 | 3,799 | |
| P (frequency in distance bin) | 0.239 | 0.539 | 0.061 | 4.0e-4 | 0.554 | 0.01 | 0.517 | 1.5e-4 | |
Note.—The table combines distance information of TEs with a significant change in expression in the hybrid to either the nearest gene (any orientation) or the nearest 5′ of any gene. Numbers of significant and nonsignificant TEs are listed for each distance bin (FDR 0.01). In the lower part, P values are given for the contribution of each distance bin to the difference between the distributions of significant and nonsignificant TEs (see Materials and Methods). Bins with significant depletion or enrichment in significant TEs are highlighted in blue and orange, respectively.
We further analyzed if changed expression of TEs and their neighboring genes in a hybrid background were correlated. Hybrid-specific changes in transcription levels of non-TE genes have already been reported (Zhu et al. 2017). We also quantified non-TE gene expression levels in parents and hybrids. Note that in our experiment, the timing of sampling accounted for the developmental differences between the fast developer A. thaliana and both A. lyrata and the hybrid, which are late flowering. Markedly fewer genes were affected by hybridization in our experiment (234 in A. thaliana and 395 in A. lyrata), yet, we observed a significant overlap in genes reported to change expression in the hybrid background in Zhu et al. (2017) (Hypergeometric test of overlap in genes changing their expression level between hybrid and parents, P < 10−17 for all comparisons). In A. lyrata 153 TEs with detectable expression were within 2 kb of a gene with hybridization-sensitive expression (FDR ≤ 0.01). Among these, 8 (5%) TEs displayed a change in expression in the hybrid background. This percentage dropped to 1% (71/6,892) for TEs with a gene neighbor showing no significant change in expression in hybrid versus parent (P < 5 × 10−5, hypergeometric test). In the A. thaliana genome, the number of expressed TEs located in the vicinity of genes was too small to calculate correlations. We conclude that hybrid-specific TE expression can coincide with a modification of the expression of nearby protein-coding genes, but this only affects a very small number of expressed loci.
Histone Marks Are Not Modified in the Hybrid Background
Two epigenetic marks are associated with transcriptional silencing in plants: the plastic mark H3K27me3 and the permanent mark H3K9me2 (Kim and Zilberman 2014; Sequeira-Mendes et al. 2014). We profiled these two histone modifications by Chromatin Immuno-Precipitation followed by sequencing (ChIP-Seq) in hybrids and A. thaliana and A. lyrata parents. Although the ChIP-seq H3K27me3 yielded a comparatively lower number of reads, especially for Col-0 (supplementary tables 2, 5, and 6, Supplementary Material online), the distribution of marks on TEs was in agreement with previous reports (fig. 3A, Seymour et al. 2014) and captured well the chromatin domains that were defined in A. thaliana (Sequeira-Mendes et al. 2014, supplementary Methods, Supplementary Material online). In our data, H3K9me2 marks showed a high correlation of normalized read counts between parental and hybrid genomes, both genome-wide (calculated across 10-kb genomic windows) and for individual TEs, TE families, and superfamilies. The genome-wide Spearman correlation coefficients of H3K9me2 read count along the genomes were 0.66 and 0.92 for A. thaliana or A. lyrata, respectively. Spearman’s rank correlation coefficients of H3K9me2 for individual TEs, TE-families, and superfamilies were 0.76, 0.94, and 0.96 for A. thaliana and 0.87, 0.97, and 0.99 for A. lyrata (fig. 3B and C). Although the high correlation values pointed to a largely additive inheritance of the mark in the hybrid on all levels of the annotation, the fact that correlation was lowest on the level of individual elements again underscores the role of element-specific responses in the hybrid.
Fig. 3.
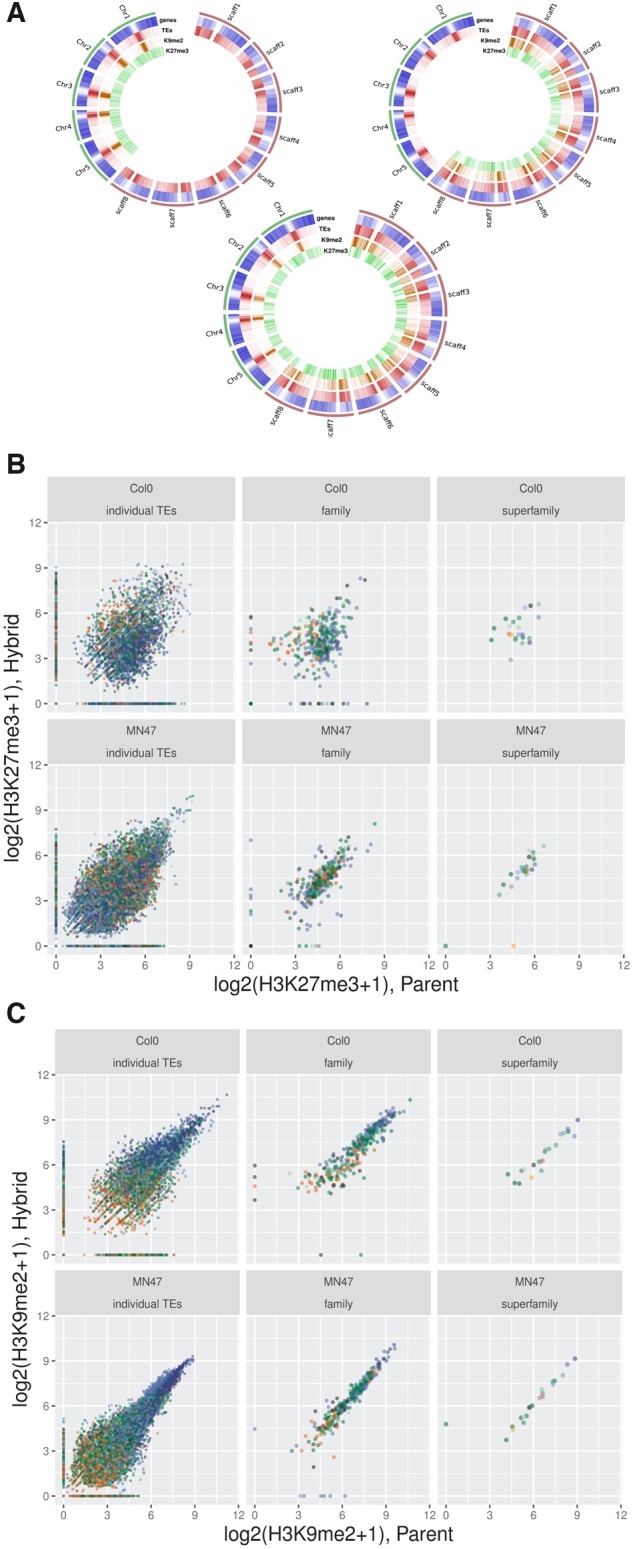
—Correlation of histone mark levels on TEs in parent and hybrid. (A) Genome-wide distribution of H3K9me2 and H3K27me2 on the parental and hybrid genomes. The circular plots represent the hybrid genome used as reference, with the five A. thaliana chromosomes in green and the eight A. lyrata chromosomes in red. Inner tracks represent gene, TE, H3K9me2 and H3K27me3 densities in 500 kb bins. Top left: Col-0, Top right: MN47, Bottom: hybrid. (B and C) Scatterplots of ChIP-seq read counts. The color code indicates superfamily membership, using the palette of figure 2. (B) H3K9me2; (C) H3K27me3.
While the function of H3K9me2 is to permanently silence TEs, H3K27me3 mainly serves to reversibly silence genes whose expression is restricted to specific conditions or developmental time windows (Gan et al. 2015). The distribution of H3K27me3 marks was strongly correlated genome-wide between parental and hybrid backgrounds (Spearman correlation coefficients: 0.63 and 0.86 for A. thaliana and A. lyrata, respectively, fig. 3A). H3K27me3 is not a typical silencing mark of TEs (Sequeira-Mendes et al. 2014). This was most apparent in our data for the LTR superfamily, which tended to be associated with the highest level of H3K9me2 and the lowest level of H3K27me3 (fig. 3B and C). Nevertheless, the correlation of H3K27me3 occupancy in parent and hybrid remained high even for TEs, in A. lyrata (Spearman’s rank correlation 0.58, 0.78, 0.92 for individual TEs, families, and superfamilies). For the A. thaliana/hybrid comparison, the correlation dropped to 0.2, 0.27, 0.21 for individual TEs, families, and superfamilies, respectively (fig. 3C). Note however that the Col-0 H3K27me3 ChIP-seq experiments yielded a comparatively lower number of reads (supplementary table 2, Supplementary Material online).
We tested then whether changes in histone mark occupancy could explain TE expression differences between parents and hybrids. TEs with a modified expression level in hybrid background generally showed similar H3K27me3 level in parent and hybrid (fig. 4). When a departure in epigenetic levels was detected between parents and hybrids, the direction of the change in expression of the TE was not necessarily in the expected direction (fig. 4). A. lyrata TEs with a modified expression level in hybrid background also generally showed similar H3K9me2 mark levels in parents and hybrids. We detected a concordant loss of H3K9me2 marks and increased expression in the hybrid for 6 A. lyrata TEs (fig. 4). But there again, we observed for 2 TEs that the change in H3K9me2 occupancy did not reflect the change in expression. Altogether, in our data, a differential deposition of H3K27me3 and H3K9me2 histone methylation marks in the hybrid is rare and generally does not coincide with hybrid-dependent changes in TE expression.
Fig. 4.

—No change in histone mark occupancy is detected for differentially expressed TEs. Shown are scatterplots of each histone marks on each of the two genomes for TEs with significantly different expression level in parent versus hybrid under standard conditions. Superfamily membership of the TEs is indicated using the palette of figure 2. Symbol shapes indicate the direction of gene expression change in hybrid versus parent (upward triangle: up, downward: down).
Robustness of TE Expression to Hybridization Is Maintained under Stress Conditions
It is assumed that transposition activity is induced by stress conditions (McClintock 1984; Cavrak et al. 2014). Dehydration stress is associated with a vast transcriptome reprogramming (Matsui et al. 2008). We analyzed the dependence of transcript abundance upon interspecific hybridization for TEs and non-TE genes under strong drought stress conditions in the same way as described for control conditions. Having found that TEs with a hybrid-specific expression change tend to be close to genes, and given the reprogramming of the gene transcriptome by the stress, we asked whether TE regulation was robust to hybridization even under stress conditions. In our data, 30–40% of the genes of both species changed expression after 3h of dehydration, with similar proportions in the parental and hybrid backgrounds. The fraction of stress responsive TEs was much smaller: 3–4% on the A. thaliana genome and 2–3% for A. lyrata (not shown). In addition, we observed no increase in the variance of TE expression in hybrid context (supplementary fig. 3, Supplementary Material online). Although the fraction of A. thaliana TEs with a significant expression difference between parent and hybrid is higher under dehydration stress than under control conditions, both the absolute numbers (4 up and 14 down) and the fraction (1.1% of all TEs with read count ≥1) remained small (table 3). The fraction of responding A. lyrata TEs decreased slightly compared with control conditions, and again the numbers remained small (33 up, 18 down, fraction 0.7%). The percentage dropped below 0.5% if an expression level of ≥10 CPM in at least one of the backgrounds was required (table 3). Like under control conditions, the correlation of TE expression levels between parents and hybrids increased at the family and superfamily levels. The Spearman’s rank correlation of the replicates’ median expression in the two backgrounds was (0.7, 0.81, 0.83) for A. thaliana, and (0.67, 0.91, 0.94) for A. lyrata, for individual, family and superfamily levels, respectively. Interestingly, we observed that more TEs were down-regulated in the hybrids compared with the parents under stress conditions than under standard conditions (fig. 5A and table 3). Only the Harbinger and LINE_L1 superfamiles were enriched among expressed A. lyrata TEs in stress conditions (fig. 5B; permutation test using TEs with ≥ 1 RNA-seq count as reference, FDR < 0.05). This further corroborates the conclusion that in our two species, TE regulation is robust to hybridization even under stress conditions.
Table 3.
Numbers and Fractions of Differentially Expressed TEs in Stress Conditions (3h) and in Response to Stress (3h specific), at Two Different False-Discovery Rates (FDR ≤0.01 or ≤0.05) and Two Different Thresholds on the Normalized Counts Per Million (CPM) (all: ≥0 CPM; ≥10 CPM)
| FDR ≤ 0.01 |
FDR ≤ 0.05 |
||||
|---|---|---|---|---|---|
| All | ≥10 CPM | All | ≥10 CPM | ||
| Arabidopsis thaliana | 3h | 4 + 14 (1.1%) | 2 + 4 (0.4%) | 7 + 26 (2.1%) | 2 + 5 (0.4%) |
| 3h specific | 3 + 12 (0.9%) | 2 + 3 (0.3%) | 4 + 23 (1.7%) | 2 + 4 (0.4%) | |
| Arabidopsis lyrata | 3h | 33 + 18 (0.7%) | 4 + 4 (0.1%) | 54 + 37 (1.3%) | 10 + 15 (0.4%) |
| 3h specific | 9 + 17 (0.4%) | 3 + 4 (0.1%) | 12 + 34 (0.7%) | 7 + 14 (0.3%) | |
Note.—Fractions were computed relative to all TEs with evidence of expression under stress conditions, discounting members of genome-specific families.
Fig. 5.
—(A) Correlation of TE expression levels in stress conditions, estimated as Median normalized counts per million over the replicates, in parent and hybrid for individual TEs, TE families, and superfamilies. TEs or (super)families with significant expression change are marked in red (significant only under control conditions), orange (significant both under standard and stress conditions), or yellow (significant only in stress conditions). (B) Observed and expected superfamily distribution of TEs whose expression is hybrid-dependent specifically in stress conditions. Colored bars show the observed numbers of TEs showing hybrid-dependent expression in each TE superfamily. Each color corresponds to one superfamily as indicated in the legend. Superfamily names are as given in the annotation of Pietzenuk et al. (2016). Names with trailing question marks were annotated as deviant/uncertain members of the respective superfamily. Transparent bars indicate expected median count for random sampling within each superfamily. Error bars indicate the 5% and 95% quantile of the 10,000 random draws. Single black stars mark superfamilies whose observed count is significantly higher than expected from random expectations. Gray stars indicate depletion (maximally 5% of the universe have counts ≤ the observed count of the respective superfamily). Double stars indicate significant enrichment or depletion that is robust to FDR correction.
Discussion
No evidence for a genome shock upon hybridization of A. lyrata and A. thaliana
The term “genomic shock,” was initially used to describe the breakage and large scale rearrangement of chromosomes in maize (McClintock 1946, 1984). This term is thus appropriate to describe genome instability following interspecific hybridization, that is, unbalanced segregation of homoelogous chromosomes, chromosome loss or translocations (Xiong et al. 2011; Chester et al. 2012; Henry et al. 2014; Spoelhof et al. 2017). Over the years, the term “genome shock” has also been used to describe the anticipated alteration in genome activity immediately following interspecific hybridization or allopolyploidization, although transposon reactivation has seldom been rigorously documented immediately after genome merging (Parisod et al. 2010). In plants, DNA cytosine methylation (mC) and the histone mark H3K9me2 embed TEs in a chromatin context unfavorable for transcription (Kim and Zilberman 2014). The establishment and maintenance of these marks is not only inherited through DNA replication, it is also guided by small interfering RNAs (siRNAs) (see Kim and Zilberman 2014 for a review of plant TE silencing mechanisms). Because of their potential to exert genome-wide trans effects via base pair homology of the short 24 nt siRNAs, the population of siRNAs expressed by a genome must be adapted to its gene content to ensure proper gene expression. Interspecific hybridization, therefore, brings together trans-regulators of TE expression that are not mutually adapted and could deeply disorganize gene regulation. Gene expression modifications following interspecific hybridization or allopolyploidization have been monitored across plant genera as diverse as Gossypium, Senecio, Brassica, Tragopogon, or Oryza (e.g., Hegarty et al. 2006; Buggs et al. 2009;Yoo et al. 2013). Many studies have concluded that epigenome incompatibilities are important contributors to hybrid sterility (Parisod et al. 2009; Ghani et al. 2014; Wang et al. 2014; Xu et al. 2014; Senerchia et al. 2015; Wu et al. 2016). In Aegilops hybrids, for example, parental divergence in TE content was associated with the emergence of new TE copies in hybrids (Senerchia et al. 2015). Allopolyploidy is also believed to be associated with drastic epigenetic reorganization although evidence for a significant reactivation of TE transposition remains scant (Parisod et al. 2010).
Our analysis contradicts the conclusions of these previous reports. Comparing the TE expression and epigenetic profile of the A. lyrata and A. thaliana genomes in parental and hybrid context, we find no evidence for a major transcriptomic or epigenomic “shock” driven by inappropriate TE silencing and epigenetic mechanisms. Both TE transcription and the decoration of TEs by the silencing histone mark H3K9me2 are globally unaffected in the hybrid. We observe that only a handful of specific TEs show a change in expression in hybrids. In addition, the changes in TE expression are generally of small magnitude. We further show that the distribution of silencing epigenetic marks is strongly correlated in parents and hybrids, and its variation does not match the changes in TE expression. Finally, under severe dehydration stress, a large number of genes change their transcriptional activity (Matsui et al. 2008). This broad genome-wide remodeling of transcriptional activity, however, far from inducing a severed genome shock, tends to increase the robustness of TE expression patterns to hybridization. These findings thus suggest that stress does not increase the impact of hybridization, further supporting the idea that the control of TE elements is robust to genome merging. Although these observations partially contradict previous reports, they are not isolated. In recent years, an increasing number of studies, mostly conducted in the Brassicaceae family, has begun to report cases where hybridization does not massively alter gene regulation (Akama et al. 2014; Shi et al. 2015; He et al. 2016; Zhang et al. 2016; Zhu et al. 2017). In addition, in tomato, transgressive expression of small RNAs was associated with the dysregulation of stress responsive genes, but not with that of TEs (Shivaprasad et al. 2012).
The contradiction between these studies and previous reports may have various explanations. First, it is striking to note that many of the studies that conclude on the absence of a global gene expression dysregulation were conducted in species with well-known genomes, where gene expression levels can be quantified with the best accuracy. They were not conducted in polyploids with large and imperfectly assembled genomes. Second, biological and experimental variance between replicates affects the strength of the correlation of expression in parental versus hybrid backgrounds, and thus the apparent global impact of hybridization. Finally, it is necessary to contrast diverse sources of information to test whether hybridization has a broad impact on gene regulation. In the allotetraploid A. suecica, modification in siRNA levels was not associated with nonadditive gene expression in newly formed allopolyploids, suggesting that changes in siRNA expression were not the result of epigenetic disorganization in the hybrid (Ha et al. 2009). Similarly, in Arabidopsis homoploid hybrids, we observed that hybrid-specific TE expression changes do not coincide with obvious epigenetic changes, a pattern also reported for protein-coding genes (Zhu et al. 2017). Taken together, the different molecular data sets used in this study show that TE expression and the distribution of epigenetic marks, which control gene expression, are not massively disorganized in hybrids.
trans- and cis-Acting Differences between Species Can Manifest in F1 Hybrids
The comparative analysis of gene expression in hybrids and their parents reflects the variability of trans- and cis-regulatory mechanisms controlling gene expression in the two species (Wittkopp et al. 2004). After hybridization, changes in total expression will simply reflect the number and magnitude of trans-regulatory differences between species, whereas the specific cis-regulatory variant of each transcript will result in allelic expression differences. These differences will manifest in hybrids on a restricted number of genes or elements, which does not imply that there is a major “genomic shock”.
Only trans-acting differences will have nonadditive consequences on expression. In fungi and cotton allopolyploid, >50% of homeologous genes inherit the expression pattern of the parents (Yoo et al. 2013; Cox et al. 2014). In rice, allelic expression bias in hybrids between Oryza sativa and O. japonica also reflects mostly interspecific differences. In hybrids between the diploid O. sativa and the tetraploid O. punctata, only 16% of the genes showed an expression pattern different from that of the parents (Wu et al. 2016). The analysis of protein-coding gene expression in A. thaliana × A.lyrata hybrids reported a larger proportion of nonadditive gene expression changes for the A. thaliana genome compared with the A. lyrata genome (Zhu et al. 2017). This pattern, like in our study, could not be associated with chromatin modifications induced by hybridization or any other genome-wide change in gene regulatory mechanisms (Zhu et al. 2017). However, the A. thaliana parental genotype Col-0 is missing an active allele of the major developmental regulator FRIGIDA. In fact, the A. thaliana allele of FLC, which is up-regulated by FRIGIDA, is one of the alleles most strongly impacted by hybridization (supplementary table 3, Supplementary Material online; Zhu et al. 2017). Interspecific differences in trans-acting factor activity will therefore determine the extent to which gene expression changes upon hybridization. If F1 hybrids are fertile, these changes can segregate in subsequent F2 generations as in, for example, Shivaprasad et al. (2012).
Here, focusing on the trans-acting effect of the passage from a native parental background to a hybrid background on TE expression, we show that at most 1–2% of expressed TEs have expression that is differentially controlled in trans. Several elements indicate that these trans-acting effects may be associated with interspecific differences in mechanisms of gene regulation in euchromatic regions. When combined in a hybrid, these trans-acting differences modify the regulatory environment of a few TE subfamilies. Indeed, the small proportion of TEs with differential expression in hybrids tend to be located in the vicinity of genes. Interspecific differences in the regulation of euchromatic regions are however likely to be minor, given the small number of elements affected.
Interspecific differences in gene regulation controlled in cis can also create a pattern of genome dominance in hybrids, with a majority of orthologous transcripts showing a transcription bias towards one parental genome (Yoo et al. 2013; Cheng et al. 2016; Zhang et al. 2016). For example, in A. thaliana × A. lyrata hybrids, a bias towards preferential expression of the A. lyrata allele has been reported by independent studies (He et al. 2012, 2016; Zhu et al. 2017). Cis-effects have not been examined in this study, because orthologous relationships cannot be established for most TEs, but for the subset of TEs that are located in orthologous positions in both species, the A. lyrata TE copy tended to be more highly expressed than the A. thaliana copy in the hybrid (He et al. 2012).
Incompatibilities Can Also Have an Oligogenic Basis
It may seem at first surprising that TE silencing shows so little disruption in interspecific A. thaliana × A. lyrata hybrids, given the estimated 6 My of divergence and their vastly different TE content (Hu et al. 2011; Hohmann et al. 2015). The probability to observe specific disruptions in gene expression in a hybrid background, however, does not necessarily scale with evolutionary distance. The proportion of genes with hybrid-specific changes in expression is not greater for hybrids between the closely related subspecies O. sativa and O. japonica than between O. sativa and the more distant species O. punctata (Wu et al. 2016). A simple oligogenic combination of alleles, in fact, can cause what is known as a Dobzhansky–Muller incompatibility, even in the absence of large evolutionary distances between parents. Dobzhansky–Muller incompatibilities, which can cause major disruption at the phenotypic and, sometimes at the transcriptome level, are not seldom within species, and were even detected within populations (Alcázar et al. 2010; Bomblies and Weigel 2010; Durand et al. 2012; Wright et al. 2013; Chae et al. 2014). We conclude that the term “shock” is not appropriate to describe the transcriptional consequences of the merging of divergent genomes in a viable F1 hybrid. Our data rather supports a less dramatic scenario, where the merging of two genomes creates a new trans-acting background, in which oligogenic Dobzhansky–Muller incompatibilities can manifest. Such incompatibilities are known to affect endosperm function (Rebernig et al. 2015) and will also occasionally affect the regulation of specific TEs and favor new bouts of transposition. Without a global robustness of TE regulation to genome merging, hybridization and polyploidization would probably be much less important for plant diversification (Alix et al. 2017). We note, however, that the impact of hybridization on TE transposition rate has not been examined in this study. It thus remains possible that TEs with undetectable expression level transpose at a rate that is higher in F1 interspecific hybrids than within species. Within A. thaliana, elements of about half of the TE families were shown to have recently transposed (Quadrana et al. 2016).
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank Franziska Turck (Max Planck Institute for Plant Breeding Research) for sharing her ChIP-seq protocol and experience. This research was supported by the Deutsche Forschung Gesellschaft (DFG) with grant ME2742/2-1 and ME2742/6-1 in the realm of the special study program SPP1529, as well as by the European Research Council with Grant 648617 “AdaptoSCOPE.” The raw data has been deposited in NCBI Sequence Read Archive (SRA) and are accessible through SRA study number SRP148881 and SRP148853 for RNA-seq and ChIP-seq data, respectively.
Literature Cited
- Akama S, Shimizu-Inatsugi R, Shimizu KK, Sese J.. 2014. Genome-wide quantification of homeolog expression ratio revealed nonstochastic gene regulation in synthetic allopolyploid Arabidopsis. Nucleic Acids Res. 426:e46.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcázar R, et al. , . 2010. Natural variation at Strubbelig Receptor Kinase 3 drives immune-triggered incompatibilities between Arabidopsis thaliana accessions. Nat Genet. 4212:1135–1139. [DOI] [PubMed] [Google Scholar]
- Alix K, et al. , . 2017. Polyploidy and interspecific hybridization: partners for adaptation, speciation and evolution in plants. Ann Bot. 1202:183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y.. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc B. 571:289–300. [Google Scholar]
- Berardini TZ, et al. , . 2015. The Arabidopsis Information Resource: making and mining the “gold standard” annotated reference plant genome. Genesis 538:474–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomblies K, Weigel D. 2007. Hybrid necrosis: autoimmunity as a potential gene-flow barrier in plant species. Nat. Rev. Genet. 85:382. [DOI] [PubMed] [Google Scholar]
- Bomblies K, Weigel D.. 2010. Arabidopsis and relatives as models for the study of genetic and genomic incompatibilities. Philos Trans R Soc B Biol Sci. 3651547:1815–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buggs RJ, et al. , . 2009. Gene loss and silencing in Tragopogon miscellus (Asteraceae): comparison of natural and synthetic allotetraploids. Heredity 1031:73–81. [DOI] [PubMed] [Google Scholar]
- Carroll TS, Liang Z, Salama R, Stark R, de Santiago I.. 2014. Impact of artifact removal on ChIP quality metrics in ChIP-seq and ChIP-exo data. Front Genet. 5:75.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavrak VV, et al. , . 2014. How a retrotransposon exploits the plant’s heat stress response for its activation. PLoS Genet. 101:e1004115.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae E, et al. , . 2014. Species-wide genetic incompatibility analysis identifies immune genes as hot spots of deleterious epistasis. Cell 1596:1341–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F, et al. , . 2016. Epigenetic regulation of subgenome dominance following whole genome triplication in Brassica rapa. New Phytol. 2111:288–299. [DOI] [PubMed] [Google Scholar]
- Chester M, et al. , . 2012. Extensive chromosomal variation in a recently formed natural allopolyploid species, Tragopogon miscellus (Asteraceae). Proc Natl Acad Sci U S A. 1094:1176–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox MP, et al. , . 2014. An interspecific fungal hybrid reveals cross-kingdom rules for allopolyploid gene expression patterns. PLoS Genet. 103:e1004180.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Chaux N, Tsuchimatsu T, Shimizu KK, Wagner A.. 2012. The predominantly selfing plant Arabidopsis thaliana experienced a recent reduction in transposable element abundance compared to its outcrossing relative Arabidopsis lyrata. Mobile DNA 31:2.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Meaux J, Pop A, Mitchell-Olds T.. 2006. Cis-regulatory evolution of Chalcone-Synthase expression in the genus Arabidopsis. Genetics 1744:2181–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, et al. , . 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 291:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand S, Bouché N, Perez Strand E, Loudet O, Camilleri C.. 2012. Rapid establishment of genetic incompatibility through natural epigenetic variation. Curr Biol. 224:326–331. [DOI] [PubMed] [Google Scholar]
- Gan ES, Xu Y, Ito T.. 2015. Dynamics of H3K27me3 methylation and demethylation in plant development. Plant Signal Behav. 109:e1027851.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghani MA, et al. , . 2014. The role of small RNAs in wide hybridisation and allopolyploidisation between Brassica rapa and Brassica nigra. BMC Plant Biol. 14:272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha M, et al. , . 2009. Small RNAs serve as a genetic buffer against genomic shock in Arabidopsis interspecific hybrids and allopolyploids. Proc Natl Acad Sci U S A. 10642:17835–17840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, et al. , . 2012. Widespread interspecific divergence in cis-regulation of transposable elements in the Arabidopsis genus. Mol Biol Evol. 293:1081–1091. [DOI] [PubMed] [Google Scholar]
- He F, et al. , . 2016. The footprint of polygenic adaptation on stress-responsive cis-regulatory divergence in the Arabidopsis genus. Mol Biol Evol. 338:2088–2101. [DOI] [PubMed] [Google Scholar]
- Hegarty MJ, et al. , . 2006. Transcriptome shock after interspecific hybridization in senecio is ameliorated by genome duplication. Curr Biol. 1616:1652–1659. [DOI] [PubMed] [Google Scholar]
- Henry IM, et al. , . 2014. The BOY NAMED SUE quantitative trait locus confers increased meiotic stability to an adapted natural allopolyploid of Arabidopsis. Plant Cell 261:181–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann N, Wolf EM, Lysak MA, Koch MA.. 2015. A time-calibrated road map of Brassicaceae species radiation and evolutionary history. Plant Cell 2710:2770–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollister JD, Gaut BS.. 2009. Epigenetic silencing of transposable elements: a trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res. 198:1419–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollister JD, et al. , . 2011. Transposable elements and small RNAs contribute to gene expression divergence between Arabidopsis thaliana and Arabidopsis lyrata. Proc Natl Acad Sci U S A. 1086:2322–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu TT, et al. , . 2011. The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat Genet. 435:476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Tam OH, Paniagua E, Hammell M.. 2015. TEtranscripts: a package for including transposable elements in differential expression analysis of RNA-seq datasets. Bioinformatics 3122:3593–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitonov VV, Jurka J.. 2004. Harbinger transposons and an ancient HARBI1 gene derived from a transposase. DNA Cell Biol. 235:311–324. [DOI] [PubMed] [Google Scholar]
- Kharchenko PK, Tolstorukov MY, Park PJ.. 2008. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat Biotechnol. 2612:1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MY, Zilberman D.. 2014. DNA methylation as a system of plant genomic immunity. Trends Plants Sci. 195:320–326. [DOI] [PubMed] [Google Scholar]
- Lafon-Placette C, Köhler C.. 2015. Epigenetic mechanisms of postzygotic reproductive isolation in plants. Curr Opin Plant Biol. 23:39–44. [DOI] [PubMed] [Google Scholar]
- Landt SG, et al. , . 2012. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 229:1813–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods. 94:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. , . 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2516:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockton S, Gaut BS.. 2010. The evolution of transposable elements in natural populations of self-fertilizing Arabidopsis thaliana and its outcrossing relative Arabidopsis lyrata. BMC Evol Biol. 10:10.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui A, et al. , . 2008. Arabidopsis transcriptome analysis under drought, cold, high-salinity and ABA treatment conditions using a tiling array. Plant Cell Physiol. 498:1135–1149. [DOI] [PubMed] [Google Scholar]
- McCarthy DJ, Chen Y, Smyth GK.. 2012. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 4010:4288–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock B. 1984. The significance of responses of the genome to challenge. Science 2264676:792–801. [DOI] [PubMed] [Google Scholar]
- Nordberg H, et al. , . 2014. The genome portal of the department of energy joint genome institute: 2014 updates. Nucleic Acids Res. 42:D26–D31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisod C, et al. , . 2009. Rapid structural and epigenetic reorganization near transposable elements in hybrid and allopolyploid genomes in Spartina. New Phytol. 1844:1003–1015. [DOI] [PubMed] [Google Scholar]
- Parisod C, et al. , . 2010. Impact of transposable elements on the organization and function of allopolyploid genomes. New Phytol. 1861:37–45. [DOI] [PubMed] [Google Scholar]
- Pietzenuk B, et al. , . 2016. Recurrent evolution of heat-responsiveness in Brassicaceae COPIA elements. Genome Biol. 171:209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadrana L, et al. , . 2016. The Arabidopsis thaliana mobilome and its impact at the species level. eLife 5:e15716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangwala SH, Richards EJ.. 2010. The structure, organization and radiation of Sadhu non-long terminal repeat retroelements in Arabidopsis species. Mobile DNA 11:10.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebernig CA, et al. , . 2015. Non-reciprocal interspecies hybridization barriers in the Capsella genus are established in the endosperm. PLoS Genet. 116:e1005295.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK.. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 261:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, Oshlack A.. 2010. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senerchia N, Felber F, Parisod C.. 2015. Genome reorganization in F1 hybrids uncovers the role of retrotransposons in reproductive isolation. Proc Biol Sci. 2821804:20142874.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki M, et al. , . 2002. Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and high-salinity stresses using a full-length cDNA microarray. Plant J. 313:279–292. [DOI] [PubMed] [Google Scholar]
- Seymour DK, Koenig D, Hagmann J, Becker C, Weigel D.. 2014. Evolution of DNA methylation patterns in the Brassicaceae is driven by differences in genome organization. PloS Genet. 1011:e1004785.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequeira-Mendes J, et al. , . 2014. The functional topography of the Arabidopsis genome is organized in a reduced number of linear motifs of chromatin states. Plant Cell 266:2351–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Zhang C, Ko DK, Chen ZJ.. 2015. Genome-wide dosage-dependent and -independent regulation contributes to gene expression and evolutionary novelty in plant polyploids. Mol Biol Evol. 329:2351–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivaprasad PV, Dunn RM, Santos BA, Bassett A, Baulcombe DC.. 2012. Extraordinary transgressive phenotypes of hybrid tomato are influenced by epigenetics and small silencing RNAs. EMBO J. 312:257–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis PS, Soltis DE.. 2009. The role of hybridization in plant speciation. Annu Rev Plant Biol. 60:561–588. [DOI] [PubMed] [Google Scholar]
- Spoelhof JP, et al. , . 2017. Karyotypic variation and pollen stainability in resynthesized allopolyploids Tragopogon miscellus and T. mirus. Am J Bot. 10410:1484–1492. [DOI] [PubMed] [Google Scholar]
- Todesco M, et al. , . 2016. Hybridization and extinction. Evol Appl. 97:892–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, et al. , . 2014. Rapid genetic and epigenetic alterations under intergeneric genomic shock in newly synthesized Chrysanthemum morifolium × Leucanthemum paludosum hybrids (Asteraceae). Genome Biol Evol. 61:247–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittkopp PJ, Haerum BK, Clark AG.. 2004. Evolutionary changes in cis and trans gene regulation. Nature 4306995:85–88. [DOI] [PubMed] [Google Scholar]
- Wright KM, Lloyd D, Lowry DB, Macnair MR, Willis JH.. 2013. Indirect evolution of hybrid lethality due to linkage with selected locus in Mimulus guttatus. PLoS Biol. 112:e1001497.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, et al. , . 2016. Transcriptome shock in an interspecific F1 triploid hybrid of Oryza revealed by RNA sequencing. J Integr Plant Biol. 582:150–164. [DOI] [PubMed] [Google Scholar]
- Xiong Z, Gaeta RT, Pires JC.. 2011. Homoeologous shuffling and chromosome compensation maintain genome balance in resynthesized allopolyploid Brassica napus. Proc Natl Acad Sci U S A. 10819:7908–7913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, et al. , . 2014. Genome-wide disruption of gene expression in allopolyploids but not hybrids of rice subspecies. Mol Biol Evol. 31:1066–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo MJ, Szadkowski E, Wendel JF.. 2013. Homoeolog expression bias and expression level dominance in allopolyploid cotton. Heredity (Edinb) 1102:171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, et al. , . 2016. Genome-wide gene expressions respond differently to A-subgenome origins in Brassica napus synthetic hybrids and natural allotetraploid. Front Plant Sci. 7:1508.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, et al. , . 2017. Altered chromatin compaction and histone methylation drive non-additive gene expression in an interspecific Arabidopsis hybrid. Genome Biol. 181:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.